
Impact of Solvents on the Crystal Morphology of CL-20/TFAZ Cocrystals: A Predictive Study



Abstract

1. Introduction
2. The Spiral Growth Mechanism
3. Computational Details
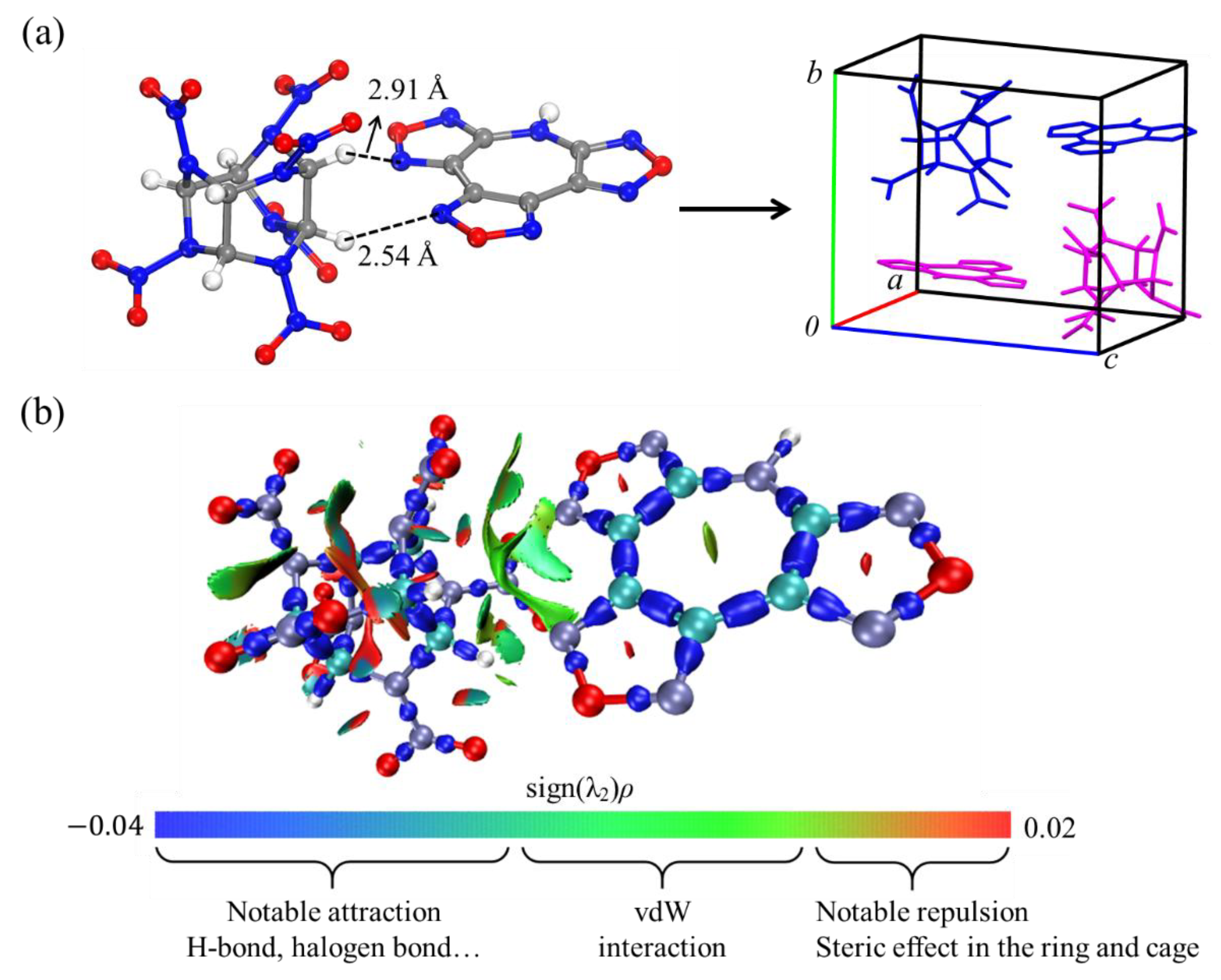
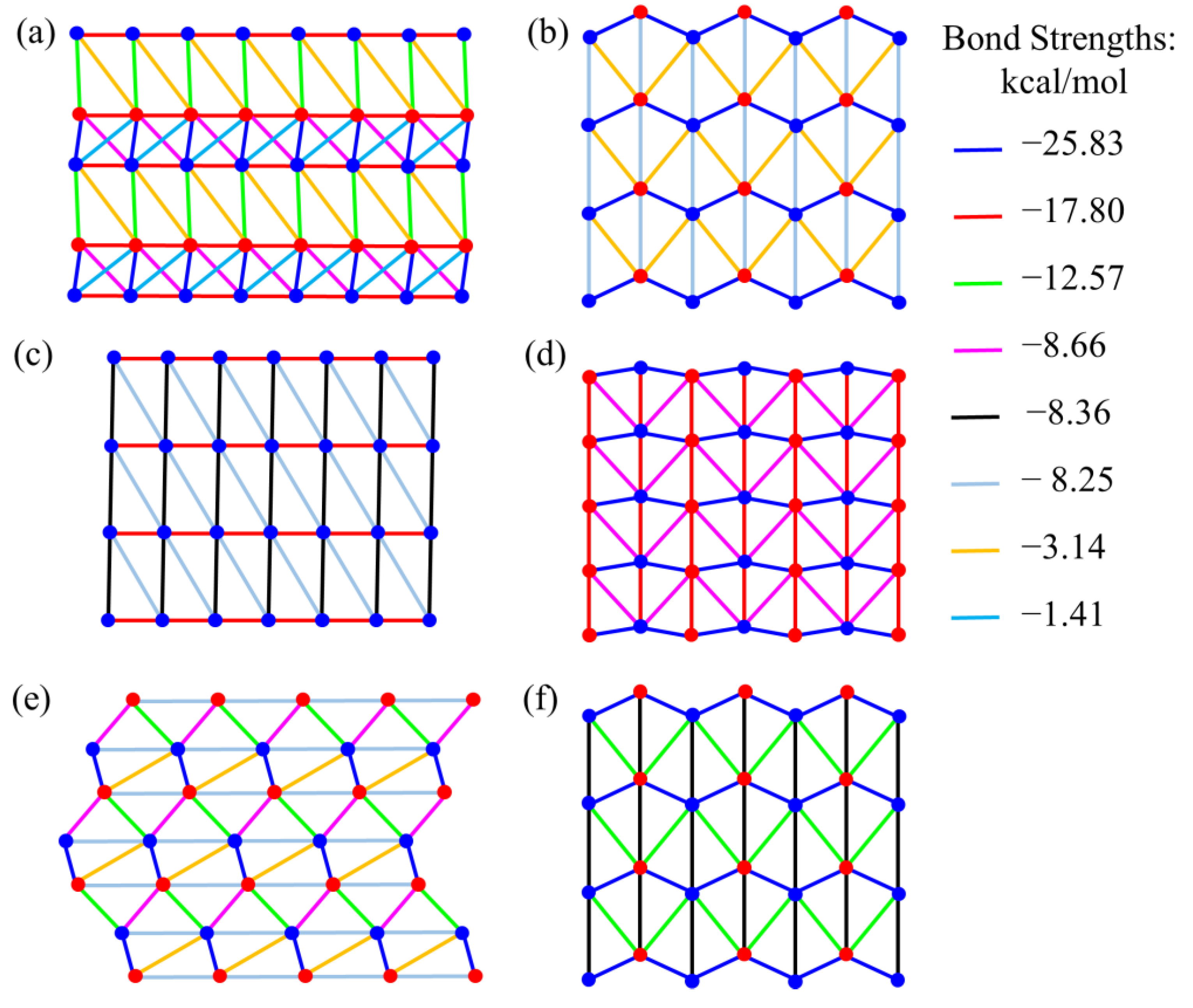
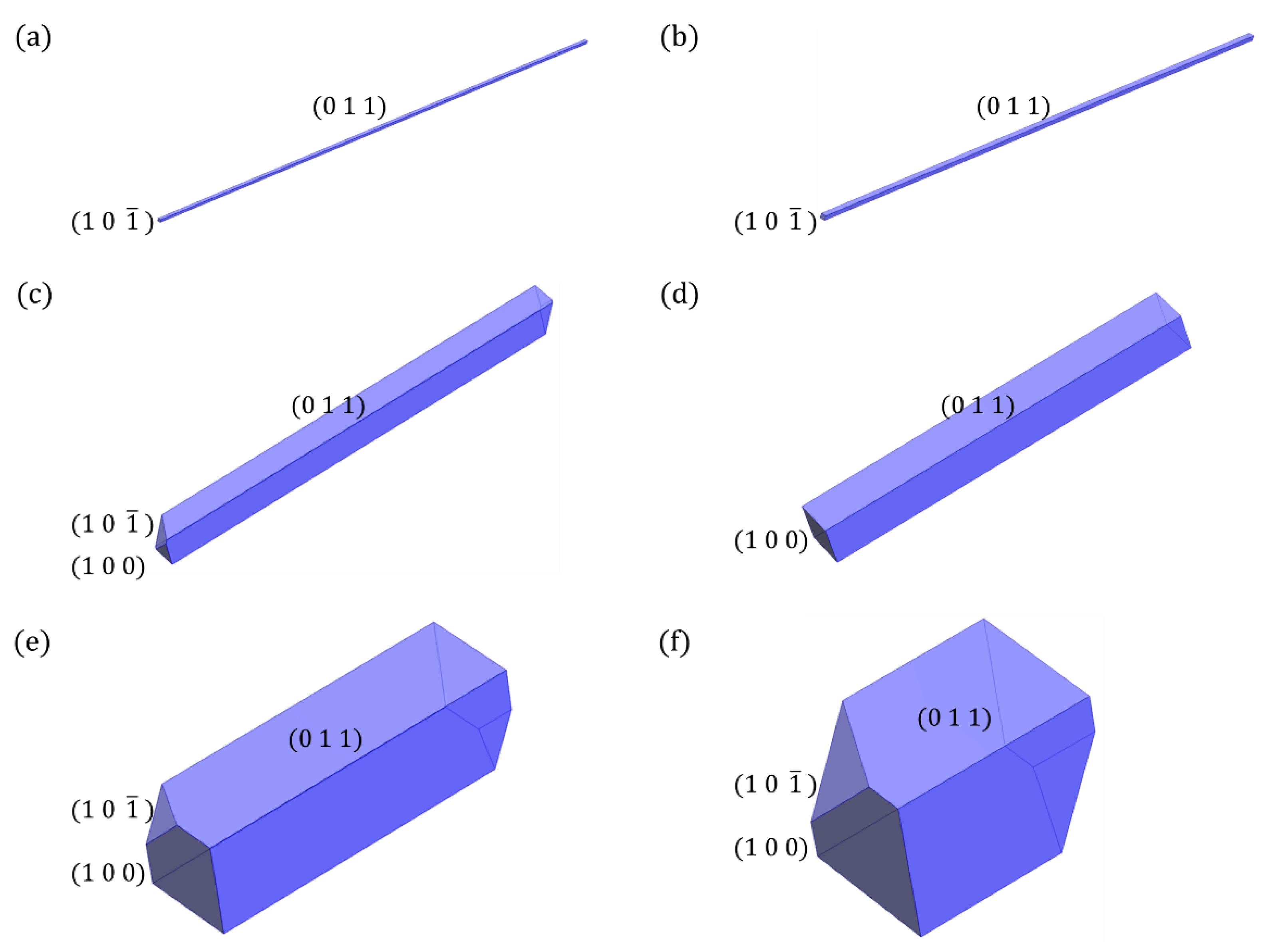
4. Results and Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, J.; Sarma, B.; Evans, J.M.B.; Myerson, A.S. Pharmaceutical Crystallization. Cryst. Growth Des. 2011, 11, 887–895. [Google Scholar] [CrossRef]
- Qiao, N.; Li, M.; Schlindwein, W.; Malek, N.; Davies, A.; Trappitt, G. Pharmaceutical cocrystals: An overview. Int. J. Pharm. 2011, 419, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wu, S.; Macaringue, E.G.J.; Zhang, T.; Gong, J.; Wang, J. Recent Progress in Continuous Crystallization of Pharmaceutical Products: Precise Preparation and Control. Org. Process Res. Dev. 2020, 24, 1785–1801. [Google Scholar] [CrossRef]
- Ejarque, D.; Calvet, T.; Font-Bardia, M.; Pons, J. Structural Landscape of α-Acetamidocinnamic Acid Cocrystals with Bipyridine-Based Coformers: Influence of Crystal Packing on Their Thermal and Photophysical Properties. Cryst. Growth Des. 2024, 24, 1746–1765. [Google Scholar] [CrossRef] [PubMed]
- Ejarque, D.; Calvet, T.; Font-Bardia, M.; Pons, J. Virtual assessment achieved two binary cocrystals based on a liquid and a solid pyridine derivative with modulated thermal stabilities. CrystEngComm 2023, 25, 4798–4811. [Google Scholar] [CrossRef]
- Ejarque, D.; Calvet, T.; Font-Bardia, M.; Pons, J. Cocrystals Based on 4,4′-bipyridine: Influence of Crystal Packing on Melting Point. Crystals 2021, 11, 191. [Google Scholar] [CrossRef]
- Landenberger, K.B.; Matzger, A.J. Cocrystal Engineering of a Prototype Energetic Material: Supramolecular Chemistry of 2,4,6-Trinitrotoluene. Cryst. Growth Des. 2010, 10, 5341–5347. [Google Scholar] [CrossRef]
- Millar, D.I.A.; Maynard-Casely, H.E.; Allan, D.R.; Cumming, A.S.; Lennie, A.R.; Mackay, A.J.; Oswald, I.D.H.; Tang, C.C.; Pulham, C.R. Crystal engineering of energetic materials: Co-crystals of CL-20. CrystEngComm 2012, 14, 3742–3749. [Google Scholar] [CrossRef]
- Zhang, X.-X.; Yang, Z.-J.; Nie, F.; Yan, Q.-L. Recent advances on the crystallization engineering of energetic materials. Energ. Mater. Front. 2020, 1, 141–156. [Google Scholar] [CrossRef]
- Pawar, N.; Saha, A.; Nandan, N.; Parambil, J.V. Solution Cocrystallization: A Scalable Approach for Cocrystal Production. Crystals 2021, 11, 303. [Google Scholar] [CrossRef]
- Heng, J.Y.Y.; Bismarck, A.; Lee, A.F.; Wilson, K.; Williams, D.R. Anisotropic Surface Energetics and Wettability of Macroscopic Form I Paracetamol Crystals. Langmuir 2006, 22, 2760–2769. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.G.; Sun, C.H.; Qiao, S.Z.; Zou, J.; Liu, G.; Smith, S.C.; Cheng, H.M.; Lu, G.Q. Anatase TiO2 single crystals with a large percentage of reactive facets. Nature 2008, 453, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.-C.; Zhou, J.-S.; Sun, J.; Qiu, Y.; Wei, D.-Z.; Shen, Y.-L. Study of the crystal shape and its influence on the anti-tumor activity of tumor necrosis factor-related apoptosis-inducing ligand (Apo2L/TRAIL). Cryst. Res. Technol. 2008, 43, 888–893. [Google Scholar] [CrossRef]
- Zhang, C.; Ji, C.; Li, H.; Zhou, Y.; Xu, J.; Xu, R.; Li, J.; Luo, Y. Occupancy Model for Predicting the Crystal Morphologies Influenced by Solvents and Temperature, and Its Application to Nitroamine Explosives. Cryst. Growth Des. 2013, 13, 282–290. [Google Scholar] [CrossRef]
- Duan, X.; Wei, C.; Liu, Y.; Pei, C. A molecular dynamics simulation of solvent effects on the crystal morphology of HMX. J. Hazard. Mater. 2010, 174, 175–180. [Google Scholar] [CrossRef]
- Bravais, A. Études Cristallographiques; Gauthier-Villars: Paris, France, 1866. [Google Scholar]
- Friedel, G. Etudes sur la loi de Bravais. Bull. Soc. Franc. Miner 1907, 30, 326–455. [Google Scholar] [CrossRef]
- Donnay, J.D.H.; Harker, D. A new law of crystal morphology extending the law of Bravais. Am. Miner. 1937, 22, 446–467. [Google Scholar]
- Hartman, P.; Perdok, W.G. On the relations between structure and morphology of crystals. I. Acta Cryst. 1955, 8, 49–52. [Google Scholar] [CrossRef]
- Hartman, P.; Perdok, W. On the relations between structure and morphology of crystals. II. Acta Cryst. 1955, 8, 521–524. [Google Scholar] [CrossRef]
- Hartman, P.; Perdok, W.G. On the relations between structure and morphology of crystals. III. Acta Cryst. 1955, 8, 525–529. [Google Scholar] [CrossRef]
- Hartman, P.; Bennema, P. The attachment energy as a habit controlling factor: I. Theoretical considerations. J. Cryst. Growth 1980, 49, 145–156. [Google Scholar] [CrossRef]
- Lu, J.J.; Ulrich, J. An improved prediction model of morphological modifications of organic crystals induced by additives. Cryst. Res. Technol. 2003, 38, 63–73. [Google Scholar] [CrossRef]
- Mazal, T.; Doherty, M.F. Modeling Morphologies of Organic Crystals via Kinetic Monte Carlo Simulations: Noncentrosymmetric Growth Units. Cryst. Growth Des. 2024, 24, 3756–3770. [Google Scholar] [CrossRef]
- Padwal, N.A.; Doherty, M.F. Step Velocity Growth Models for Molecular Crystals: Two Molecules in the Unit Cell. Cryst. Growth Des. 2024, 24, 4368–4379. [Google Scholar] [CrossRef]
- Padwal, N.A.; Mazal, T.; Doherty, M.F. Modern Modeling and Simulation Approaches for Morphology Predictions of Molecular Crystals. Ind. Eng. Chem. Res. 2024, 63, 18401–18410. [Google Scholar] [CrossRef]
- Snyder, R.C.; Doherty, M.F. Predicting crystal growth by spiral motion. Proc. R. Soc. A 2009, 465, 1145–1171. [Google Scholar] [CrossRef]
- Tilbury, C.J.; Doherty, M.F. Modeling layered crystal growth at increasing supersaturation by connecting growth regimes. AIChE J. 2017, 63, 1338–1352. [Google Scholar] [CrossRef]
- Kuvadia, Z.B.; Doherty, M.F. Spiral Growth Model for Faceted Crystals of Non-Centrosymmetric Organic Molecules Grown from Solution. Cryst. Growth Des. 2011, 11, 2780–2802. [Google Scholar] [CrossRef]
- Shim, H.-M.; Koo, K.-K. Molecular Approach to the Effect of Interfacial Energy on Growth Habit of ε-HNIW. Cryst. Growth Des. 2016, 16, 6506–6513. [Google Scholar] [CrossRef]
- Li, J.; Tilbury, C.J.; Kim, S.H.; Doherty, M.F. A design aid for crystal growth engineering. Prog. Mater. Sci. 2016, 82, 1–38. [Google Scholar] [CrossRef]
- Sun, Y.; Tilbury, C.J.; Reutzel-Edens, S.M.; Bhardwaj, R.M.; Li, J.; Doherty, M.F. Modeling Olanzapine Solution Growth Morphologies. Cryst. Growth Des. 2018, 18, 905–911. [Google Scholar] [CrossRef]
- Sun, Y.; Reutzel-Edens, S.M.; Bhardwaj, R.M.; Doherty, M.F. Crystal Morphology Modeling of Solvates and Hydrates of Organic Molecular Crystals: Olanzapine Solvate and Dihydrate. Cryst. Growth Des. 2021, 21, 4871–4877. [Google Scholar] [CrossRef]
- Nielsen, A.T.; Chafin, A.P.; Christian, S.L.; Moore, D.W.; Nadler, M.P.; Nissan, R.A.; Vanderah, D.J.; Gilardi, R.D.; George, C.F.; Flippen-Anderson, J.L. Synthesis of polyazapolycyclic caged polynitramines. Tetrahedron 1998, 54, 11793–11812. [Google Scholar] [CrossRef]
- Liu, G.; Li, H.; Gou, R.; Zhang, C. Packing Structures of CL-20-Based Cocrystals. Cryst. Growth Des. 2018, 18, 7065–7078. [Google Scholar] [CrossRef]
- Zhang, Y.; Sizemore, J.P.; Doherty, M.F. Shape evolution of 3-dimensional faceted crystals. AIChE J. 2006, 52, 1906–1915. [Google Scholar] [CrossRef]
- Chernov, A. The kinetics of the growth forms of crystals. Sov. Phys. Cryst. 1963, 7, 728–730. [Google Scholar]
- Kaischew, R.; Budevski, E. Surface processes in electrocrystallization. Contemp. Phys. 1967, 8, 489–516. [Google Scholar] [CrossRef]
- Voronkov, V.V. Dislocation mechanism of growth with a low kink density. Sov. Phys. Cryst. 1973, 18, 19–223. [Google Scholar]
- Tilbury, C.J.; Joswiak, M.N.; Peters, B.; Doherty, M.F. Modeling Step Velocities and Edge Surface Structures during Growth of Non-Centrosymmetric Crystals. Cryst. Growth Des. 2017, 17, 2066–2080. [Google Scholar] [CrossRef]
- Girifalco, L.A.; Good, R.J. A Theory for the Estimation of Surface and Interfacial Energies. I. Derivation and Application to Interfacial Tension. J. Phys. Chem. 1957, 61, 904–909. [Google Scholar] [CrossRef]
- Tilbury, C.J.; Green, D.A.; Marshall, W.J.; Doherty, M.F. Predicting the Effect of Solvent on the Crystal Habit of Small Organic Molecules. Cryst. Growth Des. 2016, 16, 2590–2604. [Google Scholar] [CrossRef]
- Van Oss, C.J.; Chaudhury, M.K.; Good, R.J. Interfacial Lifshitz-van der Waals and polar interactions in macroscopic systems. Chem. Rev. 1988, 88, 927–941. [Google Scholar] [CrossRef]
- Kaelble, D.H. Physical Chemistry of Adhesion; Wiley-Interscience: Hoboken, NJ, USA, 1971. [Google Scholar]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Crowley, M.; Walker, R.C.; Zhang, W. Amber 10; University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; et al. Gaussian 03, Revision C. 02; Gaussian: Wallingford, CT, USA, 2003. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Liu, N.; Duan, B.; Lu, X.; Zhang, Q.; Xu, M.; Mo, H.; Wang, B. Preparation of CL-20/TFAZ cocrystals under aqueous conditions: Balancing high performance and low sensitivity. CrystEngComm 2019, 21, 7271–7279. [Google Scholar] [CrossRef]
- Material Studio, version 6.1. Software for Technical Computation. Accelrys Inc.: San Diego, CA, USA, 2011.
- Zhu, S.-F.; Zhang, S.-H.; Gou, R.-J.; Wu, C.-L.; Han, G.; Jia, H.-Y. Understanding the Effect of Solvent on the Growth and Crystal Morphology of MTNP/CL-20 Cocrystal Explosive: Experimental and Theoretical Studies. Cryst. Res. Technol. 2018, 53, 1700299. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Interaction Region Indicator: A Simple Real Space Function Clearly Revealing Both Chemical Bonds and Weak Interactions. Chem. Methods 2021, 1, 231–239. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | ||||
|---|---|---|---|---|
| isopropyl acetate | 117.1 | 7.28 | 2.20 | 4.01 |
| hexane | 131.6 | 7.28 | ||
| n-heptane | 147.4 | 7.48 | ||
| n-butyl acetate | 132.5 | 7.72 | 1.81 | 3.08 |
| acetone | 74.0 | 7.58 | 5.08 | 3.42 |
| methanol | 40.7 | 7.38 | 6.01 | 10.90 |
| propionic acid | 75.0 | 7.19 | 2.59 | 6.06 |
| Face | d (Å) | (kcal/mol) |
|---|---|---|
| {0 0 1} | 11.80 | −29.58 |
| {1 0 0} | 8.29 | −38.90 |
| {0 1 1} | 8.26 | −43.70 |
| {0 1} | 8.26 | −43.70 |
| { 0 1} | 6.84 | −53.58 |
| {1 1 0} | 6.73 | −54.54 |
| {1 0} | 6.73 | −54.54 |
| {1 0 1} | 6.72 | −47.13 |
| {1} | 5.89 | −55.21 |
| {1} | 5.89 | −55.21 |
| Hexane | n-Heptane | n-Butyl Acetate | Acetone | Methanol | Propionic Acid | |
|---|---|---|---|---|---|---|
| {0 1 1} | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| {1 0 } | 115.00 | 69.70 | 11.80 | 8.63 | 3.23 | 1.40 |
| {1 0 0} | 168.00 | 98.10 | 14.40 | 9.24 | 3.45 | 1.42 |
| {0 0 1} | 496.00 | 456.00 | 91.60 | 105.00 | 56.10 | 11.70 |
| {1 1 } | 2.50 × 104 | 8.63 × 103 | 127.00 | 1.39 × 103 | 6.95 × 103 | 432.00 |
| {0 2 0} | 3.43 × 108 | 1.53 × 108 | 1.30 × 106 | 5.82 × 106 | 2.24 × 107 | 2.66 × 106 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Yu, L.; Wang, Y.; Suen, N.-T. Impact of Solvents on the Crystal Morphology of CL-20/TFAZ Cocrystals: A Predictive Study. Compounds 2025, 5, 6. https://doi.org/10.3390/compounds5010006
Sun Y, Yu L, Wang Y, Suen N-T. Impact of Solvents on the Crystal Morphology of CL-20/TFAZ Cocrystals: A Predictive Study. Compounds. 2025; 5(1):6. https://doi.org/10.3390/compounds5010006
Chicago/Turabian StyleSun, Yuanyuan, Le Yu, Yichen Wang, and Nian-Tzu Suen. 2025. "Impact of Solvents on the Crystal Morphology of CL-20/TFAZ Cocrystals: A Predictive Study" Compounds 5, no. 1: 6. https://doi.org/10.3390/compounds5010006
APA StyleSun, Y., Yu, L., Wang, Y., & Suen, N.-T. (2025). Impact of Solvents on the Crystal Morphology of CL-20/TFAZ Cocrystals: A Predictive Study. Compounds, 5(1), 6. https://doi.org/10.3390/compounds5010006