
Role of Multiple Intermolecular H-Bonding Interactions in Molecular Cluster of Hydroxyl-Functionalized Imidazolium Ionic Liquid: An Experimental, Topological, and Molecular Dynamics Study

 , , , , ,
, , , , ,
Abstract

1. Introduction
2. Experimental Section
2.1. Chemicals
Synthesis Procedure
2.2. Synthesis and Characterization of EtOHImCl
2.3. Infrared Spectroscopy
2.4. Quantum Chemical Calculation
2.4.1. Structure Calculations
2.4.2. Wavenumber Calculations
2.4.3. Binding Energy Calculation
2.4.4. Natural Bond Orbital (NBO) Analysis
2.4.5. Energy Decomposition Analysis (EDA)
2.4.6. Topology Analysis
2.4.7. Non-Covalent Interaction (NCI) Analysis
2.4.8. Ab Initio Molecular Dynamics Assay
3. Results and Discussion
3.1. Structure of Isolated Ion-Pairs and Small Clusters
3.2. FT-IR Studies
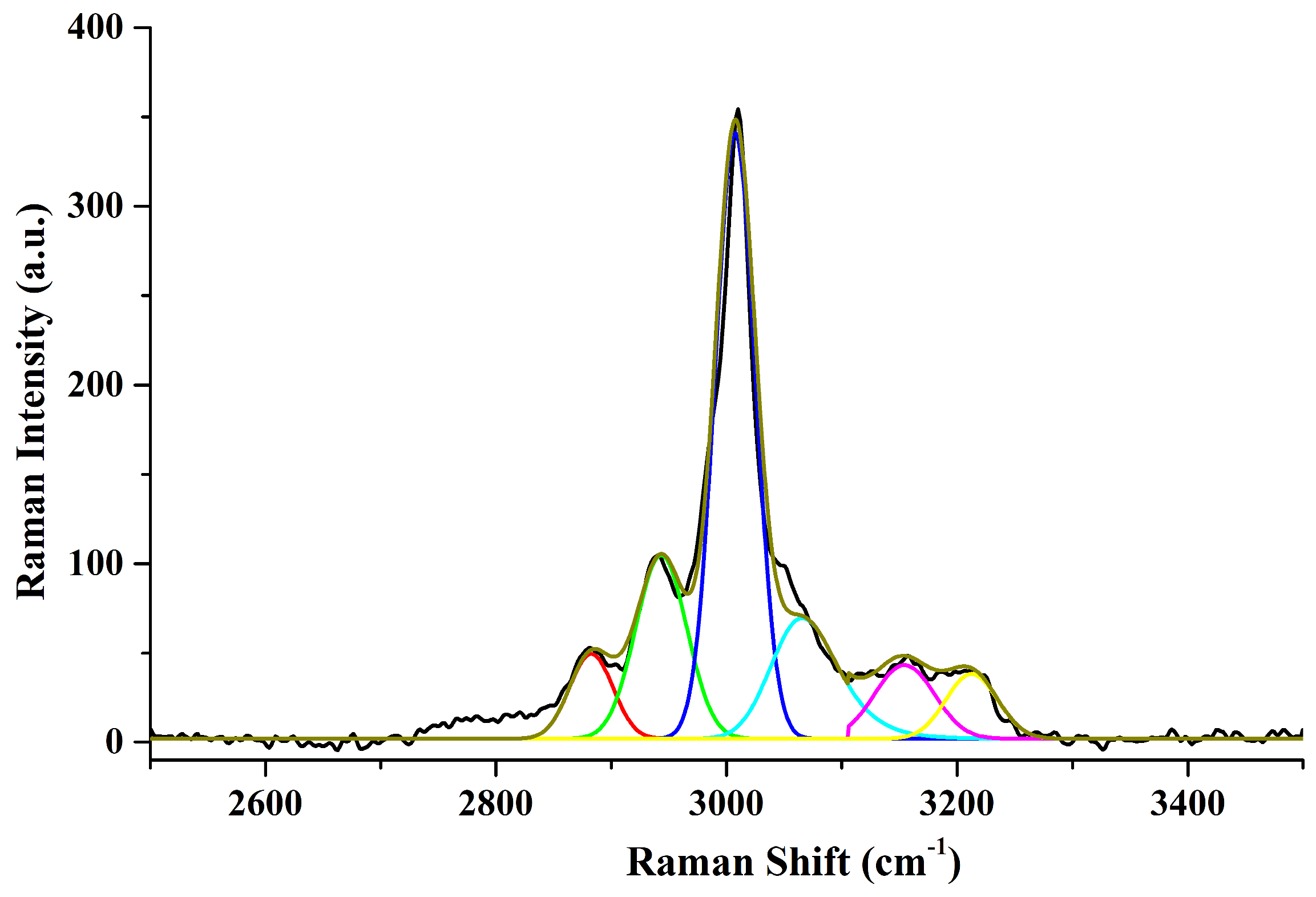
3.3. Raman Studies
3.4. Natural Bond Orbital (NBO) Analysis
3.5. Topology Analysis
3.5.1. AIM Analysis
3.5.2. Electron Localized Function (ELF) and Localized Orbital Locator (LOL) Surface Map Analysis
3.6. Non-Covalent Interaction (NCI)
3.7. Energy Decomposition Analysis (EDA)
3.8. Ab Initio Molecular Dynamics (AIMD) Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Plechkova, N.V.; Seddon, K.R. Applications of ionic liquids in the chemical industry. Chem. Soc. Rev. 2008, 37, 123–150. [Google Scholar] [CrossRef]
- Menne, S.; Pires, J.; Anouti, M.; Balducci, A. Protic ionic liquids as electrolytes for lithium-ion batteries. Electrochem. Commun. 2013, 31, 39–41. [Google Scholar] [CrossRef]
- Panja, S.K.; Saha, S. Recyclable, magnetic ionic liquid bmim[FeCl4]-catalyzed, multicomponent, solvent-free, green synthesis of quinazolines. RSC Adv. 2013, 3, 14495–14500. [Google Scholar] [CrossRef]
- Shabashini, A.; Richard, S.; Solanky, T.; Biswas, A.; Kumar, S.; Chandra Nandi, G.; Kumar Panja, S. Effect of dipolar state on J-type aggregation of acceptor group modified pyrene-based push-pull systems. J. Photochem. Photobiol. A Chem. 2024, 458, 115971. [Google Scholar] [CrossRef]
- Panja, S.K.; Dwivedi, N.; Noothalapati, H.; Shigeto, S.; Sikder, A.K.; Saha, A.; Sunkari, S.S.; Saha, S. Significance of weak interactions in imidazolium picrate ionic liquids: Spectroscopic and theoretical studies for molecular level understanding. Phys. Chem. Chem. Phys. 2015, 17, 18167–18177. [Google Scholar] [CrossRef]
- Jalili, A.H.; Mehdizadeh, A.; Shokouhi, M.; Sakhaeinia, H.; Taghikhani, V. Solubility of CO2 in 1-(2-hydroxyethyl)-3-methylimidazolium ionic liquids with different anions. J. Chem. Thermodyn. 2010, 42, 787–791. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, S.; Cheng, W.; Ren, J. Hydroxyl-functionalized ionic liquid: A novel efficient catalyst for chemical fixation of CO2 to cyclic carbonate. Tetrahedron Lett. 2008, 49, 3588–3591. [Google Scholar] [CrossRef]
- Holbrey, J.D.; Turner, M.B.; Reichert, W.M.; Rogers, R.D. New ionic liquids containing an appended hydroxyl functionality from the atom-efficient, one-pot reaction of 1-methylimidazole and acid with propylene oxide. Green Chem. 2003, 5, 731–736. [Google Scholar] [CrossRef]
- Branco, L.C.; Rosa, J.N.; Moura Ramos, J.J.; Afonso, C.A.M. Preparation and Characterization of New Room Temperature Ionic Liquids. Chem. A Eur. J. 2002, 8, 3671–3677. [Google Scholar] [CrossRef]
- Recham, N.; Dupont, L.; Courty, M.; Djellab, K.; Larcher, D.; Armand, M.; Tarascon, J.M. Ionothermal Synthesis of Tailor-Made LiFePO4 Powders for Li-Ion Battery Applications. Chem. Mater. 2009, 21, 1096–1107. [Google Scholar] [CrossRef]
- Yang, X.; Fei, Z.; Zhao, D.; Ang, W.H.; Li, Y.; Dyson, P.J. Palladium Nanoparticles Stabilized by an Ionic Polymer and Ionic Liquid: A Versatile System for C−C Cross-Coupling Reactions. Inorg. Chem. 2008, 47, 3292–3297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qi, X.; Ma, X.; Lu, L.; Deng, Y. Hydroxyl Ionic Liquids: The Differentiating Effect of Hydroxyl on Polarity due to Ionic Hydrogen Bonds between Hydroxyl and Anions. J. Phys. Chem. B 2010, 114, 3912–3920. [Google Scholar] [CrossRef]
- Panja, S.K.; Srivastava, N.; Srivastava, J.; Prasad, N.E.; Noothalapati, H.; Shigeto, S.; Saha, S. Evidence of C–FP and aromatic π–F-P weak interactions in imidazolium ionic liquids and its consequences. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 194, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Fumino, K.; Reimann, S.; Ludwig, R. Probing molecular interaction in ionic liquids by low frequency spectroscopy: Coulomb energy, hydrogen bonding and dispersion forces. Phys. Chem. Chem. Phys. 2014, 16, 21903–21929. [Google Scholar] [CrossRef]
- Niemann, T.; Stange, P.; Strate, A.; Ludwig, R. When hydrogen bonding overcomes Coulomb repulsion: From kinetic to thermodynamic stability of cationic dimers. Phys. Chem. Chem. Phys. 2019, 21, 8215–8220. [Google Scholar] [CrossRef]
- Kumar, S.; Das, A. Observation of exclusively π-stacked heterodimer of indole and hexafluorobenzene in the gas phase. J. Chem. Phys. 2013, 139, 104311. [Google Scholar] [CrossRef] [PubMed]
- Fumino, K.; Peppel, T.; Geppert-Rybczyńska, M.; Zaitsau, D.H.; Lehmann, J.K.; Verevkin, S.P.; Köckerling, M.; Ludwig, R. The influence of hydrogen bonding on the physical properties of ionic liquids. Phys. Chem. Chem. Phys. 2011, 13, 14064–14075. [Google Scholar] [CrossRef]
- Fumino, K.; Wulf, A.; Ludwig, R. Strong, Localized, and Directional Hydrogen Bonds Fluidize Ionic Liquids. Angew. Chem. Int. Ed. 2008, 47, 8731–8734. [Google Scholar] [CrossRef]
- Corderí, S.; González, E.J.; Calvar, N.; Domínguez, Á. Application of [HMim][NTf2], [HMim][TfO] and [BMim][TfO] ionic liquids on the extraction of toluene from alkanes: Effect of the anion and the alkyl chain length of the cation on the LLE. J. Chem. Thermodyn. 2012, 53, 60–66. [Google Scholar] [CrossRef]
- Giraud, G.; Gordon, C.M.; Dunkin, I.R.; Wynne, K. The effects of anion and cation substitution on the ultrafast solvent dynamics of ionic liquids: A time-resolved optical Kerr-effect spectroscopic study. J. Chem. Phys. 2003, 119, 464–477. [Google Scholar] [CrossRef]
- Jin, H.; O’Hare, B.; Dong, J.; Arzhantsev, S.; Baker, G.A.; Wishart, J.F.; Benesi, A.J.; Maroncelli, M. Physical Properties of Ionic Liquids Consisting of the 1-Butyl-3-Methylimidazolium Cation with Various Anions and the Bis(trifluoromethylsulfonyl)imide Anion with Various Cations. J. Phys. Chem. B 2008, 112, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, S.A.; Zvereva, E.E.; Vidiš, A.; Dyson, P.J. Application of Density Functional Theory and Vibrational Spectroscopy Toward the Rational Design of Ionic Liquids. J. Phys. Chem. A 2007, 111, 352–370. [Google Scholar] [CrossRef]
- Li, Y.; Hu, Y.; Chen, G.; Wang, Z.; Jin, X. Rapid proton diffusion in hydroxyl functionalized imidazolium ionic liquids. Sci. China Chem. 2017, 60, 734–739. [Google Scholar] [CrossRef]
- Wong, C.Y.; Wong, W.Y.; Loh, K.S.; Lim, K.L. Protic ionic liquids as next-generation proton exchange membrane materials: Current status & future perspectives. React. Funct. Polym. 2022, 171, 105160. [Google Scholar] [CrossRef]
- Johansson, K.M.; Izgorodina, E.I.; Forsyth, M.; MacFarlane, D.R.; Seddon, K.R. Protic ionic liquids based on the dimeric and oligomeric anions: [(AcO)xHx−1]−. Phys. Chem. Chem. Phys. 2008, 10, 2972–2978. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, T.; Nakamori, Y.; Matsumoto, K.; Hagiwara, R. Ion-Ion Interactions and Conduction Mechanism of Highly Conductive Fluorohydrogenate Ionic Liquids. J. Phys. Chem. C 2011, 115, 4324–4332. [Google Scholar] [CrossRef]
- Keutsch, F.N.; Saykally, R.J. Water clusters: Untangling the mysteries of the liquid, one molecule at a time. Proc. Natl. Acad. Sci. USA 2001, 98, 10533–10540. [Google Scholar] [CrossRef]
- Ludwig, R. Water: From Clusters to the Bulk. Angew. Chem. Int. Ed. 2001, 40, 1808–1827. [Google Scholar] [CrossRef]
- Huisken, F.; Stemmler, M. Infrared photodissociation of small methanol clusters. Chem. Phys. Lett. 1988, 144, 391–395. [Google Scholar] [CrossRef]
- Panja, S.K.; Haddad, B.; Debdab, M.; Kiefer, J.; Chaker, Y.; Bresson, S.; Paolone, A. Cluster Formation through Hydrogen Bond Bridges across Chloride Anions in a Hydroxyl-Functionalized Ionic Liquid. ChemPhysChem 2019, 20, 936–940. [Google Scholar] [CrossRef]
- Niemann, T.; Strate, A.; Ludwig, R.; Zeng, H.J.; Menges, F.S.; Johnson, M.A. Spectroscopic Evidence for an Attractive Cation–Cation Interaction in Hydroxy-Functionalized Ionic Liquids: A Hydrogen-Bonded Chain-like Trimer. Angew. Chem. Int. Ed. 2018, 57, 15364–15368. [Google Scholar] [CrossRef] [PubMed]
- Menges, F.S.; Zeng, H.J.; Kelleher, P.J.; Gorlova, O.; Johnson, M.A.; Niemann, T.; Strate, A.; Ludwig, R. Structural Motifs in Cold Ternary Ion Complexes of Hydroxyl-Functionalized Ionic Liquids: Isolating the Role of Cation–Cation Interactions. J. Phys. Chem. Lett. 2018, 9, 2979–2984. [Google Scholar] [CrossRef] [PubMed]
- Thummuru, D.N.R.; Mallik, B.S. Structure and Dynamics of Hydroxyl-Functionalized Protic Ammonium Carboxylate Ionic Liquids. J. Phys. Chem. A 2017, 121, 8097–8107. [Google Scholar] [CrossRef] [PubMed]
- Fakhraee, M.; Zandkarimi, B.; Salari, H.; Gholami, M.R. Hydroxyl-Functionalized 1-(2-Hydroxyethyl)-3-methyl Imidazolium Ionic Liquids: Thermodynamic and Structural Properties using Molecular Dynamics Simulations and ab Initio Calculations. J. Phys. Chem. B 2014, 118, 14410–14428. [Google Scholar] [CrossRef]
- Panja, S.K.; Haddad, B.; Kiefer, J. Clusters of the Ionic Liquid 1-Hydroxyethyl-3-methylimidazolium Picrate: From Theoretical Prediction in the Gas Phase to Experimental Evidence in the Solid State. ChemPhysChem 2018, 19, 3061–3068. [Google Scholar] [CrossRef]
- Knorr, A.; Ludwig, R. Cation-cation clusters in ionic liquids: Cooperative hydrogen bonding overcomes like-charge repulsion. Sci. Rep. 2015, 5, 17505. [Google Scholar] [CrossRef]
- Knorr, A.; Stange, P.; Fumino, K.; Weinhold, F.; Ludwig, R. Spectroscopic Evidence for Clusters of Like-Charged Ions in Ionic Liquids Stabilized by Cooperative Hydrogen Bonding. ChemPhysChem 2016, 17, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Strate, A.; Niemann, T.; Michalik, D.; Ludwig, R. When Like Charged Ions Attract in Ionic Liquids: Controlling the Formation of Cationic Clusters by the Interaction Strength of the Counterions. Angew. Chem. Int. Ed. 2017, 56, 496–500. [Google Scholar] [CrossRef]
- Niemann, T.; Zaitsau, D.; Strate, A.; Villinger, A.; Ludwig, R. Cationic clustering influences the phase behaviour of ionic liquids. Sci. Rep. 2018, 8, 14753. [Google Scholar] [CrossRef]
- Morais, E.M.; Abdurrokhman, I.; Martinelli, A. Solvent-free synthesis of protic ionic liquids. Synthesis, characterization and computational studies of triazolium based ionic liquids. J. Mol. Liq. 2022, 360, 119358. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed]
- Becke, A. Density-functional thermochemistry. III. The role of exact exchange Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals Self-consistent molecular orbital methods. XX. A basis set for. J. Chem. Phys. J. Chem. Phys. J. Chem. Phys. J. Chem. Phys. Hg J. Chem. Phys. J. Chem. Phys. J. Chem. Phys 1993, 981, 448799–449982. [Google Scholar]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, G.M.J. Revision A.02; Gaussian Inc.: Wallingford, UK, 2016. [Google Scholar]
- Jamróz, M.H. Vibrational Energy Distribution Analysis (VEDA): Scopes and limitations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 114, 220–230. [Google Scholar] [CrossRef]
- Kempter, V.; Kirchner, B. The role of hydrogen atoms in interactions involving imidazolium-based ionic liquids. J. Mol. Struct. 2010, 972, 22–34. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Simon, S.; Duran, M.; Dannenberg, J.J. How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? J. Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef]
- Kumar, S. Curcumin as a potential multiple-target inhibitor against SARS-CoV-2 Infection: A detailed interaction study using quantum chemical calculations. J. Serbian Chem. Soc. 2023, 4, 381–394. [Google Scholar] [CrossRef]
- Kumar, S.; Das, A. Mimicking trimeric interactions in the aromatic side chains of the proteins: A gas phase study of indole[ellipsis (horizontal)](pyrrole)2 heterotrimer. J. Chem. Phys. 2012, 136, 174302. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kaul, I.; Biswas, P.; Das, A. Structure of 7-Azaindole···2-Fluoropyridine Dimer in a Supersonic Jet: Competition between N–H···N and N–H···F Interactions. J. Phys. Chem. A 2011, 115, 10299–10308. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Singh, S.K.; Vaishnav, J.K.; Hill, J.G.; Das, A. Interplay among Electrostatic, Dispersion, and Steric Interactions: Spectroscopy and Quantum Chemical Calculations of π-Hydrogen Bonded Complexes. ChemPhysChem 2017, 18, 828–838. [Google Scholar] [CrossRef]
- Singh, S.K.; Kumar, S.; Das, A. Competition between n →π*(Ar) and conventional hydrogen bonding (N–H⋯N) interactions: An ab initio study of the complexes of 7-azaindole and fluorosubstituted pyridines. Phys. Chem. Chem. Phys. 2014, 16, 8819–8827. [Google Scholar] [CrossRef]
- Kumar, S.; Pande, V.; Das, A. π-Hydrogen Bonding Wins over Conventional Hydrogen Bonding Interaction: A Jet-Cooled Study of Indole···Furan Heterodimer. J. Phys. Chem. A 2012, 116, 1368–1374. [Google Scholar] [CrossRef]
- Kumar, S.; Mukherjee, A.; Das, A. Structure of Indole···Imidazole Heterodimer in a Supersonic Jet: A Gas Phase Study on the Interaction between the Aromatic Side Chains of Tryptophan and Histidine Residues in Proteins. J. Phys. Chem. A 2012, 116, 11573–11580. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Das, A. Effect of acceptor heteroatoms on π-hydrogen bonding interactions: A study of indole⋅⋅⋅thiophene heterodimer in a supersonic jet. J. Chem. Phys. 2012, 137, 094309. [Google Scholar] [CrossRef]
- Su, P.; Li, H. Energy decomposition analysis of covalent bonds and intermolecular interactions. J. Chem. Phys. 2009, 131, 014102. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Paschoal, V.H.; Faria, L.F.O.; Ribeiro, M.C.C. Vibrational Spectroscopy of Ionic Liquids. Chem. Rev. 2017, 117, 7053–7112. [Google Scholar] [CrossRef] [PubMed]
- Khudozhitkov, A.E.; Neumann, J.; Niemann, T.; Zaitsau, D.; Stange, P.; Paschek, D.; Stepanov, A.G.; Kolokolov, D.I.; Ludwig, R. Hydrogen Bonding Between Ions of Like Charge in Ionic Liquids Characterized by NMR Deuteron Quadrupole Coupling Constants—Comparison with Salt Bridges and Molecular Systems. Angew. Chem. Int. Ed. 2019, 58, 17863–17871. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, J.; Wagenfeld, S.; Kerlé, D. Chain length effects on the vibrational structure and molecular interactions in the liquid normal alkyl alcohols. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 189, 57–65. [Google Scholar] [CrossRef]
- Schmidt, D.A.; Miki, K. Structural Correlations in Liquid Water: A New Interpretation of IR Spectroscopy. J. Phys. Chem. A 2007, 111, 10119–10122. [Google Scholar] [CrossRef]
- Kumar, S.; Biswas, P.; Kaul, I.; Das, A. Competition between Hydrogen Bonding and Dispersion Interactions in the Indole···Pyridine Dimer and (Indole)2···Pyridine Trimer Studied in a Supersonic Jet. J. Phys. Chem. A 2011, 115, 7461–7472. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, S.K.; Calabrese, C.; Maris, A.; Melandri, S.; Das, A. Structure of saligenin: Microwave, UV and IR spectroscopy studies in a supersonic jet combined with quantum chemistry calculations. Phys. Chem. Chem. Phys. 2014, 16, 17163. [Google Scholar] [CrossRef]
- Sada, P.K.; Bar, A.; Jassal, A.K.; Kumar, P.; Srikrishna, S.; Singh, A.K.; Kumar, S.; Singh, L.; Rai, A. A Novel Rhodamine Probe Acting as Chemosensor for Selective Recognition of Cu2+ and Hg2+ Ions: An Experimental and First Principle Studies. J. Fluoresc. 2023. [Google Scholar] [CrossRef]
- Kiefer, J.; Pye, C.C. Structure of the Room-Temperature Ionic Liquid 1-Hexyl-3-methylimidazolium Hydrogen Sulfate: Conformational Isomerism. J. Phys. Chem. A 2010, 114, 6713–6720. [Google Scholar] [CrossRef]
- Wulf, A.; Fumino, K.; Ludwig, R. Comment on “New Interpretation of the CH Stretching Vibrations in Imidazolium-Based Ionic Liquids”. J. Phys. Chem. A 2010, 114, 685–686. [Google Scholar] [CrossRef]
- Grondin, J.; Lassègues, J.-C.; Cavagnat, D.; Buffeteau, T.; Johansson, P.; Holomb, R. Revisited vibrational assignments of imidazolium-based ionic liquids. J. Raman Spectrosc. 2011, 42, 733–743. [Google Scholar] [CrossRef]
- Andersson, M.P.; Uvdal, P. New Scale Factors for Harmonic Vibrational Frequencies Using the B3LYP Density Functional Method with the Triple-ζ Basis Set 6-311+G(d,p). J. Phys. Chem. A 2005, 109, 2937–2941. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, H.-O.; Ozawa, R. Structure of Ionic Liquids and Ionic Liquid Compounds: Are Ionic Liquids Genuine Liquids in the Conventional Sense? Adv. Chem. Phys. 2005, 131, 85–104. [Google Scholar]
- Ozawa, R.; Hayashi, S.; Saha, S.; Kobayashi, A.; Hamaguchi, H.-o. Rotational Isomerism and Structure of the 1-Butyl-3-methylimidazolium Cation in the Ionic Liquid State. Chem. Lett. 2003, 32, 948–949. [Google Scholar] [CrossRef]
- Kumar Panja, S.; Kumar, S. Weak intra and intermolecular interactions via aliphatic hydrogen bonding in piperidinium based ionic Liquids: Experimental, topological and molecular dynamics studies. J. Mol. Liq. 2023, 375, 121354. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO 7.0; Theoretical Chemistry Institute, University of Wisconsin-Madison: Madison, WI, USA, 2018; Available online: https://nbo6.chem.wisc.edu/biblio_css.htm (accessed on 1 September 2024).
- Kumar, V.; Kumar, R.; Kumar, N.; Kumar, S. Solvation Dynamics of Oxadiazoles as Potential Candidate for Drug Preparation. Asian J. Chem. 2023, 35, 991–998. [Google Scholar] [CrossRef]
- Prasana, J.C.; Muthu, S.; Abraham, C.S. Molecular docking studies, charge transfer excitation and wave function analyses (ESP, ELF, LOL) on valacyclovir: A potential antiviral drug. Comput. Biol. Chem. 2019, 78, 9–17. [Google Scholar]
- Kumar, S.; Panja, S.K. Intermolecular charge-transfer complex between solute and ionic liquid: Experimental and theoretical studies. Theor. Chem. Acc. 2023, 142, 126. [Google Scholar] [CrossRef]
- Kumar Panja, S.; Kumar, S.; Fazal, A.D.; Bera, S. Molecular aggregation kinetics of Heteropolyene: An Experimental, topological and solvation dynamics studies. J. Photochem. Photobiol. A Chem. 2023, 445, 115084. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FTIR Band | Vibrational Band Assignment |
|---|---|
| 651 | ω(N-H)/CH3(N) CN Str |
| 702 | CH2(N)/CH3(N)CN bend |
| 755 | ω(N-H) |
| 868 | ring CC bend |
| 944 | ρas(CH2) |
| 1069 | νC-O |
| 1166 | Ring as Str CH2(N) and CH3(N)CN Str/CC |
| 1255 | Ring ip as str |
| 1338 | Ring, ν(CH2-N) |
| 1386 | δ(N-H) ip vibration |
| 1428 | δ(O-H) ip vibration |
| 1455 | δ(CH2)/CCH, HCH as bend |
| 1475 | Str CH2(N)/CH2(N)CN Str |
| 1634 | Ring vibration C=C |
| 1636 | Ring vibration C=N |
| 2882 | νs(CH2) as Str |
| 2955 | νas(CH2) as Str |
| 3095 | ν(N)CH2 as Str |
| 3149 | νC(4/5)-H str |
| 3364 | ν(N-H)/(O-H) |
| Band Assignment | Experiment: Raman | Theory: Ion Pair | Dimeric Ion-Pair Cluster |
|---|---|---|---|
| 3140/3110 | 3140 | 3140 | |
| 3070 | 3171 | 3018 | |
| 3020 | 3125 | 3178 | |
| 2964 | 3021 | 3005 | |
| 1567 | 1590 | 1579 |
| NAOs | Donor NBO (i) | Occupancy | NAOs | Acceptor NBO (j) | Occupancy | E(2) kJ/mol |
|---|---|---|---|---|---|---|
| 27 | n(1)O1 | 1.96153 | 106 | σ*(1)N21-H34 | 0.03328 | 30.00 |
| 28 | n(2)O1 | 1.94428 | 106 | σ*(1)N21-H34 | 0.03328 | 5.10 |
| 30 | n(1)O17 | 1.96158 | 87 | σ*(1)N4-H19 | 0.03318 | 29.92 |
| 31 | n(2)O17 | 1.94426 | 87 | σ*(1)N4-H19 | 0.03318 | 4.94 |
| 35 | n(3)Cl35 | 1.95316 | 93 | σ*(1)C11-H12 | 0.05446 | 46.19 |
| 36 | n(4)Cl35 | 1.87669 | 93 | σ*(1)C11-H12 | 0.05446 | 15.90 |
| 33 | n(1)Cl35 | 1.99900 | 98 | σ*(1)O17-H18 | 0.10664 | 6.69 |
| 36 | n(4)Cl35 | 1.87669 | 98 | σ*(1)O17-H18 | 0.10664 | 134.31 |
| 37 | n(1)Cl36 | 1.99900 | 79 | σ*(1)O1-H2 | 0.10635 | 6.65 |
| 40 | n(4)Cl36 | 1.87718 | 79 | σ*(1)O1-H2 | 0.10635 | 134.01 |
| 39 | n(3)Cl36 | 1.95307 | 112 | σ*(1)C28-H29 | 0.05436 | 46.40 |
| 40 | n(4)Cl36 | 1.87718 | 112 | σ*(1)C28-H29 | 0.05436 | 15.61 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panja, S.K.; Kumar, S.; Haddad, B.; Patel, A.R.; Villemin, D.; Amine, H.-M.; Bera, S.; Debdab, M. Role of Multiple Intermolecular H-Bonding Interactions in Molecular Cluster of Hydroxyl-Functionalized Imidazolium Ionic Liquid: An Experimental, Topological, and Molecular Dynamics Study. Physchem 2024, 4, 369-388. https://doi.org/10.3390/physchem4040026
Panja SK, Kumar S, Haddad B, Patel AR, Villemin D, Amine H-M, Bera S, Debdab M. Role of Multiple Intermolecular H-Bonding Interactions in Molecular Cluster of Hydroxyl-Functionalized Imidazolium Ionic Liquid: An Experimental, Topological, and Molecular Dynamics Study. Physchem. 2024; 4(4):369-388. https://doi.org/10.3390/physchem4040026
Chicago/Turabian StylePanja, Sumit Kumar, Sumit Kumar, Boumediene Haddad, Abhishek R. Patel, Didier Villemin, Hakkoum-Mohamed Amine, Sayantan Bera, and Mansour Debdab. 2024. "Role of Multiple Intermolecular H-Bonding Interactions in Molecular Cluster of Hydroxyl-Functionalized Imidazolium Ionic Liquid: An Experimental, Topological, and Molecular Dynamics Study" Physchem 4, no. 4: 369-388. https://doi.org/10.3390/physchem4040026
APA StylePanja, S. K., Kumar, S., Haddad, B., Patel, A. R., Villemin, D., Amine, H.-M., Bera, S., & Debdab, M. (2024). Role of Multiple Intermolecular H-Bonding Interactions in Molecular Cluster of Hydroxyl-Functionalized Imidazolium Ionic Liquid: An Experimental, Topological, and Molecular Dynamics Study. Physchem, 4(4), 369-388. https://doi.org/10.3390/physchem4040026