
Assignment of the Vibrational Spectra of Diiron Nonacarbonyl, Fe2(CO)9


Abstract
:1. Introduction
2. Materials and Methods
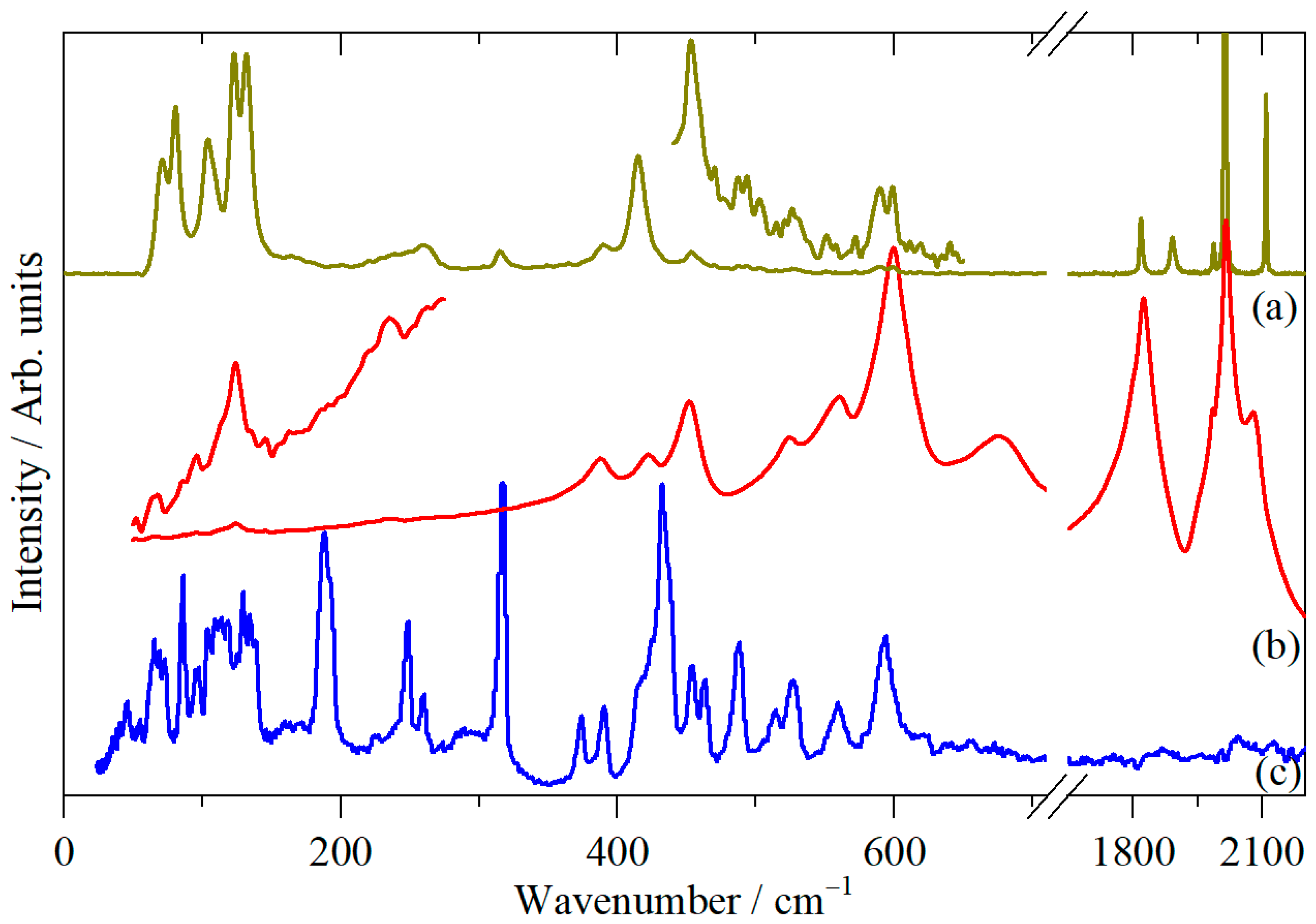
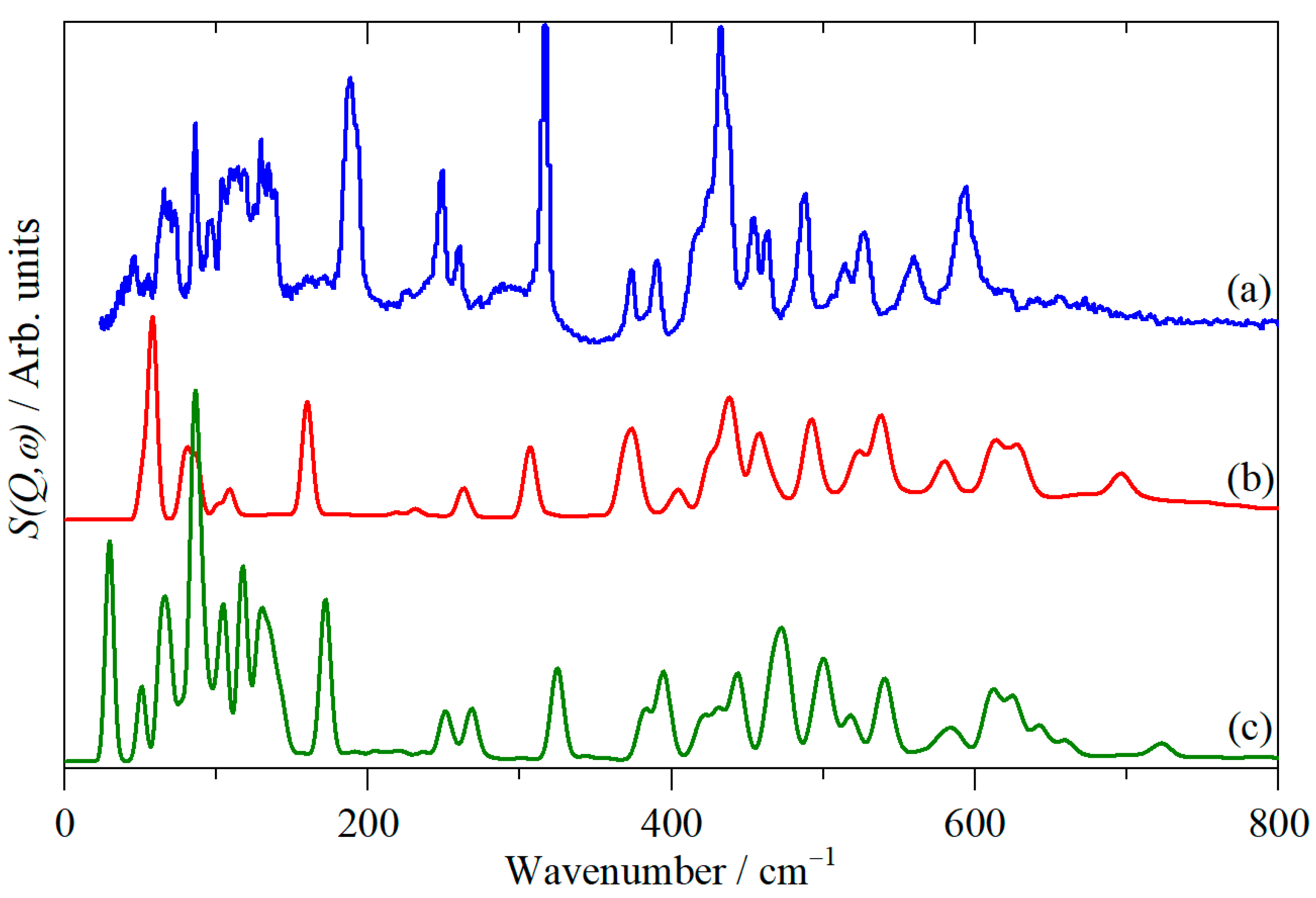
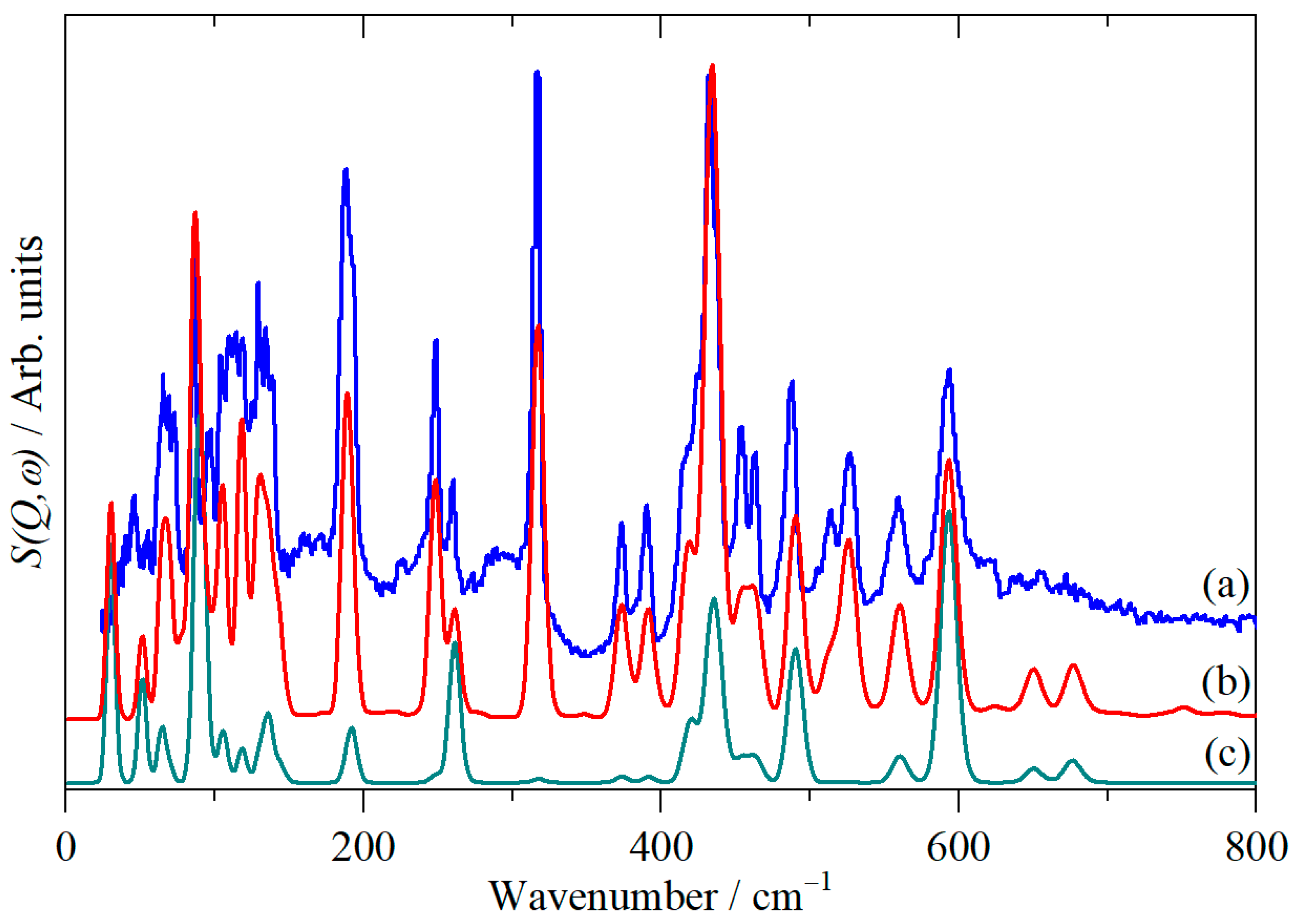
3. Results and Discussion
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dewar, J.; Jones, H.O. The physical and chemical properties of iron carbonyl. Proc. Roy. Soc. 1905, A76, 558–577. [Google Scholar] [CrossRef] [Green Version]
- Mond, L.; Langer, C.; Quincke, F. Action of carbon monoxide on nickel. J. Chem. Soc. Trans. 1890, 57, 749–753. [Google Scholar] [CrossRef] [Green Version]
- Mond, L.; Langer, C. On iron carbonyls. J. Chem. Soc. Trans. 1891, 59, 1090–1093. [Google Scholar] [CrossRef] [Green Version]
- Powell, H.M.; Ewens, R.V.G. The crystal structure of iron enneacarbonyl. J. Chem. Soc. 1939, 286–292. [Google Scholar] [CrossRef]
- Sheline, R.K.; Pitzer, K.S. The infrared spectra and structures of the iron carbonyls. J. Amer. Chem. Soc. 1950, 72, 1107. [Google Scholar] [CrossRef]
- Butler, I.S.; Kishner, S.; Plowman, K.R. Vibrational spectra of solid tri-μ-carbonyl(hexacarbonyl)di-iron(0) Fe2(CO)9. J. Mol. Struct. 1978, 43, 9. [Google Scholar] [CrossRef]
- Adams, D.M.; Taylor, I.D. Solid-state metal carbonyls Part 3.-An infrared study of Fe2(CO)9, tri-μ-carbonyl(hexacarbonyl)di-iron(0). J. Chem. Soc. Faraday Trans. 1982, 78, 1551–1559. [Google Scholar] [CrossRef]
- Jang, J.H.; Lee, J.G.; Lee, H.; Xie, Y.; Schaefer III, H.F. Molecular structures and vibrational frequencies of iron carbonyls: Fe(CO)5, Fe2(CO)9 and Fe3(CO)12. J. Phys. Chem. A 1998, 102, 5298–5304. [Google Scholar] [CrossRef]
- Mitchell, P.C.H.; Parker, S.F.; Ramirez-Cuesta, A.J.; Tomkinson, J. Vibrational Spectroscopy with Neutrons, with Applications in Chemistry, Biology, Materials Science and Catalysis; World Scientific: Singapore, 2005. [Google Scholar]
- Parker, S.F.; Fernandez-Alonso, F.; Ramirez-Cuesta, A.J.; Tomkinson, J.; Rudić, S.; Pinna, R.S.; Gorini, G.; Fernández Castañon, J. Recent and future developments on TOSCA at ISIS. J. Phys. Conf. Ser. 2014, 554, 12003. [Google Scholar] [CrossRef]
- Pinna, R.S.; Rudić, S.; Parker, S.F.; Armstrong, J.; Zanetti, M.; Škoro, G.; Waller, S.P.; Zacek, D.; Smith, C.A.; Capstick, M.J.; et al. The neutron guide upgrade of the TOSCA spectrometer. Nucl. Instrum. Methods Phys. Res. Sect. A 2018, 896, 68–74. [Google Scholar] [CrossRef] [Green Version]
- The ISIS Neutron and Muon Source. Available online: https://www.isis.stfc.ac.uk/Pages/About.aspx (accessed on 14 February 2022).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tkatchenko, A.; Scheffler, M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Refson, K.; Clark, S.J.; Tulip, P.R. Variational density-functional perturbation theory for dielectrics and lattice dynamics. Phys. Rev. B 2006, 73, 155114. [Google Scholar] [CrossRef] [Green Version]
- Dymkowski, K.; Parker, S.F.; Fernandez-Alonso, F.; Mukhopadhyay, S. AbINS: The modern software for INS interpretation. Physica B 2018, 551, 443–448. [Google Scholar] [CrossRef]
- Safa, M.; Dong, Z.; Huang, Y. Examining the structural changes in Fe2(CO)9 under high external pressures by Raman spectroscopy. Can. J. Chem. 2007, 85, 866–872. [Google Scholar] [CrossRef]
- Cotton, F.A.; Troup, J.M. Accurate determination of a classic structure in the metal carbonyl field: Nonacarbonyldi-iron. J. Chem. Soc. Dalton 1974, 800–802. [Google Scholar] [CrossRef]
- Parker, S.F.; Jayasooriya, U.A. Assignment of the solid state spectra of the group VI hexacarbonyls by inelastic neutron scattering spectroscopy. Phys. Chem. Chem. Phys. 2019, 21, 24950–24955. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, J.; Parker, S.F. Exploiting the quasi-invariance of molecular vibrational eigenvectors. Spectrochim. Acta 2011, 79A, 2017–2019. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.B., III; Onaka, S.; Shriver, D.F.; Daniels, L.; Hance, R.L.; Hutchinson, B.; Shipley, R. Metal isotope and temperature effects on the Raman spectra of diironenneacarbonyl, Fe2(CO)9, and triirondodecacarbonyl, Fe3(CO)12. Identification of Fe-Fe stretching frequencies. Inorg. Chim. Acta 1977, 24, L92. [Google Scholar] [CrossRef]
- Green, J.C.; Green, M.L.H.; Parkin, G. The occurrence and representation of three-centre two-electron bonds in covalent inorganic compounds. Chem. Commun. 2012, 48, 11481–11503. [Google Scholar] [CrossRef] [PubMed]
- Herber, R.H.; Fischer, J. Lattice-dynamics from Raman and Mössbauer studies - Extension to organometallic compound of iron. Can. J. Spectrosc. 1974, 19, 21–24. [Google Scholar]
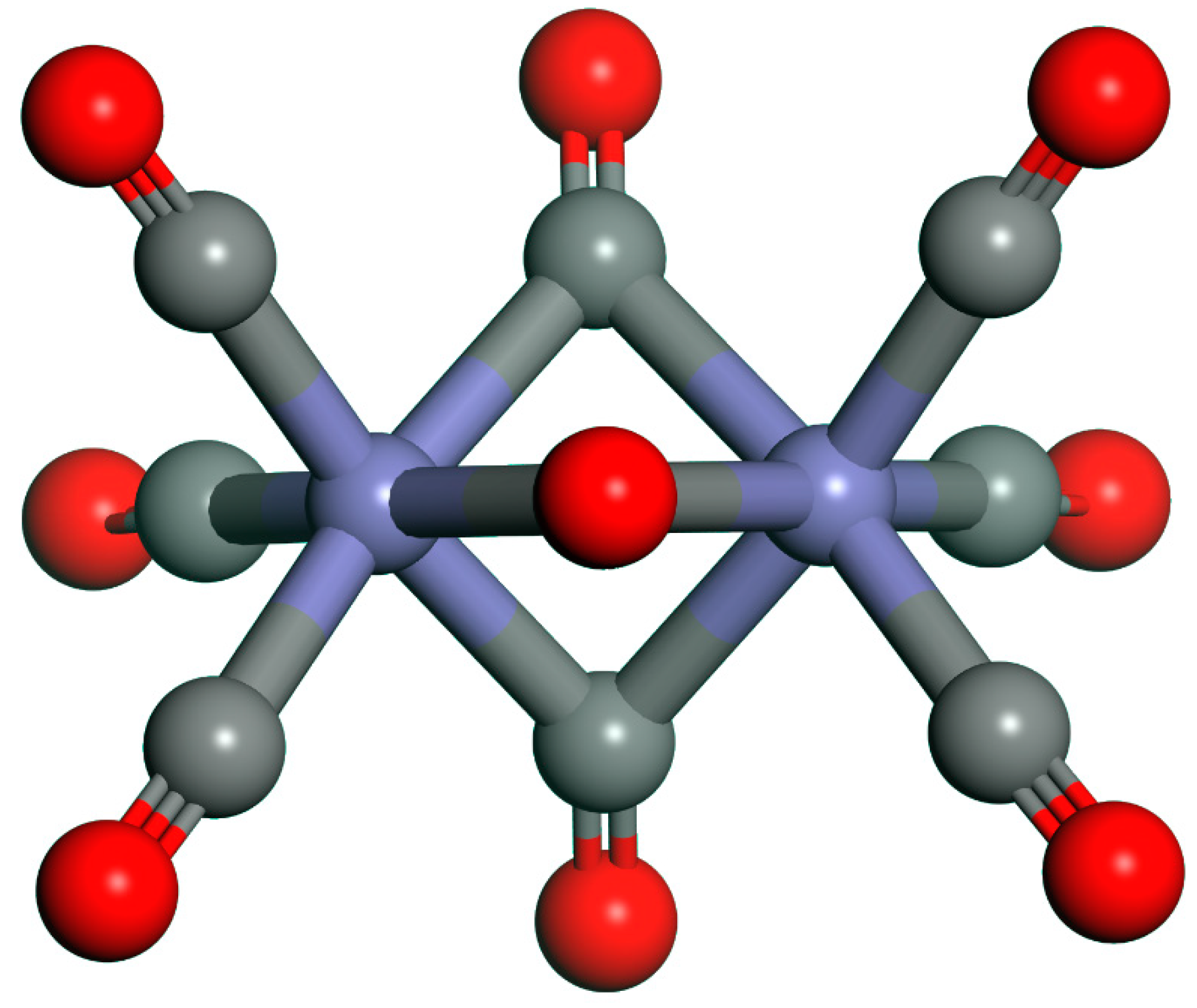
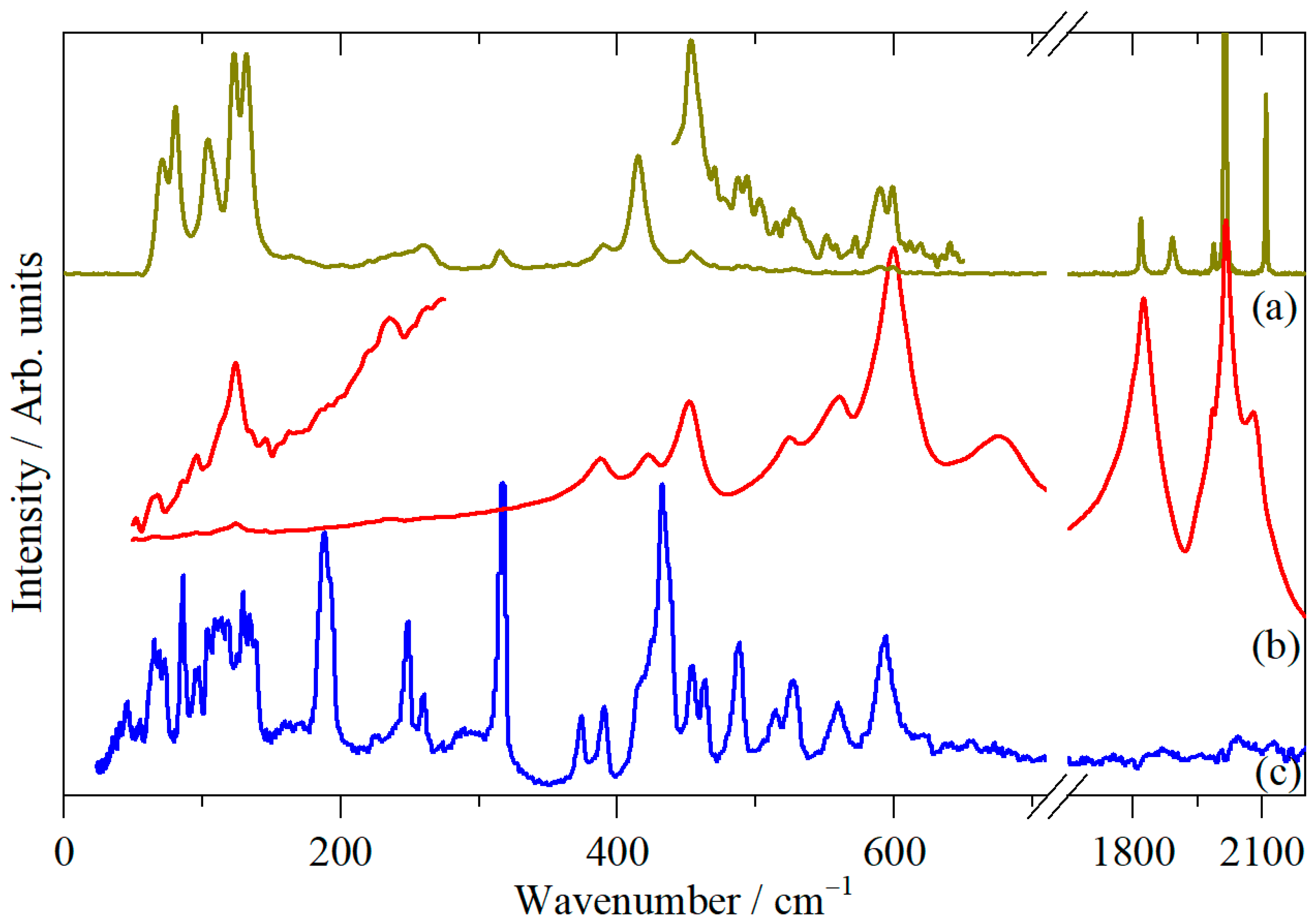
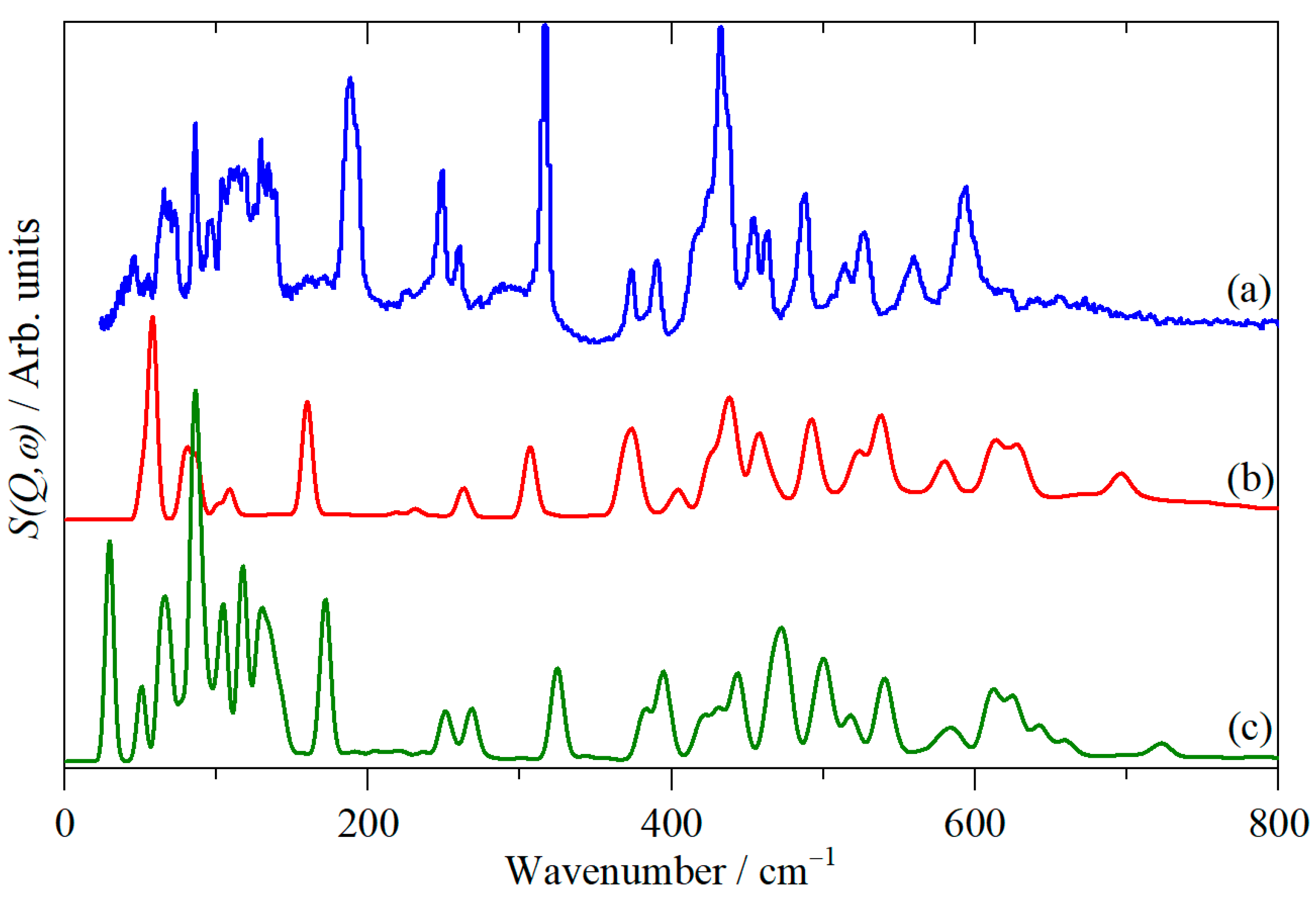


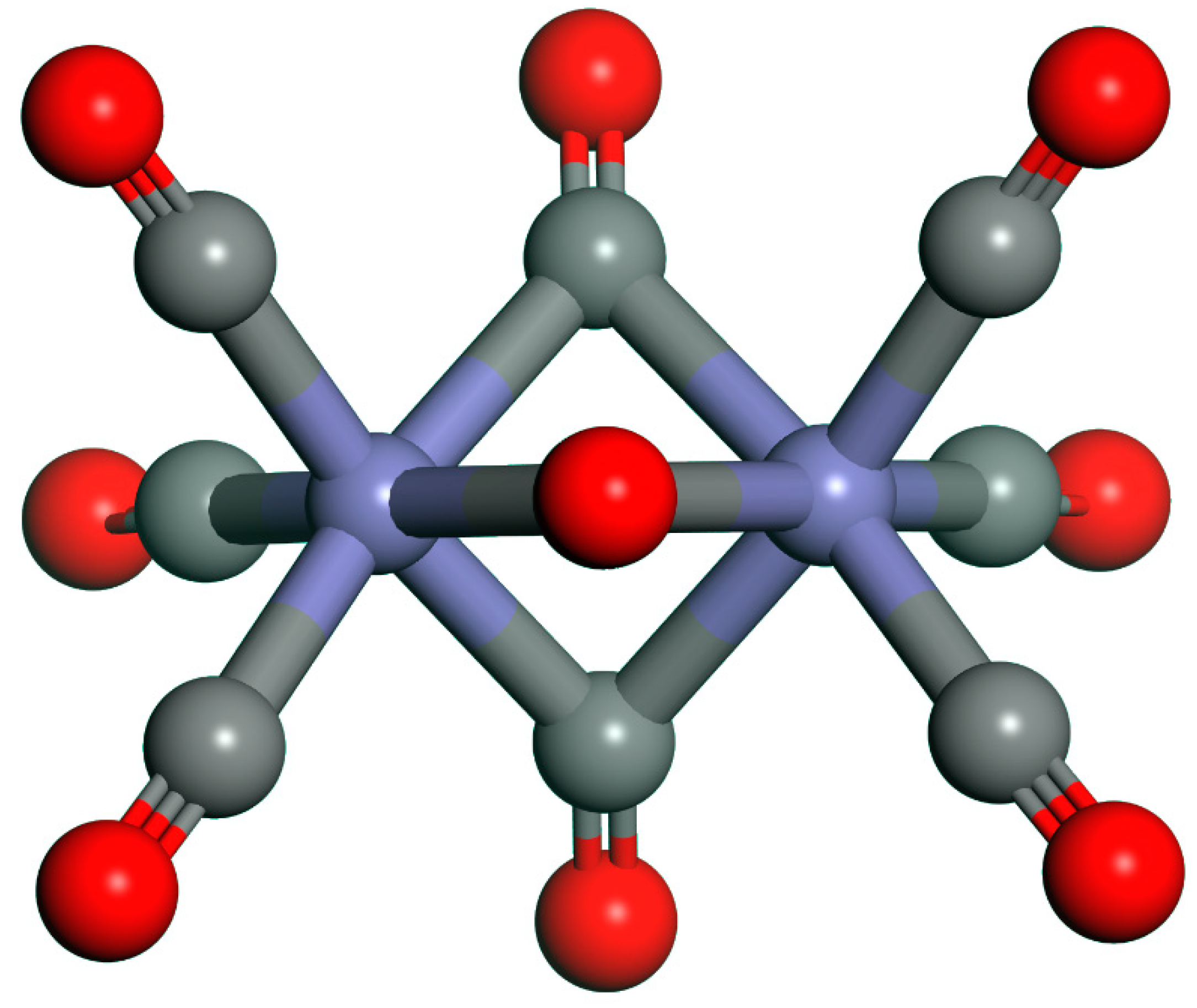{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Previous Work/cm−1 | This Work/cm−1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| D3h | Butler [6] | Adams [7] | Jang [8] a BP86/DZP | Jang [8] a B3LYP/DZP | B3LYP aug-ccVTZ | CASTEP Average b | CASTEP Range c | Final d | |
| ν1 | 2112 | 2079(0) | 2170(0) | 2183 | 2074 | 1 | 2109 | ||
| ν2 | 1891 | 1895(0) | 1957(0) | 1964 | 1875 | 0 | 1893 | ||
| ν3 | 480 | 620(0) | 624(0) | 622 | 643 | 1 | 593 | ||
| ν4 | 415 | 478(0) | 466(0) | 466 | 495 | 0 | 433 | ||
| ν5 | 260 | 399(0) | 397(0) | 405 | 420 | 0 | 415 | ||
| ν6 | 137 | 255(0) | 259(0) | 263 | 269 | 1 | 260 | ||
| ν7 | 237 | 79(0) | 83(0) | 86 | 123 | 11 | 117/128 | ||
| ν8 | 508(0) | 525(0) | 524 | 519 | 0 | 433 | |||
| ν9 | 371(0) | 376(0) | 369 | 383 | 1 | 317 | |||
| ν10 | 77(0) | 82(0) | 78 | 135 | 12 | 129/141 | |||
| ν11 | 2016 | 2020 | 2013(1322) | 2101(1527) | 2115 | 1978 | 0 | 2015 | |
| ν12 | 1814 | 1817 | 1870(682) | 1918(899) | 1924 | 1809 | 0 | 1824 | |
| ν13 | 604 | 605 | 624(134) | 631(120) | 630 | 626 | 1 | 593 | |
| ν14 | 528 | 525 | 532(4) | 545(10) | 539 | 541 | 0 | 513/526 | |
| ν15 | 390 | 390 | 461(25) | 455(40) | 457 | 475 | 0 | 438 | |
| ν16 | 451 | 454 | 433(1) | 438(7) | 438 | 444 | 1 | 391/416 | |
| ν17 | 175 | 174 | 376(0) | 374(1) | 377 | 396 | 0 | 317 | |
| ν18 | 126 | 126 | 107(1) | 110(1) | 109 | 133 | 5 | 123 | |
| ν19 | 106 | 105 | 85(0) | 88(0) | 89 | 106 | 2 | 106 | |
| ν20 | 83 | 85 | 54(0) | 58(0) | 59 | 86 | 1 | 86 | |
| ν21 | 417(0) | 427(0) | 426 | 432 | 1 | 317 | |||
| ν22 | 51(0) | 54(0) | 53 | 93 | 11 | 87/99 | |||
| ν23 | 2088 | 2038(1747) | 2120(2210) | 2134 | 2038 | 42 | 2079 | ||
| ν24 | 690 | 675(798) | 697(763) | 697 | 692 | 62 | 650/676 | ||
| ν25 | 564 | 573(98) | 583(165) | 581 | 598 | 9 | 560 | ||
| ν26 | 426 | 451(18) | 434(52) | 433 | 475 | 2 | 422 | ||
| ν27 | 130 | 225(2) | 228(1) | 232 | 251 | 2 | 192 | ||
| ν28 | 166 | 93(0) | 98(0) | 101 | 141 | 7 | 138/145 | ||
| ν29 | 1990 | 2008(0) | 2096(0) | 2110 | 1976 | 0 | 1988 | ||
| ν30 | 590 | 604(0) | 617(0) | 613 | 612 | 1 | 490 | ||
| ν31 | 493 | 486(0) | 491(0) | 492 | 502 | 1 | 453/463 | ||
| ν32 | 468 | 452(0) | 443(0) | 441 | 467 | 1 | 433 | ||
| ν33 | 315 | 314(0) | 313(0) | 308 | 325 | 0 | 248 | ||
| ν34 | 114 | 159(0) | 159(0) | 160 | 172 | 0 | 188 | ||
| ν35 | 89 | 82(0) | 78(0) | 81 | 119 | 1 | 118 | ||
| ν36 | 67 | 75(0) | 55(0) | 58 | 90 | 4 | 88/92 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parker, S.F. Assignment of the Vibrational Spectra of Diiron Nonacarbonyl, Fe2(CO)9. Physchem 2022, 2, 108-115. https://doi.org/10.3390/physchem2020008
Parker SF. Assignment of the Vibrational Spectra of Diiron Nonacarbonyl, Fe2(CO)9. Physchem. 2022; 2(2):108-115. https://doi.org/10.3390/physchem2020008
Chicago/Turabian StyleParker, Stewart F. 2022. "Assignment of the Vibrational Spectra of Diiron Nonacarbonyl, Fe2(CO)9" Physchem 2, no. 2: 108-115. https://doi.org/10.3390/physchem2020008
APA StyleParker, S. F. (2022). Assignment of the Vibrational Spectra of Diiron Nonacarbonyl, Fe2(CO)9. Physchem, 2(2), 108-115. https://doi.org/10.3390/physchem2020008