In this chapter, the results of this study are presented. The first section focuses on the data generation process, detailing the outcomes of the simulations and their reliability. The second section evaluates the predictive framework using experimental data, establishing a baseline performance score. Finally, the third section explores the impact of integrating simulated data with experimental data, assessing how this combined approach influences the predictive accuracy of the framework.
3.1. Simulated Data Results
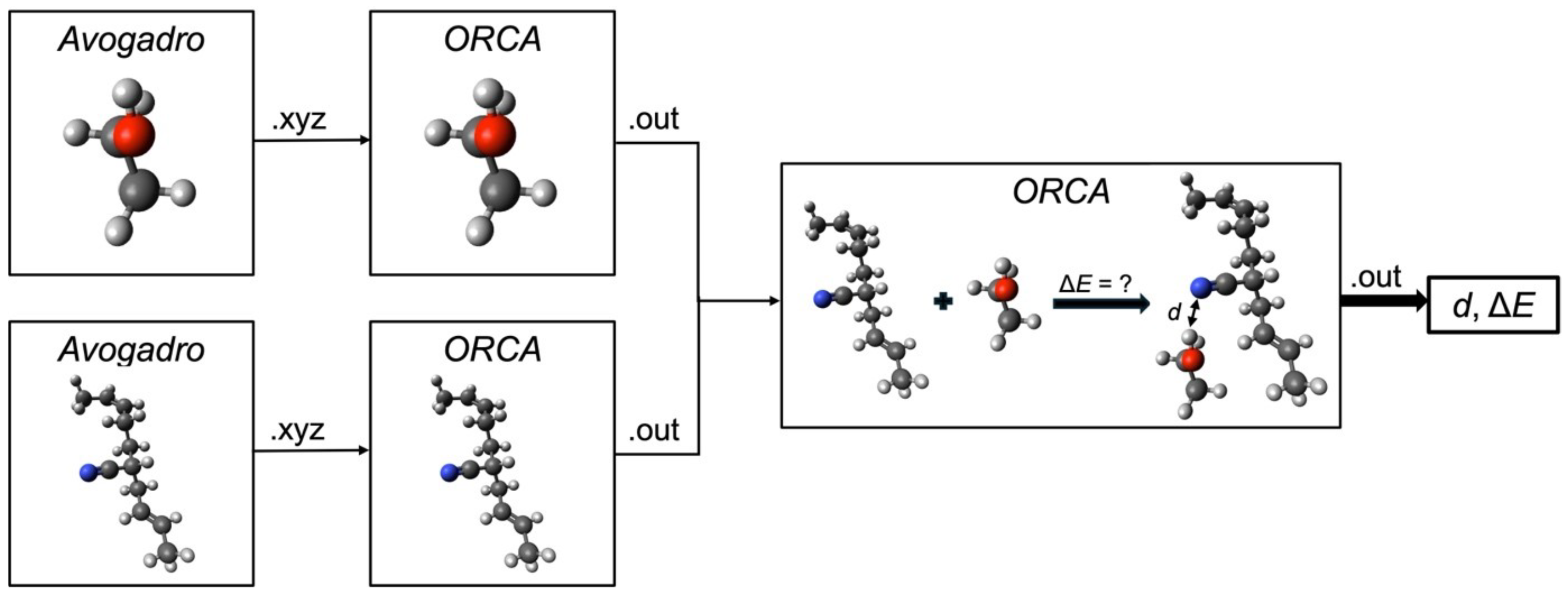
Table 3 lists all substances experimentally investigated within the FSC and their corresponding HSP and DFT values used as input features. DFT data were calculated for 28 substances, while HSP values were obtained from the literature for 47 substances. The missing DFT results are due to non-converging calculations caused by complex molecular structures. Furthermore, the elastomer volume changes (target) for all of the investigated substances are listed in
Table 3.
An initial investigation into the suitability of the gathered values for the data generation process revealed an unfavorable case for the DFT approach. Using the Pearson correlation coefficient to assess the strength of the correlation between individual values and the target variable, volume change, it becomes evident that no significant correlation exists for the DFT values. The calculated intermolecular distance
d and binding enthalpy
yielded correlation scores of
and
, respectively. In contrast, the correlation scores for the HSPs were equal or greater than
for all parameters except
(see
Table 4). However, closer examination of the DFT values highlights the limited usability of the DFT approach, at least for the functional group of alcohols. Among the 28 substances for which DFT values were calculated, 7 belonged to the functional group of alcohols. When considering only these substances, the correlation with the target values was significantly higher, suggesting that the DFT approach may still hold potential for specific functional groups (see
Table 4). However, since the approach in this study should apply to substances across all functional groups, the overall low correlation—alongside the limited number of DFT data samples—makes the DFT approach less suitable for further use in the prediction process. Hence, only HSPs predict the volume change for previously untested substances.
The poor performance of the DFT results could be due to the inherent drawbacks of DFT. While the theory of DFT is exact, the exchange-correlation functional is not known and hence all DFT functionals are approximations. Also, the KS-orbitals resulting from the applied KS-DFT approach do not have any physical meaning since they are the orbital of a fictitious system of non-interacting electrons. Thus, it must be carefully evaluated if DFT-results can be used to analyze binding. Higher-accuracy electronic structure methods, like the gold standard of CCSD(T), could be used to benchmark the applied DFT method in this case. Additional to the inherent drawbacks of the theory, there might be issues with the applied level of theory. For the basis set, an attempt was made to minimize the basis set superposition error by using a relatively large basis set. However, additional counterpoise correction might be necessary. Conformational sampling and optimization to the nearest minimum could also verify if the optimized geometry is in fact a global minimum on the potential energy surface. Also, the underlying assumption of the main interaction happening between the most positively and most negatively charged parts of the molecules might be faulty. Other atoms could be evaluated as possible points of interaction.
The volume change prediction in the data generation process emphasizes supervised regression models, including linear, lasso, neural network, and tree-based regression models. Three HSPs and the molar volume were used as input features for these models. The models were trained on a subset of 42 samples out of the 46 substances listed in
Table 3, while the remaining 5 samples were used for model validation against experimental obtained volume change data. To ensure robust performance, feature scaling, 5-fold cross-validation, and individual hyperparameter tuning were applied to all the models. An initial investigation indicated that the neural network regression model performed best with the settings presented in
Table 5. Consequently, only the results of the neural network model are presented in the following sections.
To evaluate the model’s predictive accuracy, the coefficient of determination is used as a performance metric. This metric is applied throughout the training process and the final validation of previously unseen data. An value closer to one indicates better model performance. Monitoring training and validation scores is essential for detecting overfitting or underfitting. A high training but significantly lower validation suggests overfitting, where the model memorizes the training data but fails to generalize. Conversely, low values for both indicate underfitting, meaning the model lacks the complexity needed to capture underlying patterns.
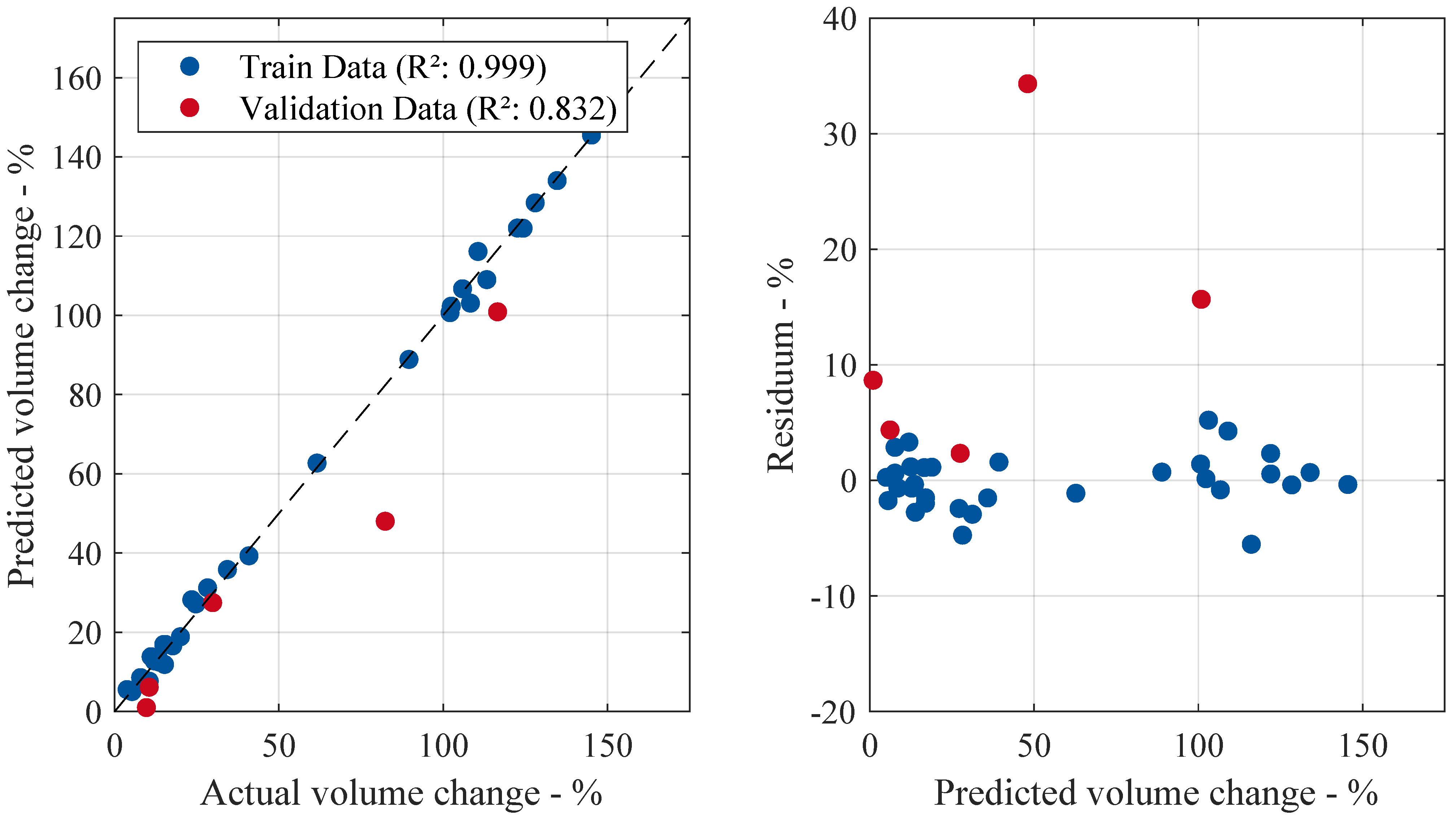
Figure 4 compares the predicted volume change to the actual values on the right-hand side during the training and validation process. Conversely, the residuals—the difference between the actual and predicted volume change—are plotted against the predicted values. A training
score of
indicates that the model was effectively trained using the available features and samples. The high validation
score of
, close to the training score, indicates good predictive accuracy and generalization ability. Both scores are visually represented in the left scatter plot, where the data points closely align with the diagonal line, indicating near-ideal predictions with an
value approaching one. Furthermore, the right-hand side of the figure shows that volume predictions changing up to around
exhibit low residuals. Beyond this threshold, the residuals tend to increase, especially for the validation case, although no clear pattern emerges.
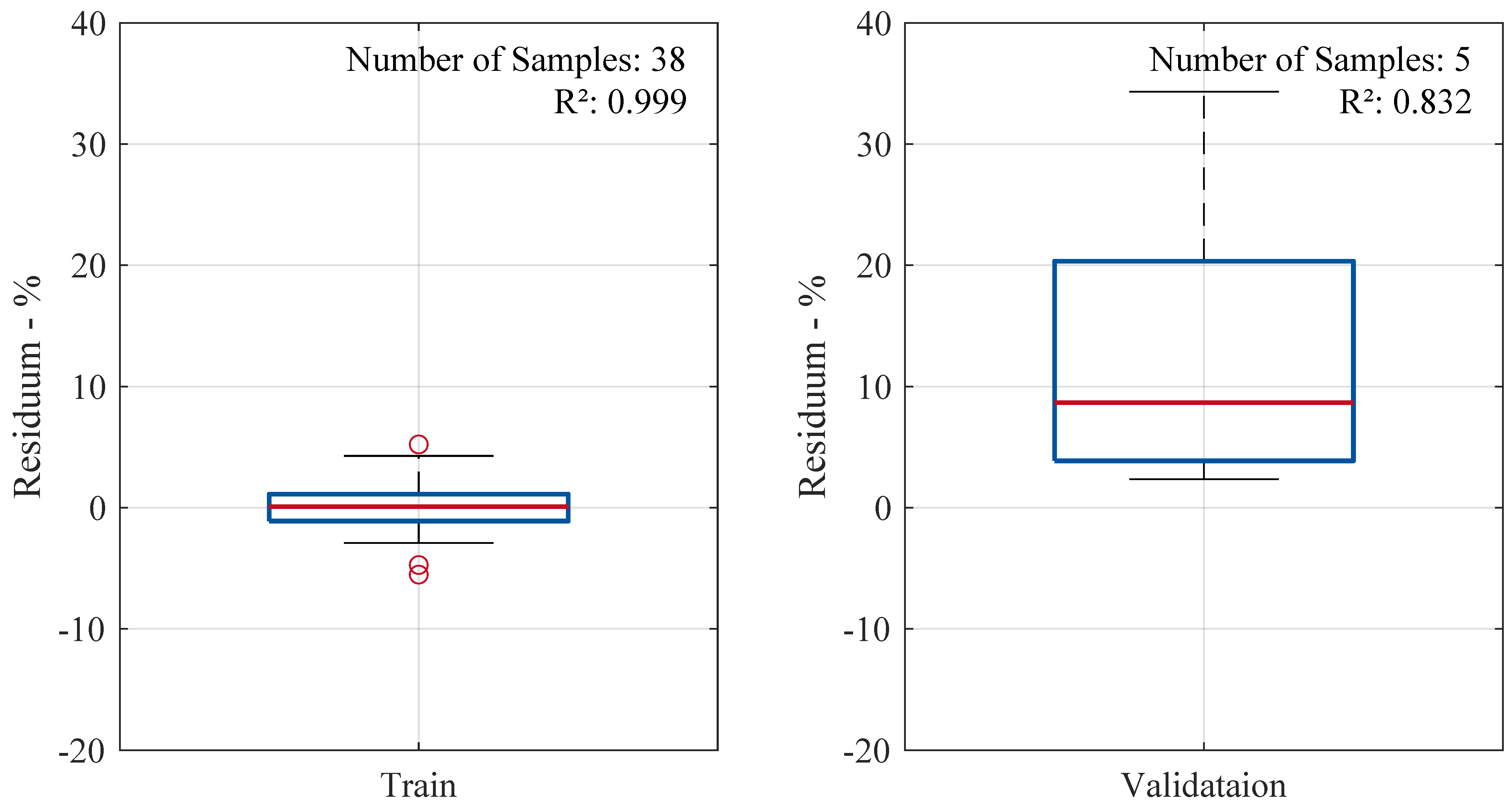
Figure 5 presents box plots of the residuals for both the training and validation datasets. The distribution of residuals in the training set appears tightly clustered around zero, indicating a well-fitted model. Most predictions deviate by less than
percentage points from the actual values. The validation residuals show a wider spread with a shift to positive values, with the median at
. This still suggests good generalization with minimal bias. However, it becomes evident that the outlier in the validation set, with an actual volume change of
(2-octanone), exhibits the highest prediction error. This is due to a lack of training samples in this range of volume change. The model demonstrates stability and reliable predictive performance within the range of sufficient training data.
Overall, the selected HSP features and the chosen regression model and architecture demonstrate promising results during training and validation. To generate synthetic data that can be incorporated into the existing experimental dataset, the trained model was applied to a set of 59 untested substances. The relevant HSP features were collected for each of these substances, and the volume change was predicted based on those features. A summary of the substances and their corresponding predicted values is provided in
Table 6 and
Table 7. It is important to note that no data validation is available at this stage, as no experimental investigations have been conducted for these substances yet.
The addition of simulation more than doubles the sample size, which can enhance the predictive accuracy of the framework presented above without the need for time-consuming immersion tests. Before exploring the impact of an expanded database on prediction accuracy, a baseline performance is established using only the experimental results.
3.2. Baseline Results
To establish a baseline score for comparison and assess whether the newly generated data impact the accuracy of the prediction framework, the model was first trained using only the available experimental data. This section presents the results of the baseline case. For this purpose, four standard regression models are evaluated: linear regression (Linear), lasso regression (Lasso), multilayer perceptron (MLP) regression, and tree-based regression (Tree). All four models are integrated into the prediction framework, with data preprocessing applied uniformly across the entire dataset. Each model is then individually optimized using halving grid search CV to determine the best hyperparameters. Finally, the optimal models are trained and evaluated using 5-fold cross-validation.
The models’ performances were evaluated during training and testing using the coefficient of determination
. Additionally, residual values—the differences between the actual and predicted volume changes—were calculated for each sample in both phases. The distribution of residuals is visualized using a box plot. As the previous chapter shows, the box plot effectively summarizes other representations, such as scatter plots of actual vs. predicted values or residuals, providing a comprehensive overview of model performance. Hence, this method of presentation is chosen for model comparison. All models are trained using 39 samples, while the remaining 10 are reserved for testing. The optimal hyperparameters, determined through hyperparameter tuning, are presented in
Table 8. Since the linear model omits adjustable hyperparameters, it is excluded from the table.
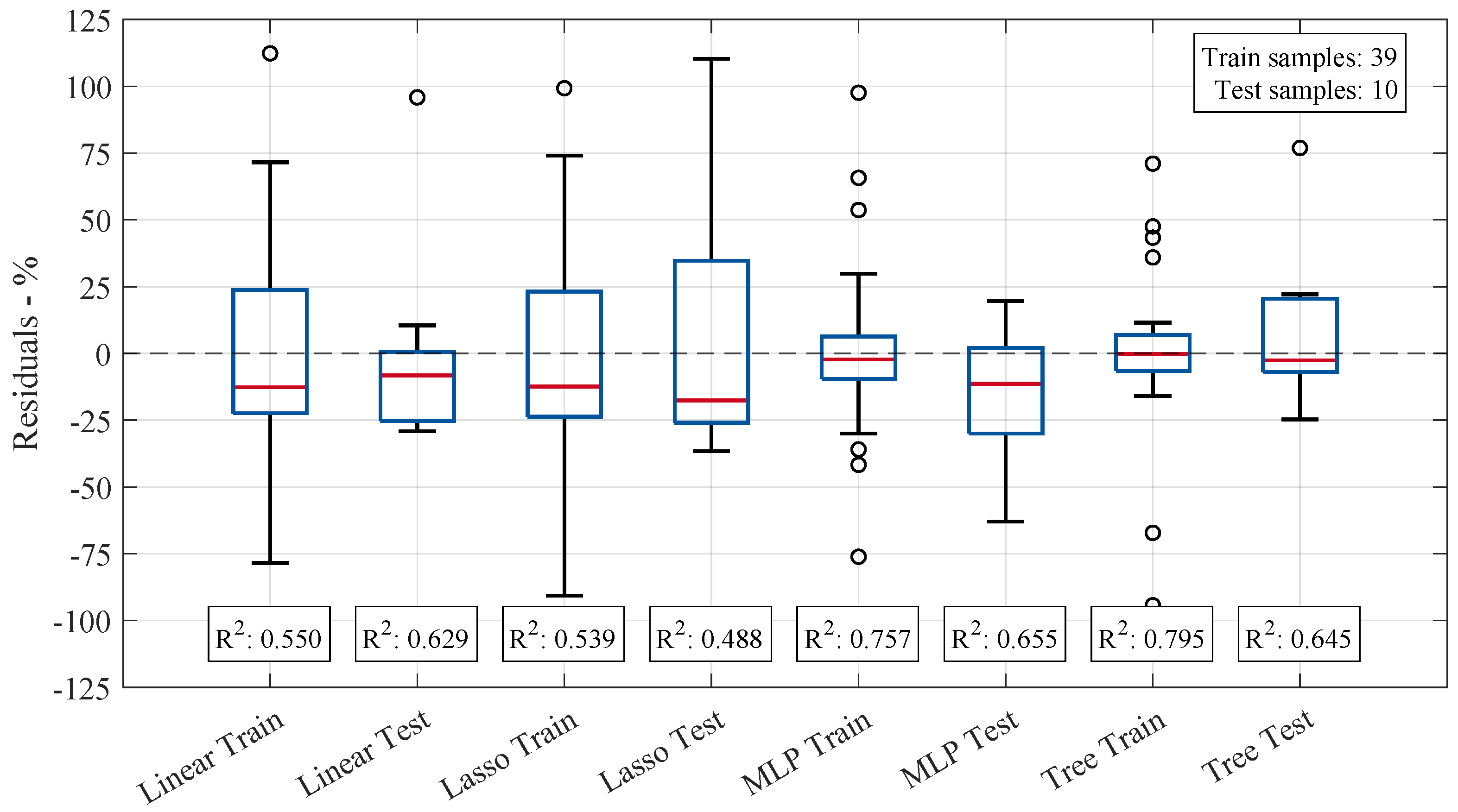
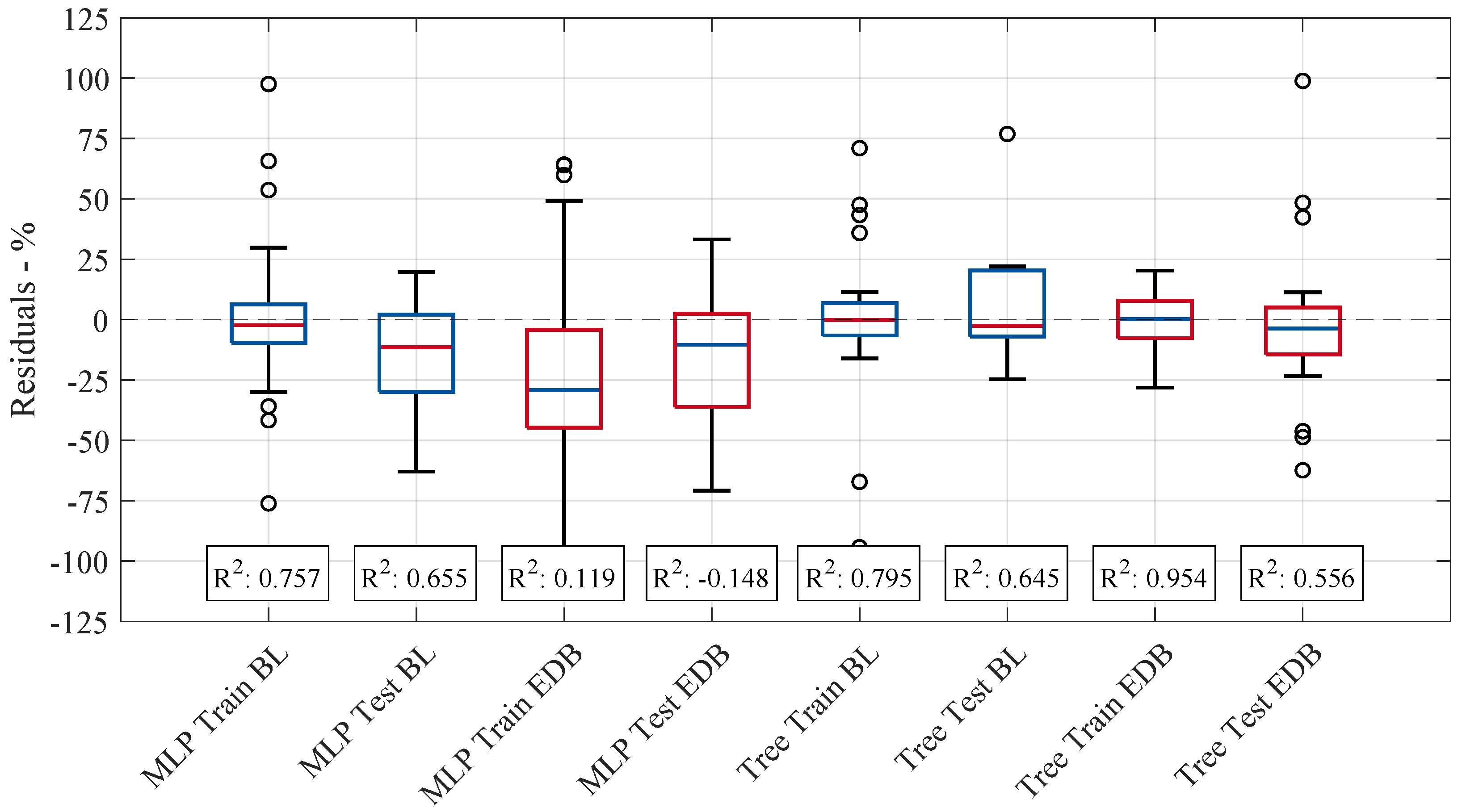
Figure 6 presents box plots of the residuals for all investigated regression models, comparing both training and testing phases. The distribution of residuals provides insight into each model’s predictive performance, with a narrower spread indicating higher accuracy. This figure highlights variations in model stability and generalization ability by visualizing the differences between the actual and predicted volume changes.
The median residuals for each model remain close to zero, indicating minimal bias towards larger prediction values. However, the variation in residual spread suggests that specific models exhibit more stability in their predictions than others. The range of residuals highlights differences in the models’ predictive reliability, with some models demonstrating more significant variation in errors across the data points, indicating less consistency in their performance. The MLP and tree-based models showed the highest values, with training scores of and , respectively, and testing scores of and , respectively. These results suggest that both models achieve a good fit for the training data and good generalization of unseen test data. However, outliers in the data still affect model performance, as their influence can lead to deviations in both the training and testing results. Despite this, the range of predicted values (minimum and maximum) remained almost consistent across all models, indicating that the models were similarly constrained in the spread of their predictions. In contrast, the linear and Lasso models yielded lower values and thus lower predictive accuracy. These models appear to underfit the data, as their relatively simple structure fails to capture the underlying complexity of the relationships in the dataset given the available describing features and number of samples. This results in less-accurate predictions on both the training and testing sets. Notably, none of the models achieved sufficiently high training values, implying that the selected molecular descriptors, alongside the limited amount of data samples, do not fully capture the underlying patterns in the data. This limitation in feature representation leads to insufficient model training, which in turn adversely affects performance on the test set. Consequently, the linear model’s better performance during testing may be attributed to chance rather than a robust generalization ability.
An increase in predictive accuracy is expected with a larger dataset. Therefore, this study explores alternative approaches to experimental testing for generating new data samples. In the following, the database is expanded by incorporating samples whose elastomer volume change has been predicted using the HSP approach and a regression model.
3.3. Expanded Database Results
This section examines the impact of a larger dataset by comparing the prediction accuracy of the expanded and baseline cases. First, the models were evaluated using the same hyperparameters as in the baseline case. In the second step, new hyperparameters optimized for the expanded dataset were applied to assess potential improvements in predictive performance. The total number of samples, including the newly generated data, amounted to 108. However, some outliers were removed due to constraints related to technical plausibility, resulting in a final dataset of 95 samples. As before, 5-fold cross-validation was performed, yielding 76 training samples and 19 testing samples per fold.
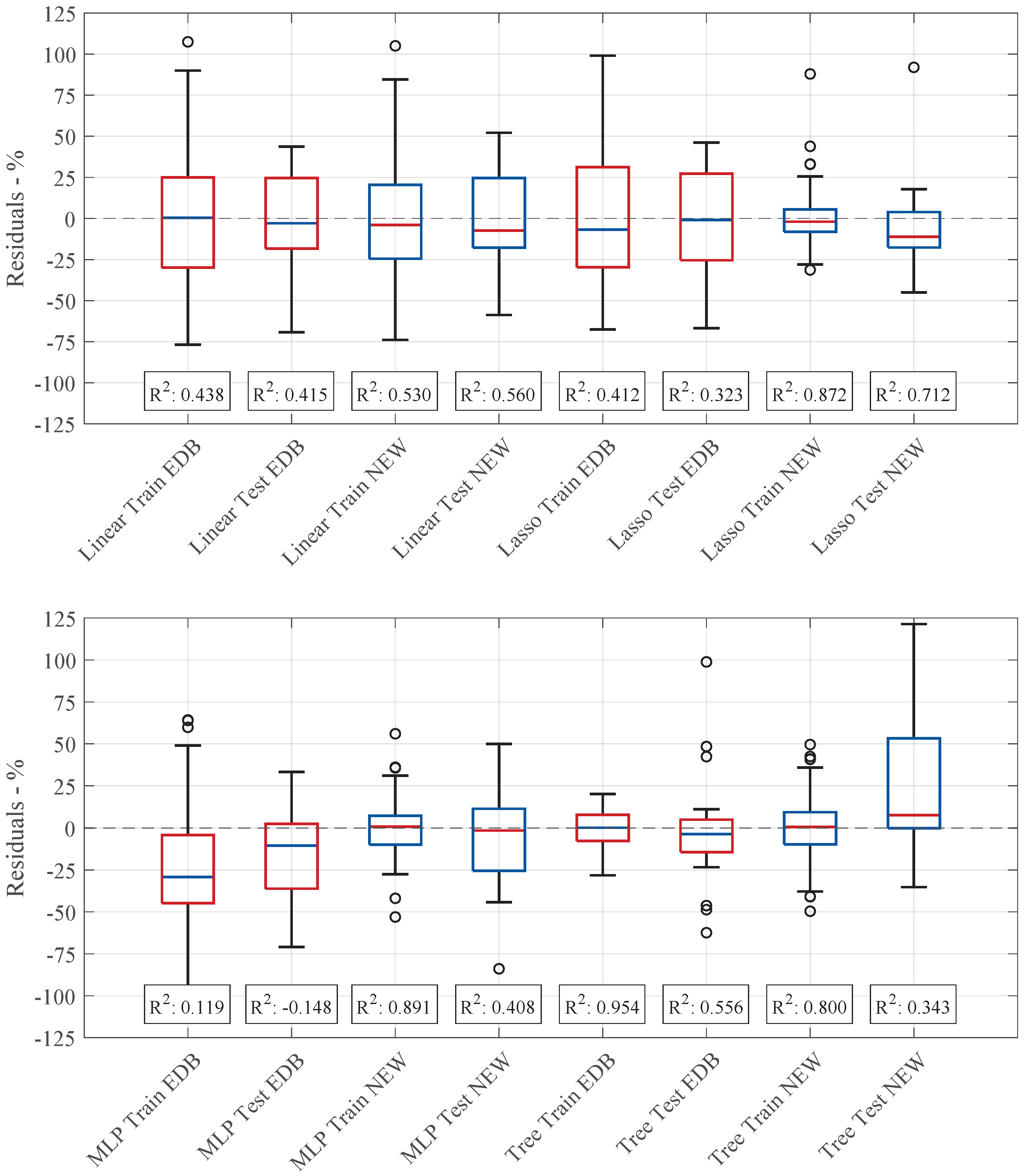
Figure 7 compares the residual values and
scores of all investigated models in the baseline case with those obtained using the expanded database. At this stage, the models in both cases share the same hyperparameters from
Table 8.
Expanding the dataset shifts the median residuals closer to zero for most models (Linear, Lasso, and tree-based) compared to the baseline case. However, across all the models, the scores decrease, except for the tree-based model in training, which slightly improves ( for BL, for EDB). This indicates a reduction in predictive accuracy for the other models. The tree-based model demonstrates greater robustness, maintaining stable or even improved performance. On the other hand, the MLP model exhibits significant instability, characterized by wider residual spreads and even a negative testing score.
The results suggest that while increased data volume can improve model training, it may also introduce complexities that negatively impact certain model architectures, particularly those more sensitive to data distribution changes. Therefore, it is essential to adjust the model architecture, where possible, to one that is optimally suited to the characteristics of the given dataset. Thus, in the following analysis, the models are re-optimized for the dataset by individually tuning their hyperparameters to improve performance and adaptability.
The following section presents the model performance results after re-optimizing the hyperparameters for the extended database case. Since the dataset size nearly doubled, each model’s architecture was individually adjusted to accommodate the increased data complexity. The resulting optimal hyperparameters are summarized in
Table 9. Furthermore, the number of input features was increased from five to seven to accommodate the larger dataset better and capture additional patterns in the data.
Figure 8 shows the residual distributions and
scores for training and testing across the different models, comparing their performance before and after hyperparameter optimization. While some models showed significant improvements, others experienced a decline in predictive accuracy, indicating that the effects of dataset expansion and parameter tuning varied depending on the model architecture.
For example, the linear model slightly improved, with a lower median residual and reduced spread in the updated configuration. This led to a modest increase in the score, rising from to in training and from to in testing. However, the overall performance remained moderate, with values of around . Since the linear model lacks adjustable hyperparameters, these improvements were primarily attributed to the increased dataset size and additional input features. The lasso regression model showed an apparent enhancement in predictive performance. The residual spread was significantly reduced, though some outliers remained present. The score improved considerably, increasing from to in training and from to in testing. Additionally, the median residual deviation in testing was close to zero, suggesting better model fit and improved generalization. The MLP regression model demonstrated the most substantial improvement. The training score rose from to , while the testing improved to . The residual distribution became more centered, with the median closer to zero, and the overall spread was reduced, particularly in training. However, despite these gains, the testing performance still exhibited a relatively wide residual distribution, indicating persistent instability in generalization. Conversely, the tree-based regression model experienced a decline in performance. While the spread of residuals increased in training, the interquartile range remained nearly unchanged. The training decreased from to , showing fewer signs of overfitting than before. However, testing performance deteriorated, shifting residual distribution toward positive values and increasing prediction errors. The testing dropped from to , indicating reduced generalization capability.
The results highlight that while hyperparameter tuning leads to substantial improvements for the MLP and lasso models, the tree-based model showed a decline in predictive performance. The linear model benefited slightly from the increased dataset, whereas the MLP model, despite achieving substantial improvements in training, still faced challenges in stability during testing. These findings emphasize the need for model-specific optimizations and careful consideration of dataset expansion effects when adjusting model architectures. No uniform statement can be made by comparing the prediction accuracy based on the score and the residual distribution of the old and new architecture.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}