Abstract
The study of radiation effects in DNA is a multidisciplinary endeavour, connecting the physical, chemical and biological sciences. Despite being mostly filtered by the ozone layer, sunlight radiation is still expected to (photo)ionise DNA in sizeable yields, triggering an electron removal process and the formation of potentially reactive cationic species. In this manuscript, photoionisation decay channels of important DNA tautomeric derivatives, 7H-adenine and 7H-guanine, are characterised with accurate CASSCF/XMS-CASPT2 theoretical methods. These simulation techniques place the onset of ionisation for 7H-adenine and 7H-guanine on average at 8.98 and 8.43 eV, in line with recorded experimental evidence when available. Cationic excited state decays are analysed next, uncovering effective barrierless deactivation routes for both species that are expected to decay to their (cationic) ground state on ultrafast timescales. Conical intersection topographies reveal that these photoionisation processes are facilitated by sloped single-path crossings, known to foster photostability, and which are predicted to enable the (VUV) photo-protection mechanisms present in these DNA tautomeric species.
1. Introduction
The study of radiation induced DNA damage is a topic of utmost interest to a broad range of disciplines as it is known to mediate in the formation of lesions [1] that lead to mutations and to healthcare concerns such as skin cancer melanoma [2]. An enhanced understanding of these processes has been achieved over the years by studying DNA excited state dynamics triggered upon radiation exposure over a wide range of derivatives: from its monomers the nucleobases (and nucleosides/tides), [3] to dimers [4,5,6], single- and double-strands [7,8] and more recently other relevant motifs like guanine quadruplexes [9,10]. This has helped uncover an intricate excited state reactivity landscape triggered in our genetic material upon light absorption that is mediated by a mixture of localised and excitonic (delocalised) states [11], and that spreads across spin multiplicities [3,12].
Work reported in the literature thus far has focused on the study of DNA reactivity upon UV-light exposure [13,14], as it is often favoured due to its relation with sunlight-mediated lesions [15,16].
Photoionisation, on the other hand, is less studied but has recently gained attention due to the development of light sources capable of generating and monitoring photo-ionised (cationic) species [17,18]. Cationic states are known to be formed not only directly upon photoionisation but also in many charge transfer and separation processes mediating DNA damage [19,20,21] and repair [22]. Moreover, recent experiments with guanine-rich sequences have revealed the formation of significant cationic yields even when irradiating with UV-B light sources [10,23], which possess longer wavelength than those required to ionise DNA nucleobases. This means the onset of ionisation might be energetically well below the ionisation potential of the nucleobases [24], making them a more widespread event than initially thought and with sizeable involvement in UV-induced DNA photophysics [19,20,21]. Uncovering how cationic species evolve and react is thus of interest to understand DNA photo-processes occurring upon radiation exposure and is the main goal of this work.
The reactivity of the ensuing DNA nucleobase cationic species still remains vastly unexplored, despite recent efforts with state-of-the-art instrumentation on this front [25,26]. Whereas pyrimidine nucleobases have been studied in relative detail [27,28,29,30], purines are less explored perhaps due to their more complex electronic structure [31], studies on their tautomeric forms being even scarcer.
DNA non-canonical nucleobases, like the 7H tautomeric forms of DNA nucleobases adenine and guanine studied in this work (see Figure 1), feature in non-negligible yields in our genetic material and possess markedly different electronic properties, which are known to affect their reactivity [32]. These species are rarely studied in the literature in their singlet form [32], and even less so in the cationic (doublet) manifold, despite being important to describe some of the photo-processes underpinning DNA damage and repair. The study of these species is however fundamental to understand how prebiotic extreme UV-light exposure in DNA nucleobases, and their ability to withstand such radiation, may have played a role in selecting our genetic lexicon [13,14,33].
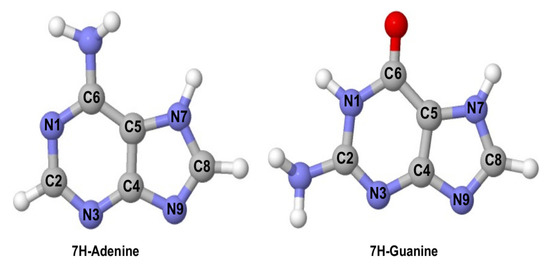
Figure 1.
Molecular structure of 7H-adenine (left) and 7H-guanine (right) and their atom labelling.
This is particularly relevant in 7H-guanine, as it has been reported to be the most stable guanine tautomer in the gas phase [34,35]. This higher stability of the 7H tautomer of guanine in the gas phase is something that has, however, been contested over the years: in [36], a comparison of theoretical estimates with photoelectron spectra appears to support a 7H-guanine assignment as the most stable tautomer [35,37,38], but experiments in molecular beams appear to favour the 9H tautomer based on recorded IR spectroscopic signals instead [36,39]. This suggests the specific conditions in which guanine is measured are critical in this tautomeric equilibrium.
In this work photoionisation processes on DNA purine tautomeric 7H species (7H-adenine and 7H-guanine, Figure 1) are studied (to our knowledge) for the first time using accurate multireference perturbation theory (CASPT2) modelling techniques. We focus on studying the 7H tautomers of both adenine and guanine, as these feature several (accessible) cationic electronic excited states upon ionisation, making the comparison between analogous tautomers featuring different purine-substituted frames more interesting in terms of understanding how chemical substitution impacts their photophysics. These systems are nevertheless of interest due to their strong resemblance with our current genetic lexicon, and studying their photo-protection mechanisms may eventually help us understand how our current nucleobases were chosen during prebiotic times and whether photostability played a critical role in it [13,14]. We analyse their ionisation potentials, where we observe systematic blue-shifts in the values for and red-shifts for states, the latter being closer to the experimental evidence, and estimate the onset of ionisation to be at 8.98 and 8.43 eV for 7H-adenine and 7H-guanine, respectively. Cationic excited state decays are predicted to be effectively barrierless and thus very short-lived (ultrafast) for both systems, though the details of the decay mechanisms are different. 7H-guanine displays localised distortions in the 5-membered right along the decay, whereas 7H-adenine features delocalised motions across the whole molecular scaffold and presents an accessible conical intersection less than a tenth of an eV from the cationic ground state, which is expected to facilitate a potential chemiexcitation channel. The characterised sloped and single-path conical intersection topographies support ultrafast deactivation channels present for both systems, which are expected to be central in fostering photostability, and that suggest (VUV) photo-protection mechanisms in DNA nucleobases might also be available to (at least some of) their tautomeric derivatives.
2. Results
The results are laid out as follows: we analyse the ionisation potentials of 7H-adenine and 7H-guanine first and move next to the study of their cationic excited state decay upon photoionisation.
2.1. Ionisation Potentials
Figure 2 displays the ionisation potentials obtained for the 7H tautomeric species considered in this study, with a comparison to recorded experimental evidence where appropriate and available.
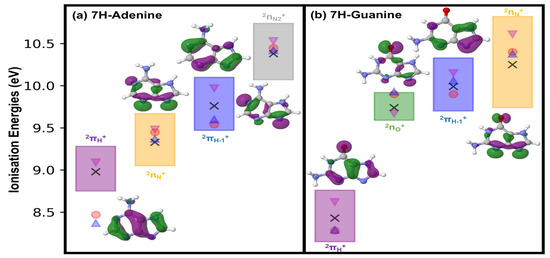
Figure 2.
Schematic representation of the gas phase vertical ionisation potentials of (a) 7H-adenine and (b) 7H-guanine, computed with a range of zeroth-order CASPT2 Hamiltonians. The uncertainty range spanned by the different zeroth-order Hamiltonians is provided. CASPT2 average values are given as black crosses, magenta inverted triangles represent XMS-CASPT2(IPEA = 0.0) estimates used in Section 2.2, red dots denote experimental evidence found in the literature for guanine [37,40] and 9H-adenine [41], and blue triangles refer to EOM-IP/CCSD/cc-pVTZ-dff//RI-MP2/cc-pVTZ-dff theoretical estimates of Bravaya et al. [35]. Purple squares denote ionisation energy ranges for , green for , blue for , orange for and grey for states. Associated singly occupied molecular orbitals (SOMOs) depicting the different cationic states are also provided. Specific values for each CASPT2 formulation are provided in Tables S1 and S2 in the Supplementary Materials.
The first cationic state in 7H-purine derivatives is characterised by the state, which corresponds to an unpaired electron in the singly occupied molecular orbital (SOMO). This is in line with what has been observed for other DNA/RNA nucleobase systems, where the SOMO is always the one embodying the first (i.e., energetically lowest) electron removal upon ionising radiation exposure [24,27,28,29,30]. This state is placed at 8.98 and 8.43 eV on average for 7H-adenine and 7H-guanine respectively, and feature similar standard deviations, with respect to the choice of CASPT2 zeroth order Hamiltonian, of 0.19 and 0.22 eV. These average ionisation values compare well with the available experimental evidence reported for guanine [37,40], which several works attribute to signals coming from the 7H tautomer [35,38]. On the other hand, it differs more significantly from estimates available for adenine that are however referred to the 9H canonical species in both theory [35] and experiment [41]. Particularly the average CASPT2 value obtained for 7H-guanine, the more comparable case, is in agreement with those reported experimentally [40] and theoretically [35] in the literature, being blue-shifted by around a tenth of an eV off those estimates.
The second ionisation potential embodies in both cases lone pair (also referred to as ) [35] states, of character for 7H-adenine and (mostly) for 7H-guanine, which are placed on average at 9.33 and 9.74 eV, respectively. As can be seen in the SOMOs provided in Figure 2, these states feature delocalisations over several lone-pair sites, which affect particularly 7H-guanine as it leads to a state of and mixed character, even if is more prominent and thus used to label the state. The associated variances arising from different CASPT2 formulations seem to differ more prominently, leading to 0.22 eV for 7H-adenine and a smaller 0.13 eV for 7H-guanine. The (averaged) ionisation potential obtained for 7H-guanine is red-shifted by ∼0.2 eV compared to experimental [40] and theoretical reference estimates [35], and red-shifted as well for 7H-adenine by ∼0.1 eV. This suggests changes in the 7H tautomer appear to affect less prominently states, as theoretical [35] and experimental [41] estimates for the 9H-tautomer seem to fit those obtained here for the 7H species.
The third cationic state corresponds to electron removal from 2 and is placed on average at 9.76 and 9.99 eV for 7H-adenine and 7H-guanine, respectively, with an associated standard deviation of 0.23 eV in both cases. Interestingly, averaged values are in this case are blue-shifted from experiment, by 0.18 eV in 7H-adenine and by 0.09 eV in 7H-guanine, being close to experimental [40,41] and reference theoretical [35] estimates.
The fourth and last cationic state analysed corresponds to ionisation in lone pair states, of character for both systems, leading to for 7H-adenine and for 7H-guanine. These are predicted to be placed (on average) at 10.38 and 10.25 eV for 7H-adenine and 7H-guanine, respectively, with associated standard deviations of 0.24 and 0.40 eV. These ionisation energies are red-shifted with respect to experimental [40,41] and theoretical [35] evidence by less than 0.1 eV in 7H-adenine, keeping in mind the experiment actually refers to 9H-adenine [41], and by ∼0.15 eV in 7H-guanine.
Upon inspection of the different estimates obtained with diverse zeroth-order Hamiltonians used (and reported in Tables S1 and S2), we select the XMS-CASPT2(IPEA = 0.0) formulation for subsequent calculations in Section 2.2: it shows qualitative agreement (slightly blue-shifted estimates) with available experiments as seen in the magenta inverted triangles in Figure 2, and it has been reported to be essential when accurately describing interstate crossings [42].
2.2. Excited State Decays
It has been recently observed that, upon ionisation, an electron might be removed from different molecular orbitals creating diverse cationic species with similar probabilities [43,44]. This makes the choice of the initial state created in our model somewhat ambiguous, as any of the states (or linear combinations of them) are in principle possible. We here assume an electron removal from the highest-lying cationic state considered as done in previous studies [29,30,45]. This enables exploring all critical structures along the potential energy surface until reaching the cationic ground state thus providing an overview of the different pathways potentially triggered in this complex photo-process.
All critical structures found along the cationic excited state decay of 7H-adenine and guanine remain in-plane. This is shared with what we and others have found for pyrimidine nucleobases and derivatives [28,29,30,46], and justifies focusing on evaluating distortions along the different bond lengths affected during the photo-process.
Figure 3 displays the potential energy profiles obtained for 7H-adenine. Upon ionisation of the highest-lying state, a rapid decay is predicted with an associated ∼0.7 eV stabilisation, with significant 0.03 Å elongations of N1-C6, C4-C5 and C8-N9 and shortening of the C8-C9 bonds until reaching . At this crossing, population is expected to transfer to the state and further decay to its shallow minimum (), which entails a slight 0.02 Å contraction of N1-C2 and an elongation of C7-N9 and that is placed at 0.64 eV adiabatically from the cationic ground state minimum ().
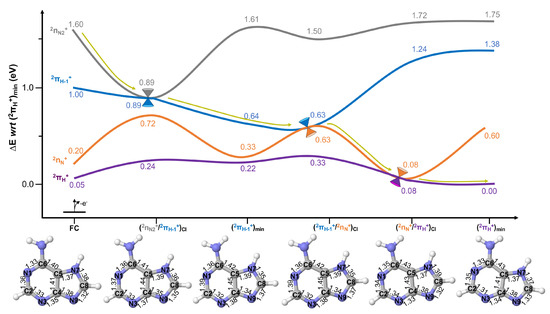
Figure 3.
Potential energy surfaces of 7H-adenine computed at the XMS-CASPT2 level of theory. All energies are given in eV with respect to (). Yellow arrows represent the evolution of the excited state population assuming initial activation of the highest-lying (grey) state. XMS-CASPT2 optimised structures are provided to display the nuclear displacements along the photo-reaction, with bond distances given in Å.
From the can be reached barrierlessly by relaxing just a further 0.01 eV, being almost identical in structure to , and displaying changes in their structure only beyond the pm. funnels the excited state population to the state, that does not feature a minimum and is thus predicted to decay swiftly to mediating the decay to the ground state. This crossing, which mediates the final part of the decay, is placed at just 0.08 eV from the cationic ground state minimum, featuring pronounced 0.05 Å N1-C2, N3-C4 and C8-N9 bond contractions and a 0.04 Å N7-C8 elongation from the previous crossing .
The population is then finally transferred to the cationic ground state , which entails a further 0.03 Å shortening of C2-N3 and 0.02 of C4-C5 and N7-C8 bonds. These small distortions encompass the strong structural resemblance with , as well as the energetic proximity: this implies re-crossings are possible just from available thermal energy potentially enabling chemiexcitation [47] events to populate back the cationic excited state.
The potential energy curves depicting photoionised excited state decay in 7H-guanine are given in Figure 4. As can be seen, upon ionisation to the highest-lying computed () state, situated at 2.07 eV adiabatically from the cationic ground state, a swift decay is predicted to reach placed at 1.80 eV and transfer population to the state. This decay is mediated by very small (∼0.02 Å) in-plane bond length rearrangements across the whole structure, which indicates the proximity of this structure to the equilibrium Franck–Condon (FC) region. It is also interesting to note that and states are practically degenerate energetically at the FC equilibrium region, and either state could thus be expected to initially be populated. However, they are both expected to lead to the population of the state, either direct or indirectly (via ) upon ionising radiation exposure.
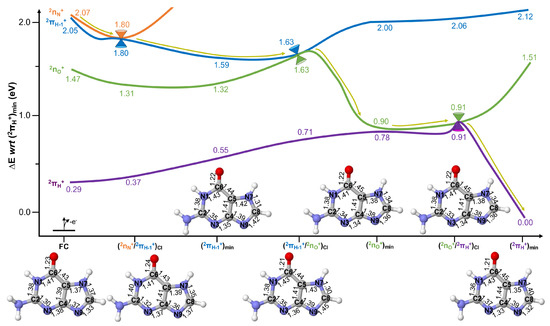
Figure 4.
Potential energy surfaces of 7H-guanine computed at the XMS-CASPT2 level of theory. All energies are given in eV with respect to (). Yellow arrows represent the evolution of the excited state population assuming initial activation of the highest-lying (orange) state. XMS-CASPT2 optimised structures are provided to display the nuclear displacements along the photo-reaction, with bond distances given in Å.
Upon populating , further relaxation is predicted until reaching , placed at 1.59 eV and encompassing significant changes that are in this case largely localised in the 5-member ring: elongations of 0.05 Å for C8-N9 and 0.03 for C4-N9 and C5-N7 bonds and shortenings of 0.04 Å for N7-C8 and 0.03 for C4-N9 bonds. This is in line with the more localised character of the SOMO describing the state over the five-member ring, which is depicted in Figure 2.
From a very small (0.04 eV) barrier is predicted to be surmounted to reach mediating population transfer to the state and placed at 1.63 eV. This crossing is characterised by a further (0.03 Å) elongation of the C8-N9 bond.
Upon populating , very significant relaxation (∼0.7 eV) is predicted until reaching , placed at 0.90 eV adiabatically from the cationic ground state. This minimum encompasses further (and very pronounced) localised distortions in the 5-member ring: C5-N7 and C8-N9 shorten by 0.05 and 0.09 Å, respectively, an an elongation of the N7-C8 bond of 0.04 Å. It is important to note that this minimum structure features a very small energy difference with the ground cationic state, which could potentially be part of the intersection seam. Indeed, this conical intersection is separated from by a mere 0.01 eV featuring an almost identical structure.
The crossing is expected to be reached almost barrierlessly (i.e., there is a negligible 0.01 eV barrier to be surmounted) and to mediate an efficient excited state decay to the cationic ground state, which is predicted to rapidly decay to where population is predicted to remain. The cationic ground state minimum encompasses significant structural rearrangements which are again partly localised in the five-member ring but that in this case expand across the molecular scaffold. This is encompassed by significant 0.06 Å elongations in N7-C8, 0.04 C2-N3 and 0.03 for the C4-C5 bonds, and shortenings of 0.04 Å for C8-N9 and 0.03 for C5-N7 bonds.
Conical Intersection Characterisation
The topography of the characterised conical intersections is analysed next, to provide complementary insights into the excited state decay mechanisms of 7H-purine DNA photoionisations.
To assess topographies of conical intersections and compare them we employ a series of parameters used by Fdez Galván et al. in a recent study [48]. Table 1 features estimates for pitch (), asymmetry (), relative tilt (), and tilt heading (), computed from the gradient difference and non-adiabatic coupling vectors obtained at the converged conical intersection geometries. For intersection analyses we use branching plane vectors so that and , as this makes them comparable to one another [48]. The values obtained are employed to classify the topography as peaked/sloped, and bifurcating/single-path, based on and values [48] which are related to those originally proposed by Ruedenberg and co-workers [49]. Values of < 1 lead to peaked topographies and >1 to sloped ones, whereas < 1 refers to bifurcating character and >1 to single-path [48].

Table 1.
Characterised conical intersection parameters for the different crossings of 7H-adenine and 7H-guanine obtained at the XMS-CASPT2 level of theory. Pitch (), asymmetry () and relative tilt () are given in atomic units, whereas tilt headings () are provided in degrees.
As can be seen in Table 1, the conical intersection mediating decay to the cationic ground state in 7H-adenine and 7H-guanine, which refer to and respectively, feature both sloped and single-path character that have been suggested to foster photostability [50,51]. These conical intersection structures have similar associated pitch values and large asymmetries while featuring negligible tilt heading angles, which supports distortions mostly arising from in-plane motions as those described above.
The second conical intersection, , which mediates → population transfer in 7H-adenine, also displays a sloped and single-path character. This crossing features a large asymmetry close to 1 (that would refer to completely asymmetric) and a slight tilt angle, with borderline and values that make it sloped and single-path. in 7H-guanine mediates → decay and strongly resembles the sloped and single-path topography displayed by . These further support ultrafast deactivation mechanisms in both 7H-adenine and 7H-guanine, which in turn support the photo-protection mechanisms associated to these species [13,14,33].
The last conical intersection considered in this study connects the initially accessed () with the state in 7H-adenine (7H-guanine), and mediates the initial part of the photoionisation process. These conical intersections present more significant changes, featuring the smallest relative tilts and leading to a peaked and bifurcating character. This suggests the emergence of potential re-crossings (and population transfers back and forth) leading to a wave packet splitting on two different directions, which is expected to embody the slower component of the decay.
3. Discussion
A few trends emerge when considering the simulation of ionisation potentials for 7H-adenine and 7H-guanine using different multireference perturbation theory approaches as in the present study: the averaged (CASPT2) values obtained are blue-shifted with respect to experimental evidence and other more correlated (EOM-IP-CCSD) theoretical approaches when considering states, while they are red-shifted for lone-pair states. There also seems to be a better agreement between the simulated states and recorded experimental evidence, which might be due to the lesser reliance of (covalent) lone-pair states on the dynamic electron correlation included in the model as compared to the strong dependence of delocalised (ionic) states [52,53,54]. It is also worth noting that the computed standard deviation of the state in 7H-guanine is twice as big as that modelled for any other state considered in this study. This is partly due to the mixed character of the state, which can be easily seen by the delocalisation of the SOMO orbital over both and sites (see Figure 2), and which leads to large off-diagonal elements in multistate and extended multistate formulations of CASPT2 [42], as well as to lower weight references in its single-state variant that also affect the overall accuracy [55]. The values of the off-diagonal elements, together with the leading configuration state functions characterising the different wave functions, are provided in Tables S3–S8 in the supporting information. Significant couplings are observed between and states, which can also be rationalised by the different weights contributing to the diverse cationic states studied featuring a 50%/20% / character at the multistate (as well as single-state) level but that is uncoupled upon employing the extended multistate variant. This, together with the closer ionisation potentials obtained for XMS-CASPT2(IPEA = 0.0) with respect to both CASPT2 average and the recorded evidence (see Table S2 in the Supplementary Materials) justify its choice as the method used throughout for optimisations.
It should be stated that more accurate approaches than those of Bravaya et al. [35] used here as reference, have been reported for modelling the ionisation potentials of these species in the literature: the work of Ortiz and co-workers employing the P3 method [56] in 9H-adenine [57] and guanine [38] has been shown to match experiments within under a tenth of an eV. The EOM-IP-CCSD approach of Bravaya et al. [35] was however deemed a better comparison for this study as it is grounded on many body perturbation theory [58,59], as is the CASPT2 method [60], while including more correlation as well as featuring a larger basis set. This reveals a lack of correlation in the model when describing (ionic) states, which has been shown to be partially corrected upon increasing the zeroth-order Hamiltonian (i.e., the active space size) [61,62], and that explains the larger deviations registered when comparing with experiment particularly for states. Additionally, it should be noted that more recent experimental results were found compared to those of Lin et al. [40] used as reference in Figure 2: the recorded evidence by Zaytseva et al. [37] measured almost four decades later, qualitatively and almost quantitatively (all photoelectron measurements being within 0.05 eV on average with the largest deviation being below 0.1 eV) agrees with the original measures of Lin et al. [40] thus validating the experimental reference employed.
Whereas all values used (as well as those computed) in this study refer to the 7H tautomer of guanine, we could not find data on the ionisation potentials of 7H-adenine so we used those available for 9H-adenine instead [41], which makes comparisons with the computed 7H-adenine data much less straight forward. As can be seen in Figure 2, differences in the ionisation potentials of 7H- (average CASPT2 values in black crosses) and 9H-adenine (blue triangles (theory) [35] and red circles (experiment) [41]) seem to be small for all but the lowest-lying state, where a significant ∼0.5 eV difference is observed. This is however in line with previous theoretical estimates found in the literature using density functional theory and the B3LYP functional, which predict a ∼0.4 eV blue-shift on the state when going from 9H- to 7H-adenine [63]. This large deviation between the computed values and the experimental evidence [41] thus arises due to different tautomeric species being measured, rather than the lack of accuracy of the model.
An aspect not considered in this study is how the solvent affects ionisation in these species. Experimental and theoretical estimates for DNA nucleobases and nucleosides recorded in the literature point towards substantial red-shifts of the ionisation energies when in water solution [64,65]. To assess this, not only on ionisation potentials but also along the cationic excited state decays. Moreover, different mechanisms such as proton-coupled electron transfer (PCET) with the environment are expected to dominate ionisation in solution, having been summarised elsewhere [66]. More sophisticated approaches are thus required [67,68,69] to study these processes, which are out of the scope of this manuscript and that will be considered in future work.
Upon photoionisation, it is predicted that 7H-adenine will undergo an ultrafast step-wise →→ decay, which is analogous to what has been observed for other DNA species [28,29,30]. A major difference, however, is brought by the very small energies (<0.1 eV) separating and critical structures (see Figure 3), which effectively enable a ⇌ equilibrium where both states could in principle present significant populations after decay to the ground state. Given the small energies separating them, this is likely to occur even when very little thermal energy is available and despite the state not presenting a well-defined minimum, thus paving the way for a potential population of the excited cationic state via chemiexcitation [47]. It is worth noting that, due to the elevated computational cost of XMS-CASPT2 gradients and non-adiabatic couplings, a reduced active space had to be used. Upon inspection of the obtained off-diagonal elements (see Tables S3 and S7 in the Supplementary Materials), we observe a relative artificial increase of this magnitude due to the smaller zeroth-order reference function, which, however, remains small enough to not bias the ensuing optimisations.
Interestingly, the decay mechanism outlined above (aside from the chemiexcitation channel accessible at the cationic ground state minimum) is similar to what has been found recently by Zhao et al. on the deactivation of 9H-adenine: [70] all low-lying cationic states appear to be vibronically coupled, which might explain the ultrafast timescales observed in the 9H species, and that are here suggested to apply also to the 7H tautomer.
7H-guanine, on the other hand, presents a predicted step-wise and ultrafast →→→ decay to the (cationic) ground state. This swift decay route is expected despite the associated potential energy barriers predicted along and state deactivations, which are however too small (0.04 eV for the former and 0.01 eV for the latter) to hamper decay.
Interestingly, despite involving ionisations mostly localised in the carbonyl moiety (i.e., the state), 7H-guanine does not display significant C=O bond elongations, which is the main reaction coordinate in cationic pyrimidine nucleobases [27,28,29,30]. Another distinctive feature of 7H-guanine and its decay is that geometrical distortions along the deactivation, at a difference from 7H-adenine, localise mostly over the five-member ring: this is a feature that has also been observed in other guanine derivatives such as thienoguanosine while studying their (singlet) excited state decays [71].
Reactivity in the cationic manifold is markedly different from what has been observed in the neutral singlet species: whereas 7H-adenine [72,73] and 7H-guanine [74] are reported to feature a potential energy barrier along the reaction coordinate hampering decay, which reflects experimentally in longer decay times [75], cations are barrierless and thus expected to decay rapidly to the (cationic) ground state. It is also worth noting that, whereas 7H DNA purines (as well as their biologically relevant 9H counterparts) feature strong out-of-plane motions along the decay in their singlet manifold [76], cations remain planar in line with what has been previously observed for other canonical DNA species [27,28,29,30].
The optimised conical intersection topographies highlight that despite featuring different lone pair states (i.e., localised on different atoms), the lowest-lying crossings of both 7H-adenine and 7H-guanine nevertheless display very strong similarities. This is also observed for the highest-lying crossing studied, and for 7H-adenine and 7H-guanine respectively, which again entail different lone pair cationic states but that lead to analogous intersection topographies that strongly resemble one another, thus pointing at symmetry (i.e., vs ) as one of the main factor governing these crossings and thus their efficient decay [77,78].
The results also seem to point at the ability of the ground cationic state to lead to sloped single-path intersections, which are known to foster photostability [50,51], while other states like lead upon crossing with different symmetry lone-pair states to peaked and bifurcating topographies instead.
Photostability refers in this case to the unique ability shown by the nucleobases [3] to divert the (cationic) excited state wave packet created upon absorption to the ground state in ultrafast timescales, reducing the time spent in the more reactive electronic excited states that may lead to photo-damaging reactions [1]. This is supported by the topographies of the conical intersections mediating decay to the ground state, as they feature the (only and) preferential decay direction towards the regeneration of the (cationic) ground state structure thus helping avoid photodegradation [13,14]. This is a well-known feature in the singlet manifold [3,13,14,79,80,81], which is relatively unique to DNA nucleobases and that justifies their resilience to radiation, and has also been observed in other DNA cationic systems [29,30]. It is important to note, however, that ultrafast deactivations in the cationic manifold might be more frequent in organic molecular systems than in the singlet due to the smaller energy gaps between states [50,51], its role towards DNA photostability thus being less evident and a possibility that we are currently investigating.
In this study, we have favoured a thorough study of the potential energy landscape, characterising the different accessible conical intersections mediating decay, as that allowed us to employ accurate XMS-CASPT2 methodologies to uncover the qualitative traits of these photo-processes. Another option would have been to use non-adiabatic molecular dynamics, such as those based on cost-effective surface hopping approaches. Non-adiabatic molecular dynamics would thus provide temporal estimates to the photo-processes studied, but at present we would have to rely on simpler (i.e., less accurate) electronic structure potential energy surfaces due to their elevated cost.
The present study provides an adequate reference for the decay channels available to both 7H adenine and guanine species, with the caveat that only conical intersection topographies are considered and not how the intersection themselves are reached [82]. This is known to significantly influence decay [83], and is an important aspect that can be extracted from dynamics simulations and that will be explored in future work.
In summary, we observe markedly different decay pathways in 7H-adenine and 7H-guanine, which however are expected to lead in both cases to ultrafast deactivations that are ultimately (i.e., on the last step of the decay) mediated by sloped and single-path conical intersections with the cationic ground state known to foster photostability [50,51]. These decay mechanisms differ in the specifics but lead to the same conclusions as those previously characterised for pyrimidine nucleobase cations [29,30], which highlight the resilience of our genetic material against VUV light radiation that we here tentatively also extend to purine tautomeric DNA bases.
4. Computational Details
OpenMolcas [84,85] was used to model the ionisation potentials reported, making use of the complete active space self-consistent field (CASSCF) [86,87] and its second-order perturbation theory extension (CASPT2) [60]. For these simulations, an Atomic Natural Orbital (ANO) large basis set with a double- polarised contraction was used [88,89].
The active spaces for modelling the ionisation potentials contained all valence and lone pair orbitals, as well as all valence unoccupied orbitals, totalling 18 electrons in 13 orbitals for 7H-adenine (17 in 13 for cations) and 20 electrons in 14 orbitals for 7H-guanine (19 in 14 for cations). CASSCF wave functions were averaged over five doublet states and were subsequently used for single-point CASPT2 energy corrections employing the single-state (SS) [90,91], multistate (MS) [92], and extended multistate (XMS) [93] variants. An imaginary level shift of 0.2 a.u. was employed in the perturbative step to avoid the presence of intruder states [94], and IPEA shifts [95] of 0.0 and 0.25 a.u. were both tested, resulting in 6 different CASPT2 zeroth-order Hamiltonians: SS-CASPT2(IPEA = 0.0), SS-CASPT2(IPEA = 0.25), MS-CASPT2(IPEA = 0.0), MS-CASPT2(IPEA = 0.25), XMS-CASPT2(IPEA = 0.0) and XMS-CASPT2(IPEA = 0.25).
To aid the discussion of ionisation energies, we have chosen to average the six different CASPT2 formulations as this provides the mean value as well as the standard deviation expected by modifying the zeroth-order Hamiltonian.
Relaxation pathways (cationic ground and excited state minima, as well as conical intersections) were characterised using BAGEL [96,97] and a cc-pVDZ basis set with its density fitting auxiliary basis using the XMS-CASPT2(IPEA = 0.0) method. This choice was motivated by XMS-CASPT2(IPEA = 0.0) featuring the closest ionisation potential estimates to the recorded evidence, and by providing the best balance for the simultaneous description of covalent and ionic excited states [93], being more reliable at or nearby crossing regions [42]. A reduced active space had to be employed due to the elevated cost of these simulations. 7H-adenine comprised the full valence occupied space and one unoccupied virtual orbital plus two lone pair orbitals to account for states, leading to 16 electrons in nine orbitals (15 in 9 for cations). 7H-Guanine, on the other hand, featured all valence occupied orbitals minus the fully in-phase (least contributing, that is, 1.99 occupation number) orbital and one unoccupied virtual orbital plus two lone pairs ( and ) to describe and states, totalling also 16 electrons in 9 orbitals (15 in 9 for cations).
The characterised cationic ground and excited state minima, as well as the different low-lying conical intersections, were optimised with XMS-CASPT2 using the single-state single reference (i.e., fully internally contracted) algorithm and no IPEA shift corrections as implemented in BAGEL [96,97]. Minima and CI optimisations (using the projection method of Bearpark et al. [98]) made use of analytical gradients [99,100,101] and non-adiabatic couplings [102]. Orbital visualisation was performed with Molden [103].
Conical intersection parameters were obtained from the XMS-CASPT2 gradient difference and non-adiabatic coupling vectors obtained at the characterised minimum energy conical intersections. These vectors were then used as described by Galván et al. to obtain pitch (), asymmetry () and relative tilt (), as well as and parameters to define the intersection types [48].
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/photochem1020018/s1: Tables S1–S8 with estimates for the ionisation potentials with the different CASPT2 approaches and reference weights and off-diagonal terms of the MS and XMS-CASPT2 effective Hamiltonians.
Author Contributions
Conceptualization, J.S.-M. and M.J.B.; methodology, J.S.-M. and M.J.B.; software, J.S.-M.; validation, J.S.-M., S.M.N. and M.J.B.; formal analysis, J.S.-M. and S.M.N.; investigation, J.S.-M. and S.M.N.; resources, M.J.B.; data curation, J.S.-M. and S.M.N.; writing–original draft preparation, J.S.-M.; writing–review and editing, J.S.-M., S.M.N. and M.J.B.; visualization, J.S.-M. and S.M.N.; supervision, M.J.B.; project administration, J.S.-M. and M.J.B.; funding acquisition, J.S.-M. All authors have read and agreed to the published version of the manuscript.
Funding
The project that gave rise to these results received the support of a fellowship from “La Caixa” Foundation (ID 100010434) and from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 847648, fellowship code “LCF/BQ/PI20/11760022” (J.S.-M.).
Acknowledgments
We thankfully acknowledge computing resources at Tirant and technical support provided by Servei d’Informatica de la Universitat de Valencia. We also thank the use and support provided by the Imperial College Research Computing Service (DOI: 10.14469/hpc/2232).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Schreier, W.J.; Gilch, P.; Zinth, W. Early Events of DNA Photodamage. Annu. Rev. Phys. Chem. 2015, 66, 497–519. [Google Scholar] [CrossRef]
- Noonan, F.P.; Zaidi, M.R.; Wolnicka-Glubisz, A.; Anver, M.R.; Bahn, J.; Wielgus, A.; Cadet, J.; Douki, T.; Mouret, S.; Tucker, M.A.; et al. Melanoma induction by ultraviolet A but not ultraviolet B radiation requires melanin pigment. Nat. Commun. 2012, 3, 884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Improta, R.; Santoro, F.; Blancafort, L. Quantum Mechanical Studies on the Photophysics and the Photochemistry of Nucleic Acids and Nucleobases. Chem. Rev. 2016, 116, 3540–3593. [Google Scholar] [CrossRef]
- Middleton, C.T.; de La Harpe, K.; Su, C.; Law, Y.K.; Crespo-Hernández, C.E.; Kohler, B. DNA Excited-State Dynamics: From Single Bases to the Double Helix. Annu. Rev. Phys. Chem. 2009, 60, 217–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespo-Hernández, C.E.; Cohen, B.; Hare, P.M.; Kohler, B. Ultrafast Excited-State Dynamics in Nucleic Acids. Chem. Rev. 2004, 104, 1977–2020. [Google Scholar] [CrossRef] [PubMed]
- Spata, V.A.; Lee, W.; Matsika, S. Excimers and Exciplexes in Photoinitiated Processes of Oligonucleotides. J. Phys. Chem. Lett. 2016, 7, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Markovitsi, D.; Gustavsson, T.; Vayá, I. Fluorescence of DNA Duplexes: From Model Helices to Natural DNA. J. Phys. Chem. Lett. 2010, 1, 3271–3276. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, Y.; Kohler, B. Excited States in DNA Strands Investigated by Ultrafast Laser Spectroscopy. Top. Curr. Chem. 2015, 356, 39–87. [Google Scholar]
- Martínez-Fernández, L.; Esposito, L.; Improta, R. Studying the excited electronic states of guanine rich DNA quadruplexes by quantum mechanical methods: Main achievements and perspectives. Photochem. Photobiol. Sci. 2020, 19, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Balanikas, E.; Banyasz, A.; Douki, T.; Baldacchino, G.; Markovitsi, D. Guanine Radicals Induced in DNA by Low-Energy Photoionization. Acc. Chem. Res. 2020, 53, 1511–1519. [Google Scholar] [CrossRef]
- Gustavsson, T.; Markovitsi, D. Fundamentals of the Intrinsic DNA Fluorescence. Acc. Chem. Res. 2021, 54, 1226–1235. [Google Scholar] [CrossRef]
- Kleinermanns, K.; Nachtigallová, D.; de Vries, M.S. Excited state dynamics of DNA bases. Int. Rev. Phys. Chem. 2013, 32, 308–342. [Google Scholar] [CrossRef]
- Beckstead, A.A.; Zhang, Y.; de Vries, M.S.; Kohler, B. Life in the light: Nucleic acid photoproperties as a legacy of chemical evolution. Phys. Chem. Chem. Phys. 2016, 18, 24228–24238. [Google Scholar] [CrossRef] [PubMed]
- Boldissar, S.; de Vries, M.S. How nature covers its bases. Phys. Chem. Chem. Phys. 2018, 20, 9701–9716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifer, G.P.; Besaratinia, A. UV wavelength-dependent DNA damage and human non-melanoma and melanoma skin cancer. Photochem. Photobiol. Sci. 2012, 11, 90–97. [Google Scholar] [CrossRef] [Green Version]
- Cadet, J.; Mouret, S.; Ravanat, J.L.; Douki, T. Photoinduced Damage to Cellular DNA: Direct and Photosensitized Reactions†. Photochem. Photobiol. 2012, 88, 1048–1065. [Google Scholar] [CrossRef] [PubMed]
- Nisoli, M.; Decleva, P.; Calegari, F.; Palacios, A.; Martín, F. Attosecond Electron Dynamics in Molecules. Chem. Rev. 2017, 117, 10760–10825. [Google Scholar] [CrossRef]
- Calegari, F.; Trabattoni, A.; Palacios, A.; Ayuso, D.; Castrovilli, M.C.; Greenwood, J.B.; Decleva, P.; Martín, F.; Nisoli, M. Charge migration induced by attosecond pulses in bio-relevant molecules. J. Phys. B At. Mol. Opt. Phys. 2016, 49, 142001. [Google Scholar] [CrossRef]
- Bucher, D.B.; Pilles, B.M.; Carell, T.; Zinth, W. Charge separation and charge delocalization identified in long-living states of photoexcited DNA. Proc. Natl. Acad. Sci. USA 2014, 111, 4369–4374. [Google Scholar] [CrossRef] [Green Version]
- Takaya, T.; Su, C.; de La Harpe, K.; Crespo-Hernández, C.E.; Kohler, B. UV excitation of single DNA and RNA strands produces high yields of exciplex states between two stacked bases. Proc. Natl. Acad. Sci. USA 2008, 105, 10285–10290. [Google Scholar] [CrossRef] [Green Version]
- Vayá, I.; Gustavsson, T.; Douki, T.; Berlin, Y.; Markovitsi, D. Electronic Excitation Energy Transfer between Nucleobases of Natural DNA. J. Am. Chem. Soc. 2012, 134, 11366–11368. [Google Scholar] [CrossRef] [PubMed]
- Bucher, D.B.; Kufner, C.L.; Schlueter, A.; Carell, T.; Zinth, W. UV-Induced Charge Transfer States in DNA Promote Sequence Selective Self-Repair. J. Am. Chem. Soc. 2016, 138, 186–190. [Google Scholar] [CrossRef]
- Banyasz, A.; Ketola, T.; Martínez-Fernández, L.; Improta, R.; Markovitsi, D. Adenine radicals generated in alternating AT duplexes by direct absorption of low-energy UV radiation. Faraday Discuss. 2018, 207, 181–197. [Google Scholar] [CrossRef]
- Roca-Sanjuán, D.; Rubio, M.; Merchán, M.; Serrano-Andrés, L. Ab initio determination of the ionization potentials of DNA and RNA nucleobases. J. Chem. Phys. 2006, 125, 084302. [Google Scholar] [CrossRef] [Green Version]
- Wolf, T.J.A.; Gühr, M. Photochemical pathways in nucleobases measured with an X-ray FEL. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2019, 377, 20170473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, T.J.A.; Holzmeier, F.; Wagner, I.; Berrah, N.; Bostedt, C.; Bozek, J.; Bucksbaum, P.; Coffee, R.; Cryan, J.; Farrell, J.; et al. Observing Femtosecond Fragmentation Using Ultrafast X-ray-Induced Auger Spectra. Appl. Sci. 2017, 7, 681. [Google Scholar] [CrossRef]
- Assmann, M.; Köppel, H.; Matsika, S. Photoelectron Spectrum and Dynamics of the Uracil Cation. J. Phys. Chem. A 2015, 119, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Assmann, M.; Weinacht, T.; Matsika, S. Surface hopping investigation of the relaxation dynamics in radical cations. J. Chem. Phys. 2016, 144, 034301. [Google Scholar] [CrossRef] [Green Version]
- Segarra-Martí, J.; Tran, T.; Bearpark, M.J. Ultrafast and radiationless electronic excited state decay of uracil and thymine cations: Computing the effects of dynamic electron correlation. Phys. Chem. Chem. Phys. 2019, 21, 14322–14330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segarra-Martí, J.; Tran, T.; Bearpark, M.J. Computing the Ultrafast and Radiationless Electronic Excited State Decay of Cytosine and 5-methyl-cytosine Cations: Uncovering the Role of Dynamic Electron Correlation. ChemPhotoChem 2019, 3, 856–865. [Google Scholar] [CrossRef]
- Crespo-Hernández, C.E.; Martínez-Fernández, L.; Rauer, C.; Reichardt, C.; Mai, S.; Pollum, M.; Marquetand, P.; González, L.; Corral, I. Electronic and Structural Elements That Regulate the Excited-State Dynamics in Purine Nucleobase Derivatives. J. Am. Chem. Soc. 2015, 137, 4368–4381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, M.S. Tautomer-Selective Spectroscopy of Nucleobases, Isolated in the Gas Phase. In Tautomerism; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2013; Chapter 7; pp. 177–196. [Google Scholar] [CrossRef]
- Serrano-Andrés, L.; Merchán, M. Are the five natural DNA/RNA base monomers a good choice from natural selection?: A photochemical perspective. J. Photochem. Photobiol. C Photochem. Rev. 2009, 10, 21–32. [Google Scholar] [CrossRef]
- Huang, Y.; Kenttamaa, H. Theoretical Estimations of the 298 K Gas-Phase Acidities of the Purine-Based Nucleobases Adenine and Guanine. J. Phys. Chem. A 2004, 108, 4485–4490. [Google Scholar] [CrossRef]
- Bravaya, K.B.; Kostko, O.; Dolgikh, S.; Landau, A.; Ahmed, M.; Krylov, A.I. Electronic Structure and Spectroscopy of Nucleic Acid Bases: Ionization Energies, Ionization-Induced Structural Changes, and Photoelectron Spectra. J. Phys. Chem. A 2010, 114, 12305–12317. [Google Scholar] [CrossRef] [Green Version]
- Marian, C.M. The Guanine Tautomer Puzzle: Quantum Chemical Investigation of Ground and Excited States. J. Phys. Chem. A 2007, 111, 1545–1553. [Google Scholar] [CrossRef]
- Zaytseva, I.L.; Trofimov, A.B.; Schirmer, J.; Plekan, O.; Feyer, V.; Richter, R.; Coreno, M.; Prince, K.C. Theoretical and Experimental Study of Valence-Shell Ionization Spectra of Guanine. J. Phys. Chem. A 2009, 113, 15142–15149. [Google Scholar] [CrossRef]
- Dolgounitcheva, O.; Zakrzewski, V.G.; Ortiz, J.V. Electron Propagator Theory of Guanine and Its Cations: Tautomerism and Photoelectron Spectra. J. Am. Chem. Soc. 2000, 122, 12304–12309. [Google Scholar] [CrossRef]
- Mons, M.; Piuzzi, F.; Dimicoli, I.; Gorb, L.; Leszczynski, J. Near-UV Resonant Two-Photon Ionization Spectroscopy of Gas Phase Guanine: Evidence for the Observation of Three Rare Tautomers. J. Phys. Chem. A 2006, 110, 10921–10924. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Yu, C.; Peng, S.; Akiyama, I.; Li, K.; Lee, L.K.; LeBreton, P.R. Ultraviolet photoelectron studies of the ground-state electronic structure and gas-phase tautomerism of hypoxanthine and guanine. J. Phys. Chem. 1980, 84, 1006–1012. [Google Scholar] [CrossRef]
- Trofimov, A.B.; Schirmer, J.; Kobychev, V.B.; Potts, A.W.; Holland, D.M.P.; Karlsson, L. Photoelectron spectra of the nucleobases cytosine, thymine and adenine. J. Phys. B At. Mol. Opt. Phys. 2005, 39, 305–329. [Google Scholar] [CrossRef]
- Shiozaki, T.; Woywod, C.; Werner, H.J. Pyrazine excited states revisited using the extended multi-state complete active space second-order perturbation method. Phys. Chem. Chem. Phys. 2013, 15, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Kotur, M.; Zhou, C.; Matsika, S.; Patchkovskii, S.; Spanner, M.; Weinacht, T.C. Neutral-Ionic State Correlations in Strong-Field Molecular Ionization. Phys. Rev. Lett. 2012, 109, 203007. [Google Scholar] [CrossRef] [Green Version]
- Kotur, M.; Weinacht, T.; Zhou, C.; Matsika, S. Following Ultrafast Radiationless Relaxation Dynamics With Strong Field Dissociative Ionization: A Comparison Between Adenine, Uracil and Cytosine. IEEE J. Sel. Top. Quantum Electron. 2012, 18, 187–194. [Google Scholar] [CrossRef]
- Segarra-Martí, J.; Bearpark, M.J. Modelling photoionisation in isocytosine: Potential formation of longer-lived excited state cations in its keto form. ChemPhysChem 2021. [Google Scholar] [CrossRef]
- Matsika, S. Two- and three-state conical intersections in the uracil cation. Chem. Phys. 2008, 349, 356–362. [Google Scholar] [CrossRef]
- Brash, D.E.; Goncalves, L.C.; Bechara, E.J. Chemiexcitation and Its Implications for Disease. Trends Mol. Med. 2018, 24, 527–541. [Google Scholar] [CrossRef]
- Fdez. Galván, I.; Delcey, M.G.; Pedersen, T.B.; Aquilante, F.; Lindh, R. Analytical State-Average Complete-Active-Space Self-Consistent Field Nonadiabatic Coupling Vectors: Implementation with Density-Fitted Two-Electron Integrals and Application to Conical Intersections. J. Chem. Theory Comput. 2016, 12, 3636–3653. [Google Scholar] [CrossRef] [Green Version]
- Atchity, G.J.; Xantheas, S.S.; Ruedenberg, K. Potential energy surfaces near intersections. J. Chem. Phys. 1991, 95, 1862–1876. [Google Scholar] [CrossRef]
- Hall, K.F.; Boggio-Pasqua, M.; Bearpark, M.J.; Robb, M.A. Photostability Via Sloped Conical Intersections: A Computational Study of the Excited States of the Naphthalene Radical Cation. J. Phys. Chem. A 2006, 110, 13591–13599. [Google Scholar] [CrossRef]
- Tokmachev, A.M.; Boggio-Pasqua, M.; Bearpark, M.J.; Robb, M.A. Photostability via Sloped Conical Intersections: A Computational Study of the Pyrene Radical Cation. J. Phys. Chem. A 2008, 112, 10881–10886. [Google Scholar] [CrossRef]
- Segarra-Martí, J.; Garavelli, M.; Aquilante, F. Multiconfigurational second-order perturbation theory with frozen natural orbitals extended to the treatment of photochemical problems. J. Chem. Theory Comput. 2015, 11, 3772–3784. [Google Scholar] [CrossRef] [PubMed]
- Segarra-Martí, J.; Francés-Monerris, A.; Roca-Sanjuán, D.; Merchán, M. Assessment of the Potential Energy Hypersurfaces in Thymine within Multiconfigurational Theory: CASSCF vs. CASPT2. Molecules 2016, 21, 1666. [Google Scholar] [CrossRef] [Green Version]
- Segarra-Martí, J.; Mukamel, S.; Garavelli, M.; Nenov, A.; Rivalta, I. Towards Accurate Simulation of Two-Dimensional Electronic Spectroscopy. Top. Curr. Chem. 2018, 376, 24. [Google Scholar] [CrossRef]
- Serrano-Andrés, L.; Merchán, M.; Lindh, R. Computation of conical intersections by using perturbation techniques. J. Chem. Phys. 2005, 122, 104107. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Tinoco, M.; Dolgounitcheva, O.; Zakrzewski, V.G.; Ortiz, J.V. Composite electron propagator methods for calculating ionization energies. J. Chem. Phys. 2016, 144, 224110. [Google Scholar] [CrossRef] [PubMed]
- Dolgounitcheva, O.; Zakrzewski, V.G.; Ortiz, J.V. Vertical Ionization Energies of Adenine and 9-Methyl Adenine. J. Phys. Chem. A 2009, 113, 14630–14635. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Musiał, M. Coupled-cluster theory in quantum chemistry. Rev. Mod. Phys. 2007, 79, 291–352. [Google Scholar] [CrossRef] [Green Version]
- Krylov, A.I. Equation-of-Motion Coupled-Cluster Methods for Open-Shell and Electronically Excited Species: The Hitchhiker’s Guide to Fock Space. Annu. Rev. Phys. Chem. 2008, 59, 433–462. [Google Scholar] [CrossRef] [Green Version]
- Roca-Sanjuán, D.; Aquilante, F.; Lindh, R. Multiconfiguration second-order perturbation theory approach to strong electron correlation in chemistry and photochemistry. WIRES Comput. Mol. Sci. 2012, 2, 585–603. [Google Scholar] [CrossRef]
- Giussani, A.; Segarra-Martí, J.; Nenov, A.; Rivalta, I.; Tolomelli, A.; Mukamel, S.; Garavelli, M. Spectroscopic fingerprints of DNA/RNA pyrimidine nucleobases in third-order nonlinear electronic spectra. Theor. Chem. Acc. 2016, 135, 121. [Google Scholar] [CrossRef]
- Segarra-Martí, J.; Zvereva, E.; Marazzi, M.; Brazard, J.; Dumont, E.; Assfeld, X.; Haacke, S.; Garavelli, M.; Monari, A.; Léonard, J.; et al. Resolving the Singlet Excited State Manifold of Benzophenone by First-Principles Simulations and Ultrafast Spectroscopy. J. Chem. Theory Comput. 2018, 14, 2570–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Close, D.M.; Crespo-Hernández, C.E.; Gorb, L.; Leszczynski, J. Ionization Energy Thresholds of Microhydrated Adenine and Its Tautomers. J. Phys. Chem. A 2008, 112, 12702–12706. [Google Scholar] [CrossRef]
- Slavíček, P.; Winter, B.; Faubel, M.; Bradforth, S.E.; Jungwirth, P. Ionization Energies of Aqueous Nucleic Acids: Photoelectron Spectroscopy of Pyrimidine Nucleosides and ab Initio Calculations. J. Am. Chem. Soc. 2009, 131, 6460–6467. [Google Scholar] [CrossRef]
- Pluhařová, E.; Jungwirth, P.; Bradforth, S.E.; Slavíček, P. Ionization of Purine Tautomers in Nucleobases, Nucleosides, and Nucleotides: From the Gas Phase to the Aqueous Environment. J. Phys. Chem. B 2011, 115, 1294–1305. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sevilla, M.D. Proton-Coupled Electron Transfer in DNA on Formation of Radiation-Produced Ion Radicals. Chem. Rev. 2010, 110, 7002–7023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altavilla, S.F.; Segarra-Martí, J.; Nenov, A.; Conti, I.; Rivalta, I.; Garavelli, M. Deciphering the photochemical mechanisms describing the UV-induced processes occurring in solvated guanine monophosphate. Front. Chem. 2015, 3, 29. [Google Scholar] [CrossRef] [Green Version]
- Weingart, O.; Nenov, A.; Altoè, P.; Rivalta, I.; Segarra-Martí, J.; Dokukina, I.; Garavelli, M. COBRAMM 2.0—A software interface for tailoring molecular electronic structure calculations and running nanoscale (QM/MM) simulations. J. Mol. Model. 2018, 24, 271. [Google Scholar] [CrossRef] [Green Version]
- Tomaník, L.; Muchová, E.; Slavíček, P. Solvation energies of ions with ensemble cluster-continuum approach. Phys. Chem. Chem. Phys. 2020, 22, 22357–22368. [Google Scholar] [CrossRef]
- Zhao, H.Y.; Lau, K.C.; Garcia, G.A.; Nahon, L.; Carniato, S.; Poisson, L.; Schwell, M.; Al-Mogren, M.M.; Hochlaf, M. Unveiling the complex vibronic structure of the canonical adenine cation. Phys. Chem. Chem. Phys. 2018, 20, 20756–20765. [Google Scholar] [CrossRef]
- Martinez-Fernandez, L.; Gavvala, K.; Sharma, R.; Didier, P.; Richert, L.; Segarra Martì, J.; Mori, M.; Mely, Y.; Improta, R. Excited-State Dynamics of Thienoguanosine, an Isomorphic Highly Fluorescent Analogue of Guanosine. Chem. Eur. J. 2019, 25, 7375–7386. [Google Scholar] [CrossRef]
- Serrano-Andrés, L.; Merchán, M.; Borin, A.C. Adenine and 2-aminopurine: Paradigms of modern theoretical photochemistry. Proc. Natl. Acad. Sci. USA 2006, 103, 8691–8696. [Google Scholar] [CrossRef] [Green Version]
- Barbatti, M. Photorelaxation Induced by Water–Chromophore Electron Transfer. J. Am. Chem. Soc. 2014, 136, 10246–10249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Andrés, L.; Merchán, M.; Borin, A.C. A Three-State Model for the Photophysics of Guanine. J. Am. Chem. Soc. 2008, 130, 2473–2484. [Google Scholar] [CrossRef]
- Cohen, B.; Hare, P.M.; Kohler, B. Ultrafast Excited-State Dynamics of Adenine and Monomethylated Adenines in Solution: Implications for the Nonradiative Decay Mechanism. J. Am. Chem. Soc. 2003, 125, 13594–13601. [Google Scholar] [CrossRef] [PubMed]
- Barbatti, M.; Aquino, A.J.A.; Szymczak, J.J.; Nachtigallová, D.; Hobza, P.; Lischka, H. Relaxation mechanisms of UV-photoexcited DNA and RNA nucleobases. Proc. Natl. Acad. Sci. USA 2010, 107, 21453–21458. [Google Scholar] [CrossRef] [Green Version]
- Yarkony, D.R. Nonadiabatic Quantum Chemistry—Past, Present, and Future. Chem. Rev. 2012, 112, 481–498. [Google Scholar] [CrossRef] [PubMed]
- Domcke, W.; Yarkony, D.R. Role of Conical Intersections in Molecular Spectroscopy and Photoinduced Chemical Dynamics. Annu. Rev. Phys. Chem. 2012, 63, 325–352. [Google Scholar] [CrossRef]
- Blancafort, L. Photochemistry and Photophysics at Extended Seams of Conical Intersection. ChemPhysChem 2014, 15, 3166–3181. [Google Scholar] [CrossRef] [Green Version]
- Pepino, A.J.; Segarra-Martí, J.; Nenov, A.; Improta, R.; Garavelli, M. Resolving Ultrafast Photoinduced Deactivations in Water-Solvated Pyrimidine Nucleosides. J. Phys. Chem. Lett. 2017, 8, 1777–1783. [Google Scholar] [CrossRef]
- Pepino, A.J.; Segarra-Martí, J.; Nenov, A.; Rivalta, I.; Improta, R.; Garavelli, M. UV-induced long-lived decays in solvated pyrimidine nucleosides resolved at the MS-CASPT2/MM level. Phys. Chem. Chem. Phys. 2018, 20, 6877–6890. [Google Scholar] [CrossRef]
- Schuurman, M.S.; Stolow, A. Dynamics at Conical Intersections. Annu. Rev. Phys. Chem. 2018, 69, 427–450. [Google Scholar] [CrossRef]
- Schalk, O.; Boguslavskiy, A.E.; Stolow, A. Substituent Effects on Dynamics at Conical Intersections: Cyclopentadienes. J. Phys. Chem. A 2010, 114, 4058–4064. [Google Scholar] [CrossRef] [Green Version]
- Fdez. Galván, I.; Vacher, M.; Alavi, A.; Angeli, C.; Aquilante, F.; Autschbach, J.; Bao, J.J.; Bokarev, S.I.; Bogdanov, N.A.; Carlson, R.K.; et al. OpenMolcas: From Source Code to Insight. J. Chem. Theory Comput. 2019, 15, 5925–5964. [Google Scholar] [CrossRef] [PubMed]
- Aquilante, F.; Autschbach, J.; Baiardi, A.; Battaglia, S.; Borin, V.A.; Chibotaru, L.F.; Conti, I.; De Vico, L.; Delcey, M.; Fdez. Galván, I.; et al. Modern quantum chemistry with [Open]Molcas. J. Chem. Phys. 2020, 152, 214117. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J. The CASSCF method: A perspective and commentary. Int. J. Quantum Chem. 2011, 111, 3267–3272. [Google Scholar] [CrossRef]
- Robb, M.A. Theoretical Chemistry for Electronic Excited States; Theoretical and Computational Chemistry Series; The Royal Society of Chemistry: London, UK, 2018; pp. P001–P225. [Google Scholar] [CrossRef] [Green Version]
- Widmark, P.O.; Malmqvist, P.Å.; Roos, B.O. Density matrix averaged atomic natural orbital (ANO) basis sets for correlated molecular wave functions. I. First row atoms. Theor. Chim. Acta 1990, 77, 291. [Google Scholar] [CrossRef]
- Widmark, P.O.; Persson, B.J.; Roos, B.O. Density matrix averaged atomic natural orbital (ANO) basis sets for correlated molecular wave functions. II. Second row atoms. Theor. Chim. Acta 1991, 79, 419. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.A.; Roos, B.O.; Sadlej, A.J.; Wolinski, K. Second-order perturbation theory with a CASSCF reference function. J. Phys. Chem. 1990, 94, 5483–5488. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.Å.; Roos, B.O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. [Google Scholar] [CrossRef]
- Finley, J.; Malmqvist, P.Å.; Roos, B.O.; Serrano-Andrés, L. The multi-state CASPT2 method. Chem. Phys. Lett. 1998, 288, 299–306. [Google Scholar] [CrossRef]
- Granovsky, A.A. Extended multi-configuration quasi-degenerate perturbation theory: The new approach to multi-state multi-reference perturbation theory. J. Chem. Phys. 2011, 134, 214113. [Google Scholar] [CrossRef]
- Forsberg, N.; Malmqvist, P.Å. Multiconfiguration perturbation theory with imaginary level shift. Chem. Phys. Lett. 1997, 274, 196–204. [Google Scholar] [CrossRef]
- Ghigo, G.; Roos, B.O.; Malmqvist, P.Å. A modified definition of the zeroth-order Hamiltonian in multiconfigurational perturbation theory (CASPT2). Chem. Phys. Lett. 2004, 396, 142–149. [Google Scholar] [CrossRef]
- Shiozaki, T. BAGEL: Brilliantly Advanced General Electronic-structure Library. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1331. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Al-Saadon, R.; MacLeod, M.K.; Shiozaki, T.; Vlaisavljevich, B. Multireference Electron Correlation Methods: Journeys along Potential Energy Surfaces. Chem. Rev. 2020, 120, 5909. [Google Scholar] [CrossRef]
- Bearpark, M.J.; Robb, M.A.; Schlegel, H.B. A direct method for the location of the lowest energy point on a potential surface crossing. Chem. Phys. Lett. 1994, 223, 269–274. [Google Scholar] [CrossRef]
- Shiozaki, T.; Gyorffy, W.; Celani, P.; Werner, H.J. Communication: Extended multi-state complete active space second-order perturbation theory: Energy and nuclear gradients. J. Chem. Phys. 2011, 135, 081106. [Google Scholar] [CrossRef]
- MacLeod, M.K.; Shiozaki, T. Communication: Automatic code generation enables nuclear gradient computations for fully internally contracted multireference theory. J. Chem. Phys. 2015, 142, 051103. [Google Scholar] [CrossRef] [Green Version]
- Vlaisavljevich, B.; Shiozaki, T. Nuclear Energy Gradients for Internally Contracted Complete Active Space Second-Order Perturbation Theory: Multistate Extensions. J. Chem. Theory Comput. 2016, 12, 3781–3787. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Shiozaki, T. Analytical Derivative Coupling for Multistate CASPT2 Theory. J. Chem. Theory Comput. 2017, 13, 2561–2570. [Google Scholar] [CrossRef] [Green Version]
- Schaftenaar, G.; Noordik, J. Molden: A pre- and post-processing program for molecular and electronic structures*. J. Comput. Aided Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).