
Proposed Framework for Comparison of Continuous Probabilistic Genotyping Systems amongst Different Laboratories

 , ,
, , {kind=link}
Abstract
1. Introduction
- No possibility of drop in and drop out;
- No possibility of drop in but some possibility of drop out;
- No possibility of drop out but with artificial alleles added to mimic the possibility of drop in.
2. The Likelihood Ratio Produced by Probabilistic Genotyping
3. A Reproducible Subset of Likelihood Ratios from Probabilistic Genotyping
- wi → 0 (i.e., uncertainty is minimised between genotype sets with all alleles belonging to contributors and no others and those with at least one allele not belonging to contributors or without all contributor alleles present) and;
- H1 is true (i.e., H1 corresponds with the contributors only).
4. Conditions for Achieving Reproducible LRs from Probabilistic Genotyping
- The same standard mixtures should be examined.
- The same propositions should be considered.
- The same loci should be employed.
- The same population allele frequencies should be employed.
- The same population genetic model and sub-structure correction, θ, should be employed (e.g., θ = 0).
- No allele and allele drop out;
- A (low peak height) contributor allele and allele drop in;
- A (low peak height) contributor allele and a stutter peak;
- A single allele and shared (“stacked”) alleles, either of which may or may not include allele drop in and stutter peaks.
- 6.
- The DNA template from true donors should be maximised to a point within the linear range and below saturation of the epg.
- 7.
- Each laboratory is presented with aliquots of the same dilution series of DNA solutions which then undergo analyses to produce epgs for each solution according to each laboratory’s standard practice (according to which the PG system was validated in that laboratory).
- 8.
- All known donors are present in equal proportion by DNA template amount.
- 9.
- The trial should be blinded. Laboratories presented with a dilution series of DNA solutions to be analysed should not know which is which.
- 10.
- The trial should be facilitated by an entity not associated with the PG systems under comparison.
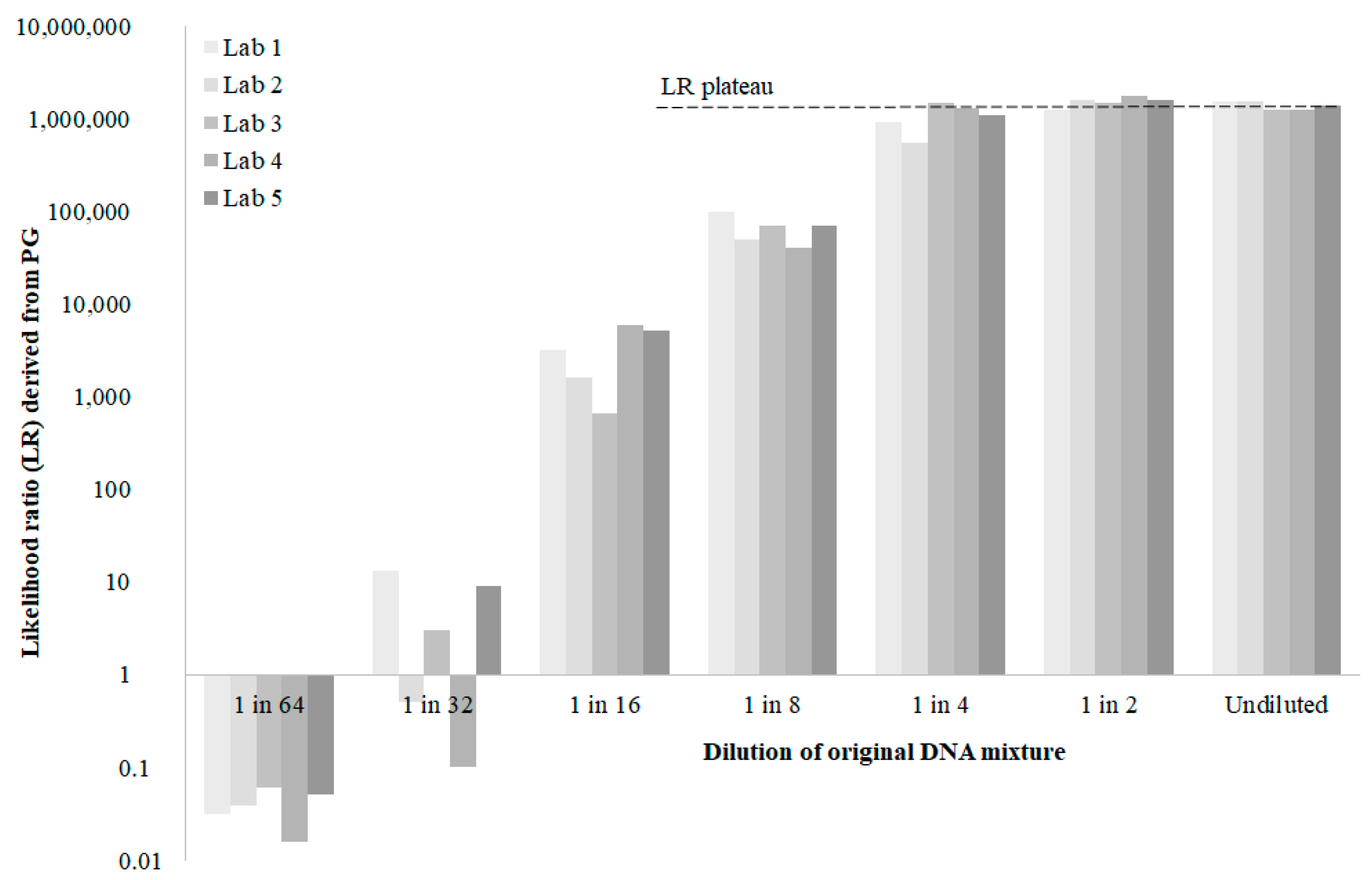
5. An Inter-Laboratory Comparison
- The position of the plateaued, maximum LR from any laboratory within the theoretical range defined by Equation (12). This is a measure of performance, if not accuracy.
- The range of plateaued, maximum LRs reported by laboratories. This is an indication of the credible interval for LRs reported under the best possible conditions designed to minimise variance in LRs. This credible interval would suggest a minimum as we would expect the variance amongst laboratories to increase the further they are from conditions 1 to 8.
- Outlier laboratories. This would provide guidance on which laboratories (if any) might need to re-validate their PG system.
- Outlier PG systems. This would provide guidance on which PG systems (if any) do not model allele peak height variance adequately according to the procedures in a particular laboratory.
- The minimum template amounts at which fortuitous LRs are encountered for any laboratory (LR > 1 for a non-contributor, LR < 1 for a contributor). As DNA template amounts decrease in the dilution series, LRs for contributors and non-contributors will approach 1 but may actually overshoot.
- Identify participating laboratories. They are required not to communicate with each other concerning the trial.
- Identify reported loci in common amongst participating laboratories. Longer loci, where Equation (7) might not be expected to hold, could also be excluded (with agreement). These excluded loci should not be used either to estimate parameters such as mixture proportions or to calculate LRs. In practice, any laboratory could nominate a locus to be excluded. A comparison between PG systems could, theoretically, be made with as little as one locus but, of course, more loci will increase the stringency of any trial.
- Identify a trial facilitator not associated with any of the PG systems to be used. This could be a university, a centre of excellence or a national forensic regulator, for example.
- The trial facilitator collects samples from reference cell lines or consenting volunteers and performs DNA extraction and quantitation for each sample.
- The DNA concentration for each sample is normalised according to the quantitation results and assessed as being of a suitable (high) quantity and quality.
- A single source STR profile for each donor is generated according to best practice. These are the contributor reference profiles. Non-contributor reference profiles can also be generated.
- Equal volume and equal concentration aliquots of high abundance DNA are combined from various donors to create mixtures of 2, 3, 4,… and N contributors in equal proportion by DNA amount.
- For each mixture, a dilution series is created (e.g., undiluted, 1 in 2, 1 in 4, 1 in 8, etc.).
- Aliquots of the various dilution series (one dilution series per mixture) are distributed to the participating laboratories, labelled randomly such that the laboratory does not know the concentration of DNA in any sample. For one, two, three, four and five contributors each at seven different dilutions, for example, a total of 35 samples would be supplied.
- Each participating laboratory produces an STR epg for each aliquot according to the standard procedures for that laboratory.
- The participating laboratories are also supplied with the following:
- Reference profiles.
- Allele frequencies from a defined population.
- The following propositions are also provided to each of the participating laboratories:
- H1: The donor of reference profile X is a contributor to the mixture which also consists of N other known but unrelated contributors (where all N+1 reference profiles are supplied);
- H2: The donor of reference profile X is not a contributor to the mixture which consists of an unrelated, random member of the (defined) population and N other known but unrelated contributors.
These can be applied to both contributor and non-contributor reference profiles. - Each laboratory is asked to provide a LR according to Equation (1). The laboratories are instructed to use the allele frequencies provided from the defined population without any population substructure corrections and using a consistent population genetic model (e.g., Hardy–Weinberg proportions).
- The LRs are collated and compared by the trial facilitator.
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van Oorschot, R.A.H.; Szkuta, B.; Meakin, G.E.; Kokshoorn, B.; Goray, M. DNA transfer in forensic science: A review. Forensic Sci. Int. Genet. 2019, 38, 140–166. [Google Scholar] [CrossRef]
- Perlin, M.W. Inclusion probability for DNA mixtures is a subjective one-sided match statistic unrelated to identification information. J. Pathol. Inf. 2015, 6, 59. [Google Scholar] [CrossRef]
- Bieber, F.R.; Buckleton, J.S.; Budowle, B.; Butler, J.M.; Coble, M.D. Evaluation of forensic DNA mixture evidence: Protocol for evaluation, interpretation, and statistical calculations using the combined probability of inclusion. BMC Genet. 2016, 17, 125. [Google Scholar] [CrossRef] [PubMed]
- Curran, J.M.; Buckleton, J. Inclusion probabilities and dropout. J. Forensic Sci. 2010, 55, 1171–1173. [Google Scholar] [CrossRef]
- Coble, M.D.; Bright, J.-A. Probabilistic genotyping software: An overview. Forensic Sci. Int. Genet. 2019, 38, 219–224. [Google Scholar] [CrossRef]
- Brenner, C.H. DNA·VIEW User’s Manual. Charles Brenner, UC Berkeley, 6801 Thornhill Drive Oakland, California, USA. 2019. Available online: http://dna-view.com/downloads/documents/manuals/DNAVIEW%202019%20US.pdf (accessed on 8 June 2021).
- Perlin, M.W.; Legler, M.M.; Spencer, C.E.; Smith, J.L.; Allan, W.P.; Belrose, J.L.; Duceman, B.W. Validating TrueAllele® DNA mixture interpretation. J. Forensic Sci. 2011, 56, 1430–1447. [Google Scholar] [CrossRef]
- Perlin, M.W.; Sinelnikov, A. An information gap in DNA evidence interpretation. PLoS ONE 2009, 4, e8327. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Bright, J.-A.; Buckleton, J. The interpretation of single source and mixed DNA profiles. Forensic Sci. Int. Genet. 2013, 7, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Bleka, Ø.; Storvik, G.; Gill, P. EuroForMix: An open source software based on a continuous model to evaluate STR DNA profiles from a mixture of contributors with artefacts. Forensic Sci. Int. Genet. 2016, 21, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Benschop, C.C.G.; Hoogenboom, J.; Hovers, P.; Slagter, M.; Kruise, D.; Parag, R.; Steensma, K.; Slooten, K.; Nagel, J.H.A.; Dieltjes, P.; et al. DNAxs/DNAStatistX: Development and validation of a software suite for the data management and probabilistic interpretation of DNA profiles. Forensic Sci. Int. Genet. 2019, 42, 81–89. [Google Scholar] [CrossRef]
- Cowell, R.G.; Graversen, T.; Lauritzen, S.L.; Mortera, J. Analysis of forensic DNA mixtures with artefacts. J. R. Stat. Soc. Ser. C 2015, 64, 1–48. [Google Scholar] [CrossRef]
- Get More Information from DNA Mixtures with TrueAllele® Casework. Available online: https://www.cybgen.com/products/casework.shtml (accessed on 16 May 2021).
- STRmix™. Empowering Forensic Science. Available online: https://www.strmix.com/ (accessed on 16 May 2021).
- Brenner, C. What is DNA•VIEW®? An Integrated Software Package for DNA Identification. Available online: http://dna-view.com/dnaview.htm (accessed on 16 May 2021).
- Butler, J.M.; Kline, M.C.; Coble, M.D. NIST interlaboratory studies involving DNA mixtures (MIX05 and MIX13): Variation observed and lessons learned. Forensic Sci. Int. Genet. 2018, 37, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, H.; Qureshi, M.O.; Grgicak, C.M.; Duffy, K.; Lun, D.S. Four model variants within a continuous forensic DNA mixture interpretation framework: Effects on evidential inference and reporting. PLoS ONE 2018, 13, e0207599. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, H.; Garg, A.; Grgicak, C.M.; Medard, M.; Lun, D.S. CEESIt: A computational tool for the interpretation of STR mixtures. Forensic Sci. Int. Genet. 2016, 22, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.; Bleka, Ø.; Hansson, O.; Benschop, C.; Haned, H. Forensic Practitioner’s Guide to the Interpretation of Complex DNA Profiles; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Taylor, D.; Bright, J.-A.; Buckleton, J.S. The continuous model. In Forensic DNA Evidence Interpretation, 2nd ed.; Buckleton, J.S., Bright, J.-A., Taylor, D., Eds.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Association of Forensic Science Providers. Standards for the formulation of evaluative forensic science expert opinion. Sci. Justice 2009, 49, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Bright, J.-A.; Evett, I.W.; Taylor, D.; Curran, J.M.; Buckleton, J. A series of recommended tests when validating probabilistic DNA profile interpretation software. Forensic Sci. Int. Genet. 2015, 14, 125–131. [Google Scholar] [CrossRef]
- You, Y.; Balding, D. A comparison of software for the evaluation of complex DNA profiles. Forensic Sci. Int. Genet. 2019, 40, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Manabe, S.; Morimoto, C.; Hamano, Y.; Fujimoto, S.; Tamaki, K. Development and validation of open-source software for DNA mixture interpretation based on a quantitative continuous model. PLoS ONE 2017, 12, e0188183. [Google Scholar] [CrossRef]
- Riman, S.; Iyer, H.; Vallone, P.M. Exploring DNA interpretation software using the PROVEDIt dataset. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 724–726. [Google Scholar] [CrossRef]
- Buckleton, J.S.; Bright, J.-A.; Cheng, K.; Budowle, B.; Coble, M.D. NIST interlaboratory studies involving DNA mixtures (MIX13): A modern analysis. Forensic Sci. Int. Genet. 2018, 37, 172–179. [Google Scholar] [CrossRef]
- Bright, J.-A.; Cheng, K.; Kerr, Z.; McGovern, C.; Kelly, H.; Moretti, T.R.; Smith, M.A.; Bieber, F.R.; Budowle, B.; Coble, M.D.; et al. STRmix™ collaborative exercise on DNA mixture interpretation. Forensic Sci. Int. Genet. 2019, 40, 1–8. [Google Scholar] [CrossRef]
- Benschop, C.C.G.; Hoogenboom, J.; Bargeman, F.; Hovers, P.; Slagter, M.; van der Linden, J.; Parag, R.; Kruise, D.; Drobnic, K.; Klucevsek, G.; et al. Multi-laboratory validation of DNAxs including the statistical library DNAStatistX. Forensic Sci. Int. Genet. 2020, 49, 102390. [Google Scholar] [CrossRef]
- Alladio, E.; Omedei, M.; Cisana, S.; D’Amico, G.; Caneparo, D.; Vincenti, M.; Garofano, P. DNA mixtures interpretation—A proof-of-concept multi-software comparison highlighting different probabilistic methods’ performances on challenging samples. Forensic Sci. Int. Genet. 2018, 37, 143–150. [Google Scholar] [CrossRef]
- Eduardoff, M.; Santos, C.; de la Puente, M.; Gross, T.E.; Fondevila, M.; Strobl, C.; Sobrino, B.; Ballard, D.; Schneider, P.M.; Carracedo, Á.; et al. Inter-laboratory evaluation of SNP-based forensic identification by massively parallel sequencing using the Ion PGM™. Forensic Sci. Int. Genet. 2015, 17, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Steensma, K.; Ansell, R.; Clarisse, L.; Connolly, E.; Kloosterman, A.D.; McKenna, L.G.; van Oorschot, R.A.H.; Szkuta, B.; Kokshoorn, B. An inter-laboratory comparison study on transfer, persistence and recovery of DNA from cable ties. Forensic Sci. Int. Genet. 2017, 31, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Köcher, S.; Müller, P.; Berger, B.; Bodner, M.; Parson, W.; Roewer, L.; Willuweit, S. Inter-laboratory validation study of the ForenSeq™ DNA Signature Prep Kit. Forensic Sci. Int. Genet. 2018, 36, 77–85. [Google Scholar] [CrossRef] [PubMed]
- President’s Council of Advisors on Science and Technology. Forensic Science in Criminal Courts: Ensuring Scientific Validity of Feature-Comparison Methods; Executive Office of the President of the United States: Washington, DC, USA, 2016. [Google Scholar]
- Butler, J.M. Forenisc DNA Typing, 2nd ed.; Academic Press: Cambridge, MA, USA, 2005. [Google Scholar]
- McNevin, D.; Wright, K.; Chaseling, J.; Barash, M. Commentary on: Bright et al. (2018) Internal validation of STRmix™—A multi laboratory response to PCAST, Forensic Science International: Genetics, 34: 11–24. Forensic Sci. Int. Genet. 2019, 41, e14–e17. [Google Scholar] [CrossRef]
- Buckleton, J.S.; Bright, J.-A.; Ciecko, A.; Kruijver, M.; Mallinder, B.; Magee, A.; Malsom, S.; Moretti, T.; Weitz, S.; Bille, T.; et al. Response to: Commentary on: Bright et al. (2018) Internal validation of STRmix™—A multi laboratory response to PCAST, Forensic Science International: Genetics, 34: 11–24. Forensic Sci. Int. Genet. 2020, 44. [Google Scholar] [CrossRef]
- Bright, J.-A.; Stevenson, K.E.; Curran, J.M.; Buckleton, J.S. The variability in likelihood ratios due to different mechanisms. Forensic Sci. Int. Genet. 2015, 14, 187–190. [Google Scholar] [CrossRef]
- Ramos, D.; Gonzalez-Rodriguez, J. Reliable support: Measuring calibration of likelihood ratios. Forensic Sci. Int. 2013, 230, 156–169. [Google Scholar] [CrossRef]
- Bright, J.-A.; Jones Dukes, M.; Pugh, S.N.; Evett, I.W.; Buckleton, J.S. Applying calibration to LRs produced by a DNA interpretation software. Aust. J. Forensic Sci. 2021, 53, 147–153. [Google Scholar] [CrossRef]
- Kelly, H.; Bright, J.-A.; Kruijver, M.; Cooper, S.; Taylor, D.; Duke, K.; Strong, M.; Beamer, V.; Buettner, C.; Buckleton, J. A sensitivity analysis to determine the robustness of STRmix™ with respect to laboratory calibration. Forensic Sci. Int. Genet. 2018, 35, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Moretti, T.R.; Just, R.S.; Kehl, S.C.; Willis, L.E.; Buckleton, J.S.; Bright, J.-A.; Taylor, D.A.; Onorato, A.J. Internal validation of STRmix™ for the interpretation of single source and mixed DNA profiles. Forensic Sci. Int. Genet. 2017, 29, 126–144. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Buckleton, J.; Bright, J.-A. Factors affecting peak height variability for short tandem repeat data. Forensic Sci. Int. Genet. 2016, 21, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Buckleton, J.S.; Bright, J.-A.; Gittelson, S.; Moretti, T.R.; Onorato, A.J.; Bieber, F.R.; Budowle, B.; Taylor, D.A. The probabilistic genotyping software STRmix: Utility and evidence for its validity. J. Forensic Sci. 2019, 64, 393–405. [Google Scholar] [CrossRef]
- Bauer, D.W.; Butt, N.; Hornyak, J.M.; Perlin, M.W. Validating TrueAllele® interpretation of DNA mixtures containing up to ten unknown contributors. J. Forensic Sci. 2020, 65, 380–398. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Bright, J.-A.; Kerr, Z.; Taylor, D.; Ciecko, A.; Curran, J.; Buckleton, J. Examining the additivity of peak heights in forensic DNA profiles. Aust. J. Forensic Sci. 2020, 1–15. [Google Scholar] [CrossRef]
- Brookes, C.; Bright, J.-A.; Harbison, S.; Buckleton, J. Characterising stutter in forensic STR multiplexes. Forensic Sci. Int. Genet. 2012, 6, 58–63. [Google Scholar] [CrossRef]
- Morrison, G.S. Special Issue on Measuring and Reporting the Precision of Forensic Likelihood Ratios. Sci. Justice 2016, 56. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McNevin, D.; Wright, K.; Barash, M.; Gomes, S.; Jamieson, A.; Chaseling, J. Proposed Framework for Comparison of Continuous Probabilistic Genotyping Systems amongst Different Laboratories. Forensic Sci. 2021, 1, 33-45. https://doi.org/10.3390/forensicsci1010006
McNevin D, Wright K, Barash M, Gomes S, Jamieson A, Chaseling J. Proposed Framework for Comparison of Continuous Probabilistic Genotyping Systems amongst Different Laboratories. Forensic Sciences. 2021; 1(1):33-45. https://doi.org/10.3390/forensicsci1010006
Chicago/Turabian StyleMcNevin, Dennis, Kirsty Wright, Mark Barash, Sara Gomes, Allan Jamieson, and Janet Chaseling. 2021. "Proposed Framework for Comparison of Continuous Probabilistic Genotyping Systems amongst Different Laboratories" Forensic Sciences 1, no. 1: 33-45. https://doi.org/10.3390/forensicsci1010006
APA StyleMcNevin, D., Wright, K., Barash, M., Gomes, S., Jamieson, A., & Chaseling, J. (2021). Proposed Framework for Comparison of Continuous Probabilistic Genotyping Systems amongst Different Laboratories. Forensic Sciences, 1(1), 33-45. https://doi.org/10.3390/forensicsci1010006