Abstract
The possibility of using graphane monolayer crystals as an electrode material is becoming popular. Graphane is stable at room temperature and has a large surface area, but its chemical inertness hinders its direct interactions with Li ions. In this study, we performed density functional theory calculations to study the energetic stability and structural and electronic properties of Li on graphane with various CH divacancy configurations (v12, v13, and v14). The results show that the adsorption of the Li atom reduces the formation energy of the CH divacancy configurations. The Li-v12 is most stable with the highest binding energy of 3.25 eV/Li and relaxes to in-plane with other C atoms. Altering the Li charge state to have Li−1-v12 or Li+1-v12 affects the energetic stability and electronic characters of Li-v12. The Li−1-v12 (Li+1-v12) slightly (greatly) reduces the binding force between the Li and v12 configuration, and furthermore it improves (deteriorates) the conductivity of the structure. Further investigation of graphane with vacancies is encouraged due to these intriguing observations, as it holds promise for potential utilization as an electrode material.
1. Introduction
Lithium-ion batteries (LIBs) have been widely used as power sources for most electronic devices [1]. The LIBs are known to be non-toxic, have long discharging rates and high energy density, etc. [1,2,3]. The most-used electrode in LIBs is graphite [1]. Nevertheless, many of its properties, such as its specific capacity, are unlikely to meet the increasing future demand for high energy. The search for alternative electrode materials with distinct properties from graphite is rapidly intensifying [4,5,6]. Two-dimensional (2D) materials are emerging as promising candidates to address future energy demands due to their high electrical conductivity, reduced dimensionality, high thermodynamic stability, large surface area, etc., distinguishing them from their bulk counterparts [7,8,9]. Some of these properties make 2D materials well-suited to accommodate a greater number of Li atoms, among other advantages.
Graphane [10] monolayer material falls under materials belonging to the graphene derivatives where each carbon is bonded to hydrogen atoms in an alternating pattern. The realization of graphane was first reported by Sluiter et al. [11], based on the density functional theory (DFT) [12]. Graphane was later synthesized [13,14,15] by applying hydrogen plasma to graphene monolayer samples. The graphane samples were found to be stable at room temperatures [13]. Each carbon atom’s bonding network takes on the form of sp3 hybridization due to the presence of hydrogen atoms [14].
Experimental studies report that graphane has insulating characters [13,16,17], with some reporting an energy band gap range of 3.50–3.90 [16,17]. This was supported by the theoretical studies predicting the energy band gap of 3.50 eV (standard DFT) [10], 5.40 eV (GW) [18], and 4.21 eV (HSE06) [19]. Graphane has a large surface area, high volumetric capacity, and non-toxic samples [14,20,21]. These indicators suggest that a graphane monolayer could find utility in a range of technological applications. The focus of this work is on the mechanisms of anchoring the Li ions on the graphane monolayer for electrode exploitation.
Theoretically, an inertness [14,20,21] (under ambient conditions) of C atoms conjugating a graphane monolayer due to the sp3 bonding network could physisorb instead of chermisorbing Li ions, which will be a setback for anchoring Li atoms.Watcharinyanon et al. [22] studied the intercalation of Li ions on graphane supported by a SiC(0001) substrate using low-energy electron microscopy, micro-low-energy electron diffraction, and photoelectron spectroscopy. It was reported that instead of covalent bonding formation with graphane substrate, Li atoms form islands among themselves at a certain temperature. The DFT study by Yang et al. [23] revealed that Li ions are screened by H atoms, as no such Li chemisorption is taking place.
The modification of the graphane monolayer for the possibility of enhancing Li interaction is necessary. Several methods are known for introducing reactive sites on graphane, potentially enhancing its interaction with Li ions. These methods include structural modifications like creating vacancies, doping, and strain application, among others [14,20,21,24]. The creation of vacancies in graphane can primarily be in different forms: hydrogen (H) vacancy, carbon (C) vacancy, and carbon–hydrogen (CH) vacancy. The creation of multiple vacancies is also possible in the graphane monolayer. H divacancy, trivacancy, and CH divacancy have been created in a graphane monolayer.These vacancies can be created by applying high or low ion bombardment, and they can also occur during synthetic processes [14,20,21,24]. Numerous properties of these mentioned vacancies have been examined, ranging from their energetic stabilities to magnetic properties [14,20,21,24,25,26,27,28]. With the exception of the divacancy structure, all other types of vacancies introduce mid-gap states within the band gap and exhibit magnetic properties.
The structural and electronic behavior of CH divacancies in graphane have been studied using DFT [28]. The structural reconstruction of the two adjacent vacancies has been reported, which breaks the hexagonal symmetry and translates into the new 5-8-5 defect structure (considered to be self-healed) which is thermodynamically stable. Graphane with a 5-8-5 defect structure is characterized as a wide band gap material [28]. Considering the potential use of graphane with this defect structure as an electrode holds merit. Notably, the adsorption of Li ions onto the graphane with a 5-8-5 defect structure remains unexplored. A comprehensive understanding of how Li anchors at different charge states on graphane is imperative. This helps to understand the energetic and electronic behavior of Li during charging and discharging. The standard DFT predicts wrong band gap sizes of semiconductor materials, including 2D materials. Although the computational methods such as the GW method [29] and the screened hybrid exchange correlation functional developed by Heyd, Scuseria, and Ernzerhof (HSE06 functional) [30] can predict the band gap better, they are computationally demanding in terms of time and resources. In this study, we used the HSE06 functional for all the calculations considered.
This study intends to understand the energetic stabilities (formation energy and binding energy), structural properties (bond length), and electronic properties (density of states) of Li on the graphane with the 5-8-5 defect structure at different Li charge states (+1 or −1). This study relies on a spin-polarized density functional theory approach, and the outcomes will serve as a reference for the experimentalists to corroborate their findings during practical synthesis and characterization processes.
2. Computational Details
All the spin-polarized calculations in this study have been performed using the hybrid density functional theory approach implemented within the Vienna ab initio simulation package (VASP) code [31]. The chosen hybrid exchange-correlation functional is the Heyd, Scuseria, and Ernzerhof [30], which contains a fraction of non-local Hartree–Fock exchange. The Fock mixing parameter used is 0.25, which is known to give the correct band gap of 2D materials. For the description of pseudopotentials, the projector augmented wave (PAW) methods [32] with C atoms are represented as 2s22p2, H atoms are represented by 1s1, and the Li adatom is represented as 2s1 valence electrons.
The kinetic energy cut-off of 500 eV was used for the expansion of the plane wavefunctions. For sampling the Brillouin zone of the graphane monolayer, the 6 × 6 × 1 k-mesh grid was used, employing the Monkhorst–Pack scheme [33] method. This k-mesh grid is doubled during the density of state calculations. The atomic positions were allowed to relax until their residual forces were less than 0.01 eVÅ−1, utilizing the Hellman–Feynman theorem. The total energies of the structures were allowed to converge to within 10−7 eV during the self-consistent field calculations of each relaxation. The 6 × 6 supercell of graphane was used for all our calculations. This supercell size was chosen to ensure that the divacancy interaction between the cell images along the x and y directions is minimal. The separation spacing between the layers was set to the converged 20 Å to avoid unwanted interactions due to periodic images. It is known that the introduction of charged lithiated vacancies would lead to spurious electrostatic interactions between periodic images. In the VASP calculations, the background charge (jellium) is assumed for the charged lithiated vacancies to be neutralized in each periodic supercell.
To study the energetic stability of Li on graphane with various CH divacancy configurations (v12, v13, and v14), their formation energies were calculated employing the Zhang–Northrup expression [34] below:
where is the total energy of Li on a CH divacancy configuration in a 6 × 6 graphane supercell and is the total energy of 6 × 6 pristine graphane. The in Equation (1) represents the chemical potential of H, C, and Li atoms calculated as the total energies per atom of isolated hydrogen molecules in a large box, and graphene and Li in a bulk body-centered cube (BCC), respectively. represents the number of atomic species (H, C, and Li atoms) removed or adsorbed by the graphane monolayer system.
To assess interactions between the Li adatoms and various configurations of CH divacancies in graphane monolayer, we calculated the binding energy . The binding energy is the energy needed to remove the Li atom from the graphane supercell as opposed to adsorption energy which is the energy due to the attraction of the Li atom and graphane supercell. In this work, is given as follows:
where is the total energy of the Li adatom on a CH divacancy in a 6 × 6 graphane supercell, is the total energy of the 6 × 6 graphane with divacancy, and is the total energy per adatom of Li in a bulk body-centered cube (BCC).
The calculated values are shown in Table 1. One must bear in mind that the interpretation of results is just the opposite of the well-known adsorption energy values. The positive value suggests the attractive force between the Li atom and graphane system, whereas the negative value will reveal the repulsive force. For us to consider the results is to be able to compare our values with that of Li bulk (BCC), also considered by Chan et al. [35].
3. Results and Discussion
3.1. Proposed Structures Studied
Firstly, we demonstrate how different CH pair divacancy configurations are identified on an isolated single hexagon in a graphane monolayer. A single hexagon in a graphane monolayer is shown in Figure 1 circled with purple. The numbers 1–6, positioned around an isolated single hexagon, serve as labels for the CH pairs that should be removed during the vacancy creation in this study. Numbers 1–6 are also used to name different vacancy configurations. In this paper, we report a systematic study of different CH pair divacancy configurations presented in Figure 1.
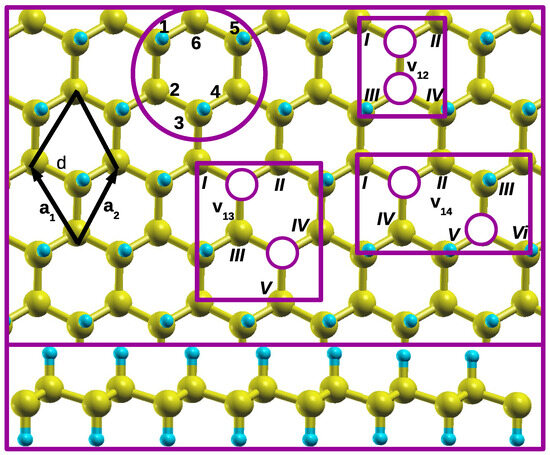
Figure 1.
Top panel present top view of the ball-and-stick model of graphane structure, whereas bottom panel presents side views of graphane structure. Different CH pair divacancy configurations v12, v13, and v14 positions are shown. The white and purple circles hide the numerically labeled CH pair vacancies that are created. The rectangular shapes that enclose vacancy configurations primarily connect the carbon atoms through their potential dangling bonds before undergoing structural relaxation. These carbon atoms are labeled with the Roman numbers. The black rhombus shape represents the primitive unit cell of graphane structure, consisting of two C and two H atoms. The vectors a1 and a2 correspond to its Bravais lattice vectors. The C and H atoms are represented by the yellow and light blue spheres, respectively.
Three distinct configurations are identified as follows: (1) v12 configuration—this configuration consists of a CH divacancy where two CH pairs are adjacent to each other. It is denoted as a v12 configuration, meaning that two adjacent CH pairs are removed from positions 1 and 2 of the single hexagon, as illustrated in Figure 1; (2) v13 divacancy configuration—in this configuration, there are two CH pair vacancies at positions 1 and 3, with the CH pair at position 2 separating them; (3) v14 configuration—this configuration represents the two CH pair vacancies at positions 1 and 4, facing each other, and separated by the two CH adjacent pairs. These configurations describe different arrangements of CH pair vacancies within the hexagonal structure.
3.2. Thermodynamic Stability and Structural Properties of Different CH Vacancy Configurations
As a common practice in first-principles work, the relative stabilities of various vacancy defect configurations in the host materials are evaluated through the formation energy analysis. In this study, we conduct a comparative analysis of the energetic stability of previously mentioned distinct divacancy configurations, namely v12, v13, and v14. This comparison is based on their calculated formation energies (EForm(v)), as illustrated in Figure 2 and indicated by the red circles. All calculations are computed at the same level of accuracy. As shown in Figure 2a, divacancy configuration v12 has the lowest formation energy of 2.71 eV/vCH. This value is 2.21 eV/vCH and 2.00 eV/vCH lower than those of v13 and v14, respectively. Perhaps this could be the reason the previous study [28] only reported properties of v12 divacancy configuration. As it is often observed in many material studies, we have observed that the thermodynamic stability of the v12, v13, and v14 configurations is significantly influenced by the structural reconstruction and the behavior of dangling bonds around the vacancy within a graphane layered structure after relaxation.
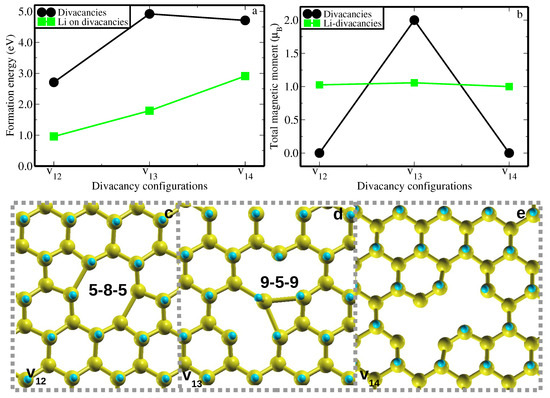
Figure 2.
(a) The formation energies of different CH divacancy configurations (v12, v13, and v14) are indicated by the circle spheres, whereas those of a Li atom on the different CH divacancy configurations (Li-v12, Li-v13, and Li-v14) are represented by the square shapes. (b) The total magnetic moments arising from the v12, v13, and v14 configurations are indicated by the circle spheres, whereas those influenced by a Li atom adsorption on the different CH divacancy configurations (Li-v12, Li-v13, and Li-v14) are represented by the square shapes. (c–e) The relaxed structures of different CH divacancy configurations (v12, v13, and v14) were created in a 6 × 6 supercell. Each divacancy configuration is reconstructed to form a peculiar type of topology defect.
In the v12 configuration (seen in Figure 2c), the pair of dangling C atoms I and as well as the C atoms and attract each other ’to a certain extent’, forming a weak bond length of dI,II = dIII,IV = 1.99 Å , after geometry optimization. This structural reconstruction or self-healing forms a ’peculiar’ vacancy defect type of 5-8-5 divacancy structure as shown in Figure 2c, which is in good agreement with the results of Ref [28], obtained using GGA-PBE functional. Such types of defects have experimentally been realized in graphene, the mother of graphane, using the high-energy ion beams to create a stable carbon chain [36]. Figure 2b shows that v12 lacks a magnetic moment, indicating that the structural reconstruction of the dangling C atoms suppresses any potential magnetic moment in agreement with Ref [28].
In a v13 divacancy configuration, a C atom having possibly two unpaid electrons (based on electronic counting) relaxes towards another two C atoms and . The equivalent bond distances dIII,II and dIII,IV of 2.42 Å are formed between C atoms and as well as between C atoms and , respectively. This reconstruction of v13 divacancy configuration yields an unusual defect structure of 9-4-9 type (see Figure 2d). It is possible that the construction of the 9-4-9 defect structure does not saturate all the dangling bonds surrounding the vacancy, and hence the magnetic moment of 1 has been achieved (Figure 2b). This could be attributed to the C atoms I and each still having an unpaired electron after structural relaxation. Figure 2e presents the relaxed geometry of a v14 divacancy configuration. The C atoms I, II, III, IV, V, and VI are unable to rearrange to form any typical vacancy defect. Based on electron counting, v14 remains with six unpaired electrons (one electron on each C atom I, II, III, IV, V, and VI) after structural relaxation. The absence of total magnetic moments in this system (Figure 2b) could be the result of the cancellation of opposite electronic spins.
3.3. Effect of Li on the Energetic Stability and Structural Properties of Different CH Vacancy Configurations
We now examine the effects of the Li atom on the energetic stabilities and structural and electronic properties of v12, v13, and v14 configurations. Initially, we adsorbed the Li atom on the biggest hollow site of each vacancy configuration, i.e., v12 (octagon site), v13 (nanogon site), and v14 (largest site). Figure 2a shows the effects of the Li atom on the energetic stability of v12, v13, and v14 configurations. It is noted that the Li atom energetically stabilizes the v12, v13, and v14 configurations through a reduction in their formation energies. The formation energy of the Li-v12 configuration, which is 0.96 eV, suggests that although it is endothermic (requiring an input of energy), it can be relatively easy to synthesize compared to the Li-v13 and Li-v14 configurations.
Figure 3a–c presents the relaxed structures of the Li atom on the different CH divacancy configurations Li-v12, Li-v13, and Li-v14, respectively. In these configurations, the Li atom relaxes closer to the hollow site, which has an impact on the local vacancy structures. Figure 3a shows that, in the case of Li-v12, the Li atom remains within the octagon hollow site leading to a height of 0.00 Å (insertion to remain in-plane with other C atoms). Consequently, this results in an increase in the bond distances dIII,II and dIII,IV to 2.63 Å, equivalently. This has led to a high amount of binding force (greater anchoring) between Li and the substrate with a binding energy of 3.25 eV. The Li-v12 interactions induced a magnetic moment of 1.00 , as shown in Figure 2b. Figure 3b shows that Li-v13 buckled up after relaxation, hence leading to a height of 0.62 Å with a binding energy of 2.07 eV. Figure 2b shows that the Li adsorption on a v13 reduces the magnetic moment from 2.00 to 1.00 . In this configuration, the vacancy defect structure of 9-4-9 is reconstructed to create a penta ring and vCH (closest to each other) separated by a newly formed bondlength dIII,II of 1.72 Å. The latter value is less than that of its counterpart in a 5-8-5 defect without a Li atom. In the Li-v14 configuration, the Li atom relaxes to a height of 1.54 Å with a binding energy of 1.99 eV. Notably, there is no significant reconstruction observed in this configuration, as depicted in Figure 3c.
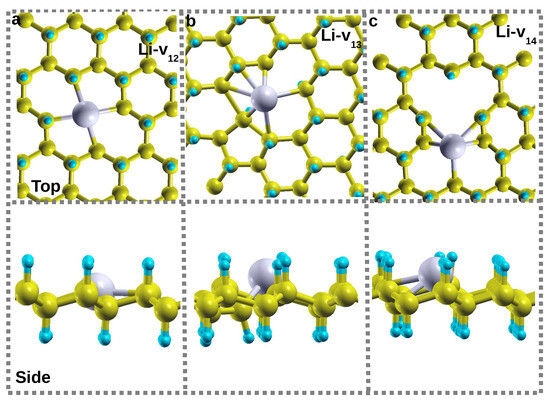
Figure 3.
The relaxed structures of Li atom on the different CH divacancy configurations: (a) Li-v12, (b) Li-v13, and (c) Li-v14 created in a 6 × 6 supercell. In some configurations, the Li atom enforces divacancy configuration reconstruction.
The binding energies for Li-v12, Li-v13, and Li-v14 configurations, respectively, are relatively larger than that of Li on pristine graphene (1.096) eV [35] and on a single vacancy (vCH) in graphane [19], calculated on the same level of accuracy. The binding energies between Li and v12, v13, and v14 configurations are more than the Li bulk cohesive energy (1.63 eV), suggesting no chances of Li cluster formation but possible short time Li charging.
3.4. Influence of Different Charge States (−1 and +1) on the Energetic Stability, Structural, and Electronic Properties of Li-v12
In this section, we examined the effects of Li charge state alteration on the energetics, local structure around the v12 vacancy, and electronic properties (DOS) and made a comparison with those of the uncharged structure. The charge states considered on Li-v12 are q = −1 and +1 of which their respective structures are denoted as Li−1-v12 and Li+1-v12. The binding energies for Li−1-v12 and Li+1-v12 structures are 3.21 eV and 2.24 eV, respectively. In comparison with the uncharged binding energy shown in Table 1, the charge doping reduces the binding force between the Li atom and v12 substrate, significantly in configuration Li+1-v12.

Table 1.
Comparing the binding energies Eb (in eV) and distances between Li atom and graphane dLi (in Å) for Li-v12, Li-v13, and Li-v14 configurations with that of Li on graphene (Li-graphene).
Table 1.
Comparing the binding energies Eb (in eV) and distances between Li atom and graphane dLi (in Å) for Li-v12, Li-v13, and Li-v14 configurations with that of Li on graphene (Li-graphene).
| Configurations | Eb | dLi | Vacancy Reconstruction |
|---|---|---|---|
| Li-v12 | 3.25 | 0.00 | 5-8-5 changes to divacancy symmetry |
| Li-v13 | 2.07 | 0.62 | 9-4-9 translates to penta ring and vCH |
| Li-v14 | 1.99 | 1.14 | None |
| Li-vCH | 1.72 a | 1.54 a | None |
| Li- | 1.096 b, 1.29 c | 1.71 b 1.69 c | None |
a Ref. [19], b Ref. [35], and c Ref. [37].
In the case of an addition of an electron (Li−1-v12), an Li atom remains within the octagon hollow site, maintaining the same in-plane level with other C atoms leading to a height of 0.00 Å as shown in Figure 4a. Figure 4a shows that the bond distances dIII,II and dIII,IV increase slightly by 0.03 Å as compared to uncharged Li-v12 (Figure 4b), which is an indication that the addition of an electron to the system (Li−1-v12) increases the repulsion force between the C atoms surrounding the Li atom. For the removal of electron (Li+1-v12), Li atoms move slightly upward to a height of 0.51 Å. Figure 4b shows that the bond distances dIII,II and dIII,IV decrease significantly by 0.25 Å as compared to uncharged Li-v12 (see Figure 4b). This is an indication that the removal of electrons from the system (Li+1-v12) reduces the attractive force between the C atoms surrounding the Li atom.
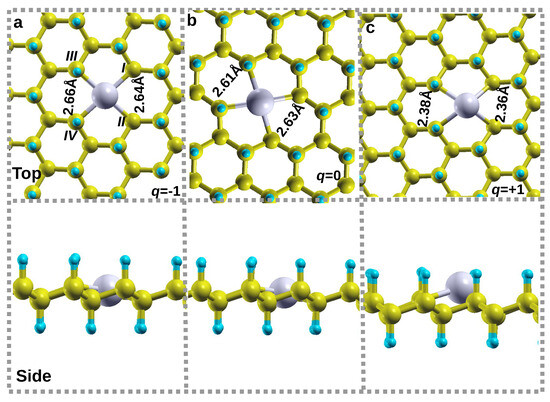
Figure 4.
The relaxed structures of Li on v12 vacancy (Li-v12) at different charge states; (a) Li−1-v12, (b) Li0-v12, and (c) Li+1-v12. The top panel of figures depicts the top view of relaxed structures, whereas the bottom panel depicts the side view of the same structures.
Figure 5 presents the density of states (DOS) for the most stable Li-v12 configuration, comparing it with that of the one without the Li counterpart (v12). Figure 5a (top panel) shows that v12 has insulating features with a wide band gap of 4.18 eV (HSE06) between the valence band maximum (VBM) and conduction band minimum (CBM). The VBM and CBM are mainly contributed by the carbon p orbital states as shown in Figure 5a (bottom panel). This is in agreement with Pujari et al. [28], reporting that the carbon atoms surrounding the divacancy rearrange to form two new bonds leading to the formation of a 5-8-5 ringed structure with a band gap of 3.00 eV (GGA-PBE). Figure 5b (top panel) shows the effects of Li atoms on the DOS of v12. It is noted that the Li atom shifts the Fermi level from the VBM towards the middle of the band gap (suggesting an excess of electrons in the system). It also introduces the occupied and unoccupied states at distinct positions with some crossing the Fermi level in a spin-up channel, suggesting a metallic character. The observed states are due to the hybridization of carbon p orbital and Li s orbital states (Figure 5b (bottom panel)). An addition of electron into the system (Li−1-v12) shifts the Fermi level further towards the CBM Figure 5c. It is interesting to realize that the partially filled mid-gap states, as shown in Figure 5b, become fully occupied in Figure 5c. The removal of electrons from the system (Li+1-v12) shifts the Fermi level back towards the VBM (Figure 5d), retaining the metallic character of the system with the partially filled states crossing the Fermi level. The alteration of charge states of the Li atom changes the electronic behavior of the Li-v12 structure. We propose that graphane with Li-v12 may serve as a suitable electrode material for LIBs. This suitability arises from the potential enhancement of electronic transmission performance mechanisms facilitated by the newly identified Li states.
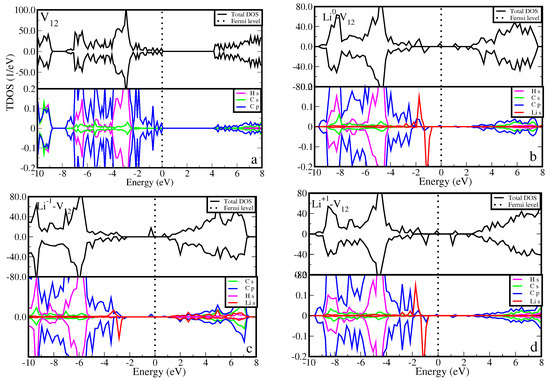
Figure 5.
The calculated density of states for (a) v12, (b) Li-v12, (c) Li−1-v12, and (d) Li+1-v12. For each sub-figure, the top panel represents the total density of states, whereas the bottom panel represents the partial density of states. The Fermi level is set to 0.00 eV and marked by the vertical dashed line.
4. Conclusions
The adsorption mechanisms of Li on graphane with various CH divacancy (v12, v13, and v14) configurations were studied using a DFT approach. Firstly, we compared the energetic stability (formation energies) and structural aspects (bond lengths) for v12, v13, and v14 configurations. Employing structural optimization calculations, it was established that there is a vacancy reconstruction leading to a new defect configuration surrounded by the hexagonal rings in graphane. The v12 was translated to a 5-8-5 defect configuration and is the most stable CH divacancy configuration. Furthermore, we adsorbed Li on v12, v13, and v14 configurations. The formation energy analysis revealed that Li tends to stabilize these CH vacancies, with Li-v12 being the most stable. The Li atom relaxes to the same in-plane level as C atoms on the octagon ring. Li interacts strongly with v12 at the highest binding energy of 3.25 eV/Li, more than the Li bulk cohesive energy of 1.63 eV/Li. The charge doping Li−1-v12 or Li+1-v12 alter the energetic stability, structural properties, and electronic characters of Li-v12. The Li−1-v12 configuration has stronger binding strength than the Li+1-v12 configuration. The electronic density of the state plots for Li−1-v12 reveals an abundance of electrons and an increase in conductivity. These interesting observations encourage further studies on designing graphane with vacancies and its characterization for LIBs.
Author Contributions
Conceptualization, R.E.M., S.P.K., H.M. and E.I.; methodology, R.E.M.; software, R.E.M.; validation, R.E.M., S.P.K., H.M. and E.I.; formal analysis, R.E.M., S.P.K., H.M. and E.I.; investigation, R.E.M., S.P.K., H.M. and E.I.; resources, R.E.M.; data curation, R.E.M.; writing—original draft preparation, R.E.M.; writing—review and editing, R.E.M., S.P.K., H.M. and E.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Acknowledgments
The authors acknowledge the University of Pretoria for financial support and availability of computational resources. We express gratitude to the National Institute for Theoretical and Computational Sciences (NITheCS) for funding. The center for high-performance computing (CHPC) in Cape Town is acknowledged for its state-of-the-art resources.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Tarascon, J.M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359. [Google Scholar] [CrossRef] [PubMed]
- Oyama, N.; Tatsuma, T.; Sato, T.; Sotomura, T. Dimercaptan–polyaniline composite electrodes for lithium batteries with high energy density. Nature 1995, 374, 196. [Google Scholar] [CrossRef]
- Dubal, D.P.; Ayyad, O.; Ruiz, V.; Gómezromero, P. Hybrid energy storage: The merging of battery and supercapacitor chemistries. Chem. Soc. Rev. 2015, 44, 1777. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cui, L.F.; Yang, Y.; Sanchez, C.H.; Robinson, J.T. Mn3O4-graphene hybrid as a high-capacity anode material for lithium ion batteries. J. Am. Chem. Soc. 2010, 132, 13978. [Google Scholar] [CrossRef] [PubMed]
- Zou, F.; Hu, X.; Li, Z.; Long, Q.; Hu, C. MOF-derived porous ZnO/ZnFe2O4/C octahedra with hollow interiors for high-rate lithium-ion batteries. Adv. Mater. 2014, 26, 6622. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Si, W.; Liu, X.; Deng, J.; Xi, L. Multifunctional Ni/NiO hybrid nanomembranes as anode materials for high-rate Li-ion batteries. Nano Energy 2014, 9, 168. [Google Scholar] [CrossRef]
- Chen, K.-S.; Balla, I.; Luu, N.S.; Hersam, M.C. Emerging opportunities for two- dimensional materials in lithium-ion batteries. ACS Energy Lett. 2017, 2, 2026–2034. [Google Scholar] [CrossRef]
- Peng, L.; Zhu, Y.; Chen, D.; Ruoff, R.S.; Yu, G. Two-dimensional materials for beyond- lithium-ion batteries. Adv. Energ Mater. 2016, 6, 1600025. [Google Scholar] [CrossRef]
- Li, H.; Shi, Y.; Chiu, M.-H.; Li, L.-J. Emerging energy applications of two-dimensional layered transition metal dichalcogenides. Nano Energy 2015, 18, 293–305. [Google Scholar] [CrossRef]
- Sofo, J.O.; Chaudhari, A.S.; Barber, G.D. Graphane: A two-dimensional hydrocarbon. Phys. Rev. B 2007, 75, 153401. [Google Scholar] [CrossRef]
- Sluiter, M.H.F.; Kawazoe, Y. Cluster expansion method for adsorption: Application to hydrogen chemisorption on graphene. Phys. Rev. B 2003, 68, 085410. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Density functional theory (DFT). Phys. Rev. 1964, B864, 136. [Google Scholar]
- Elias, D.C.; Nair, R.R.; Mohiuddin, T.M.G.; Morozov, S.V.; Blake, P.; Ferrari, M.P.H.A.C.; Boukhvalov, D.W.; Katsnelson, M.I.; Geim, A.K.; Novoselov, K.S. Control of graphene’s properties by reversible hydrogenation: Evidence for graphane. Science 2009, 323, 610. [Google Scholar] [CrossRef]
- Zhou, C.; Chen, S.; Lou, J.; Wang, J.; Yang, Q.; Liu, C.; Huang, D.; Zhu, T. Graphene’s cousin: The present and future of graphane. Nanoscale Res. Lett. 2014, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Shavelkina, M.B.; Amirov, R.K.; Alikhanov, N.R.; Vakhitov, I.R.; Shatalova, T.B. Continuous Synthesis of Hydrogenated Graphene in Thermal Plasma. J. Struct. Chem. 2018, 59, 773–779. [Google Scholar] [CrossRef]
- Son, J.; Lee, S.; Kim, S.J.; Park, B.C.; Lee, H.; Kim, S.; Kim, J.H.; Hong, B.H.; Hong, J. Hydrogenated monolayer graphene with reversible and tunable wide band gap and its field-effect transistor. Nat. Comm. 2016, 7, 13261. [Google Scholar] [CrossRef] [PubMed]
- Betti, M.G.; Placidi, E.; Izzo, C.; Blundo, E.; Polimeni, A.; Sbroscia, M.; Avila, J.; Dudin, P.; Hu, K.; Ito, Y.; et al. Gap Opening in Double-Sided Highly Hydrogenated Free-Standing Graphene. Nano Lett. 2022, 22, 2971–2977. [Google Scholar] [CrossRef] [PubMed]
- Lebègue, S.; Klintenberg, M.; Eriksson, O.; Katsnelson, M.I. Accurate electronic band gap of pure and functionalized graphane from GW calculations. Phys. Rev. B. 2009, 79, 245117. [Google Scholar] [CrossRef]
- Mapasha, R.E.; Molepo, M.P.; Chetty, N. Li states on a C–H vacancy in graphane: A first-principles study. RSC Adv. 2017, 7, 39748–39757. [Google Scholar] [CrossRef]
- Sahin, H.; Leenaerts, O.; Singh, S.K.; Peeters, F.M. Graphane. Wires Comput. Mol. Sci. 2015, 5, 255–272. [Google Scholar] [CrossRef]
- Keith, E.; Whitener, J. Review Article: Hydrogenated graphene: A user’s guide. J. Vac. Sci. Technol. A 2018, 36, 05G401. [Google Scholar] [CrossRef]
- Watcharinyanon, S.; Johansson, L.; Zakharov, A.; Virojanadara, C. Studies of Li intercalation of hydrogenated graphene on SiC(0001). Surf. Scie 2012, 606, 401–406. [Google Scholar] [CrossRef]
- Yang, Y.E.; Xiao, Y.; Yan, X.H. Charge distribution of lithium-doped graphane/graphene hybrid system: Role of nearly-free electronic states. Sol. Stat. Commun. 2016, 229, 43–48. [Google Scholar] [CrossRef]
- Sahin, H.; Ataca, C.; Ciraci, S. Electronic and magnetic properties of graphane nanoribbons. Phys. Rev. B 2010, 81, 205417. [Google Scholar] [CrossRef]
- Sahin, H.; Ataca, C.; Ciraci, S. Magnetization of graphane by dehydrogenation. Appl. Phys. Lett. 2009, 95, 222510. [Google Scholar] [CrossRef]
- Berashevich, J.; Chakraborty, T. Sustained ferromagnetism induced by H-vacancies in graphane. Nanotechnology 2010, 21, 355201. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mapasha, R.E.; Molepo, M.P.; Chetty, N. Ab initio studies of isolated hydrogen vacancies in graphane. Phys. E 2016, 79, 52–58. [Google Scholar] [CrossRef]
- Pujari, B.S.; Kanhere, D.G. Density functional investigations of defect-induced mid-gap states in graphane. J. Phys. Chem. C 2009, 113, 21063. [Google Scholar] [CrossRef]
- Hedin, L. New method for calculating the one-particle Green’s function with application to the electron-gas problem. Phys. Rev. 1965, 139, A796. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef] [PubMed]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Zhang, S.B.; Northrup, J.E. Chemical potential dependence of defect formation energies in GaAs: Application to Ga self-diffusion. Phys. Rev. Lett. 1991, 67, 2339. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.T.; Neaton, J.B.; Cohen, M.L. First-principles study of metal adatom adsorption on graphene. Phys. Rev. 2008, 77, 235430. [Google Scholar] [CrossRef]
- Jin, C.; Lan, H.; Peng, L.; Suenaga, K.; Iijima, S. Deriving carbon atomic chains from graphene. Phys. Rev. Lett. 2009, 102, 205501. [Google Scholar] [CrossRef]
- Garay-Tapia, A.M.; Romero, A.H.; Barone, V. Lithium adsorption on graphene: From isolated adatoms to metallic sheets. J. Chem. Theory Comput. 2012, 8, 1064–1071. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).