
Anticoagulation for Splanchnic Vein Thrombosis in Myeloproliferative Neoplasms: The Drug and the Duration

{kind=link}
Abstract
:1. Introduction
2. Overview of MPN-SVT
2.1. Epidemiology
2.2. Pathogenesis
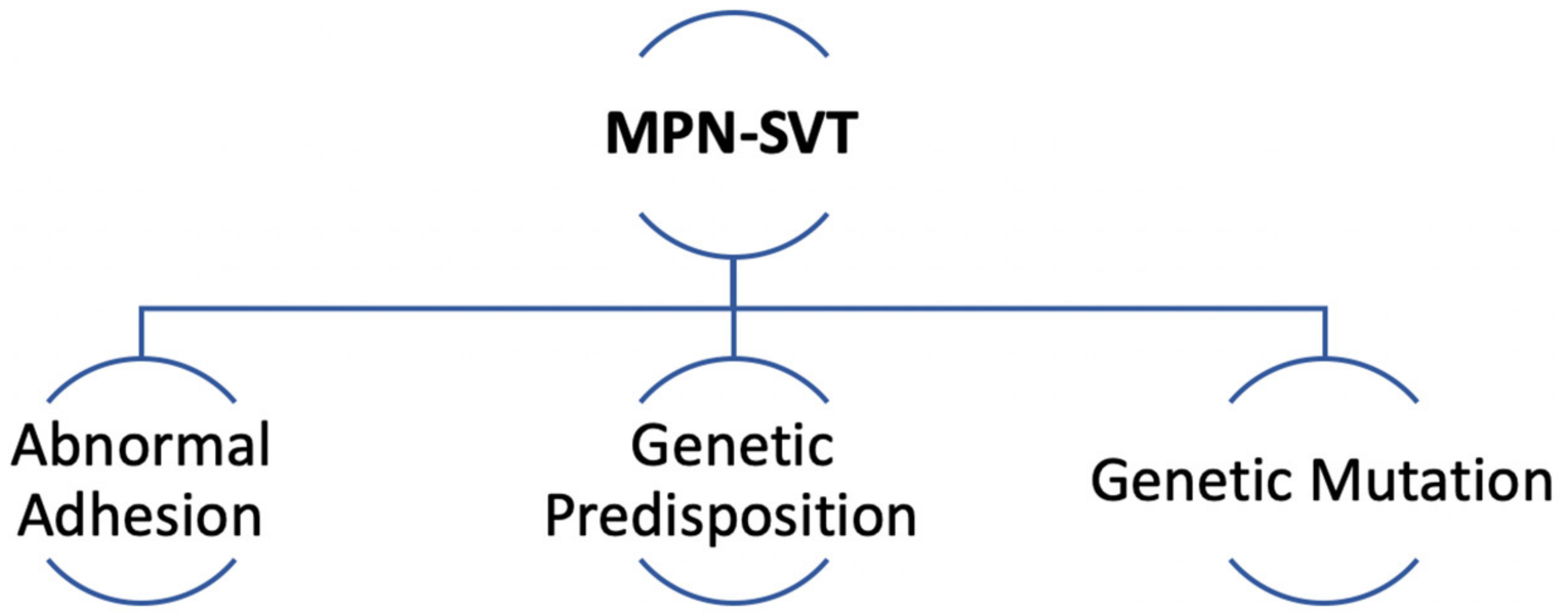
2.2.1. MPN-Thrombosis at Large
2.2.2. MPN-SVT Pathogenesis
2.3. Prognosis
3. Treatment Choices
3.1. Anticoagulation
3.2. Direct Oral Anticoagulants (DOACs)
4. Anticoagulation Treatment Duration
4.1. MPN Thrombosis at Large
4.2. MPN-SVT
5. Cytoreduction
5.1. Hematocrit Control
5.2. Hydroxyurea
5.3. Pegylated Interferon
5.4. Ruxolitinib
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Finazzi, G.; Vannucchi, A.M.; Barbui, T. Prefibrotic myelofibrosis: Treatment algorithm 2018. Blood Cancer J. 2018, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- Sant’Antonio, E.; Guglielmelli, P.; Pieri, L.; Primignani, M.; Randi, M.L.; Santarossa, C.; Rumi, E.; Cervantes, F.; Delaini, F.; Msc, A.C.; et al. Splanchnic vein thromboses associated with myeloproliferative neoplasms: An international, retrospective study on 518 cases. Am. J. Hematol. 2020, 95, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Rungjirajittranon, T.; Owattanapanich, W.; Ungprasert, P.; Siritanaratkul, N.; Ruchutrakool, T. A systematic review and meta-analysis of the prevalence of thrombosis and bleeding at diagnosis of Philadelphia-negative myeloproliferative neoplasms. BMC Cancer 2019, 19, 184. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Marchetti, M. Thrombotic disease in the myeloproliferative neoplasms. Hematol. Am. Soc. Hematol. Educ. Program. 2012, 2012, 571–581. [Google Scholar] [CrossRef]
- Barbui, T.; Falanga, A. Molecular biomarkers of thrombosis in myeloproliferative neoplasms. Thromb. Res. 2016, 140, S71–S75. [Google Scholar] [CrossRef]
- Ageno, W.; Riva, N.; Schulman, S.; Beyer-Westendorf, J.; Bang, S.M.; Senzolo, M.; Grandone, E.; Pasca, S.; Di Minno, M.N.D.; Duce, R.; et al. Long-term Clinical Outcomes of Splanchnic Vein Thrombosis: Results of an International Registry. JAMA Intern. Med. 2015, 175, 1474–1480. [Google Scholar] [CrossRef]
- Stein, B.L.; Saraf, S.; Sobol, U.; Halpern, A.; Shammo, J.; Rondelli, D.; Michaelis, L.; Odenike, O.; Rademaker, A.; Zakarija, A.; et al. Age-related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk. Lymphoma 2013, 54, 1989–1995. [Google Scholar] [CrossRef] [PubMed]
- Smalberg, J.; Arends, L.; Valla, D.; Dominique, C.; Kiladjian, J.-J.; Janssen, H.; Leebeek, F. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: A meta-analysis. Blood 2012, 120, 4921–4928. [Google Scholar] [CrossRef]
- De Stefano, V.; Za, T.; Ciminello, A.; Betti, S.; Rossi, E. Causes of adult splanchnic vein thrombosis in the mediterranean area. Mediterr. J. Hematol. Infect. Dis. 2011, 3, e2011063. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Vainchenker, W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Johnson, S. The ins and outs of calreticulin: From the ER lumen to the extracellular space. Trends Cell Biol. 2001, 11, 122–129. [Google Scholar] [CrossRef]
- Li, M.; De Stefano, V.; Song, T.; Zhou, X.; Guo, Z.; Zhu, J.; Qi, X. Prevalence of CALR mutations in splanchnic vein thrombosis: A systematic review and meta-analysis. Thromb. Res. 2018, 167, 96–103. [Google Scholar] [CrossRef] [PubMed]
- How, J.; Trinkaus, K.M.; Oh, S.T. Distinct clinical, laboratory and molecular features of myeloproliferative neoplasm patients with splanchnic vein thrombosis. Br. J. Haematol. 2017, 183, 310–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumi, E.; Pietra, D.; Guglielmelli, P.; Bordoni, R.; Casetti, I.; Milanesi, C.; Sant’Antonio, E.; Ferretti, V.; Pancrazzi, A.; Rotunno, G.; et al. Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 2013, 121, 4388–4395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, B.L.; Martin, K. From Budd-Chiari syndrome to acquired von Willebrand syndrome: Thrombosis and bleeding complications in the myeloproliferative neoplasms. Hematol. Am. Soc. Hematol. Educ. Program. 2019, 2019, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, C.M.; Manning, H.; Bennett, C.; Vasquez, L.; Severin, S.; Brain, L.; Mazharian, A.; Guerrero, J.A.; Li, J.; Soranzo, N.; et al. JAK2V617F leads to intrinsic changes in platelet formation and reactivity in a knock-in mouse model of essential thrombocythemia. Blood 2013, 122, 3787–3797. [Google Scholar] [CrossRef]
- Gangaraju, R.; Song, J.; Kim, S.J.; Tashi, T.; Reeves, B.N.; Sundar, K.M.; Thiagarajan, P.; Prchal, J.T. Thrombotic, inflammatory, and HIF-regulated genes and thrombosis risk in polycythemia vera and essential thrombocythemia. Blood Adv. 2020, 4, 1115–1130. [Google Scholar] [CrossRef] [Green Version]
- Li, S.L.; Zhang, P.J.; Sun, G.X.; Lu, Z.J. The JAK2 46/1 haplotype (GGCC) in myeloproliferative neoplasms and splanchnic vein thrombosis: A pooled analysis of 26 observational studies. Ann. Hematol. 2014, 93, 1845–1852. [Google Scholar] [CrossRef]
- Spivak, J.L. How I treat polycythemia vera. Blood 2019, 134, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Vascular bed-specific thrombosis. J. Thromb. Haemost. 2007, 5, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Sozer, S.; Fiel, M.I.; Schiano, T.; Xu, M.; Mascarenhas, J.; Hoffman, R. The presence of JAK2V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood 2009, 113, 5246–5249. [Google Scholar] [CrossRef]
- Teofili, L.; Martini, M.; Iachininoto, M.G.; Capodimonti, S.; Nuzzolo, E.R.; Torti, L.; Cenci, T.; LaRocca, L.M.; Leone, G. Endothelial progenitor cells are clonal and exhibit the JAK2V617F mutation in a subset of thrombotic patients with Ph-negative myeloproliferative neoplasms. Blood 2011, 117, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Larrán, A.; Pereira, A.; Magaz, M.; Hernández-Boluda, J.C.; Garrote, M.; Cuevas, B.; Ferrer-Marín, F.; Gómez-Casares, M.T.; García-Gutiérrez, V.; Mata-Vázquez, M.I.; et al. Natural history of polycythemia vera and essential thrombocythemia presenting with splanchnic vein thrombosis. Ann. Hematol. 2020, 99, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Lavu, S.; Szuber, N.; Mudireddy, M.; Yogarajah, M.; Gangat, N.; Pardanani, A.; Hanson, C.A.; Ketterling, R.P.; Ashrani, A.A.; Kamath, P.S.; et al. Splanchnic vein thrombosis in patients with myeloproliferative neoplasms: The Mayo clinic experience with 84 consecutive cases. Am. J. Hematol. 2018, 93, E61–E64. [Google Scholar] [CrossRef] [PubMed]
- Debureaux, P.-E.; Cassinat, B.; Soret-Dulphy, J.; Mora, B.; Verger, E.; Maslah, N.; Plessier, A.; Rautou, P.-E.; Ollivier-Hourman, I.; De Ledinghen, V.; et al. Molecular profiling and risk classification of patients with myeloproliferative neoplasms and splanchnic vein thromboses. Blood Adv. 2020, 4, 3708–3715. [Google Scholar] [CrossRef]
- De Stefano, V.; Vannucchi, A.; Ruggeri, M.; Cervantes, F.; Alvarez-Larrán, A.; Iurlo, A.; Randi, M.; Pieri, L.; Rossi, E.; Guglielmelli, P.; et al. Splanchnic vein thrombosis in myeloproliferative neoplasms: Risk factors for recurrences in a cohort of 181 patients. Blood Cancer J. 2016, 6, e493. [Google Scholar] [CrossRef]
- Kearon, C.; Akl, E.A.; Ornelas, J.; Blaivas, A.; Jimenez, D.; Bounameaux, H.; Huisman, M.; King, C.S.; Morris, T.A.; Sood, N.; et al. Antithrombotic Therapy for VTE Disease: CHEST Guideline and Expert Panel Report. Chest 2016, 149, 315–352. [Google Scholar] [CrossRef] [PubMed]
- Finazzi, G.; De Stefano, V.; Barbui, T. Splanchnic vein thrombosis in myeloproliferative neoplasms: Treatment algorithm 2018. Blood Cancer J. 2018, 8, 64. [Google Scholar] [CrossRef]
- Barbui, T.; Tefferi, A.; Vannucchi, A.M.; Passamonti, F.; Silver, R.T.; Hoffman, R.; Verstovsek, S.; Mesa, R.; Kiladjian, J.-J.; Hehlmann, R.; et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: Revised management recommendations from European LeukemiaNet. Leukemia 2018, 32, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Ianotto, J.-C.; Couturier, M.-A.; Galinat, H.; Mottier, D.; Lippert, E.; Delluc, A.; Berthou, C.; Guillerm, G. Administration of direct oral anticoagulants in patients with myeloproliferative neoplasms. Int. J. Hematol. 2017, 106, 517–521. [Google Scholar] [CrossRef]
- Huenerbein, K.; Sadjadian, P.; Becker, T.; Kolatzki, V.; Deventer, E.; Engelhardt, C.; Griesshammer, M.; Wille, K. Direct oral anticoagulants (DOAC) for prevention of recurrent arterial or venous thromboembolic events (ATE/VTE) in myeloproliferative neoplasms. Ann. Hematol. 2020. [Google Scholar] [CrossRef] [PubMed]
- De Gottardi, A.; Trebicka, J.; Klinger, C.; Plessier, A.; Seijo, S.; Terziroli, B.; Magenta, L.; Semela, D.; Buscarini, E.; Langlet, P.; et al. Antithrombotic treatment with direct-acting oral anticoagulants in patients with splanchnic vein thrombosis and cirrhosis. Liver Int. 2017, 37, 694–699. [Google Scholar] [CrossRef]
- Valeriani, E.; Riva, N.; Di Nisio, M.; Ageno, W. Splanchnic Vein Thrombosis: Current Perspectives. Vasc. Health Risk Manag. 2019, 15, 449–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, M.H.; Lavi, N.; Vannucchi, A.; Harrison, C. Treatment of thromboembolic events coincident with the diagnosis of myeloproliferative neoplasms: A physician survey. Thromb. Res. 2014, 134, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Jamieson, C.; Bhatia, R.; Deininger, M.W.; Fletcher, C.D.; Gerds, A.T.; Gojo, I.; Gotlib, J.; Gundabolu, K.; Hobbs, G.; et al. NCCN Guidelines Insights: Myeloproliferative Neoplasms, Version 2.2018. J. Natl. Compr. Cancer Netw. 2017, 15, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Boluda, J.-C.; Pereira, A.; Arellano-Rodrigo, E.; Cervantes, F.; Alvarez-Larrán, A.; Gómez, M.; Barba, P.; Mata, M.-I.; Gonzalez-Porras, J.-R.; Ferrer-Marin, F.; et al. Oral anticoagulation to prevent thrombosis recurrence in polycythemia vera and essential thrombocythemia. Ann. Hematol. 2015, 94, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Wille, K.; Sadjadian, P.; Becker, T.; Kolatzki, V.; Horstmann, A.; Fuchs, C.; Griesshammer, M. High risk of recurrent venous thromboembolism in BCR-ABL-negative myeloproliferative neoplasms after termination of anticoagulation. Ann. Hematol. 2018, 98, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Kreher, S.; Ochsenreither, S.; Trappe, R.U.; Pabinger, I.; Bergmann, F.; Petrides, P.E.; Koschmieder, S.; Matzdorff, A.; Tiede, A.; Griesshammer, M.; et al. Prophylaxis and management of venous thromboembolism in patients with myeloproliferative neoplasms: Consensus statement of the Haemostasis Working Party of the German Society of Hematology and Oncology (DGHO), the Austrian Society of Hematology and Oncology (ÖGHO) and Society of Thrombosis and Haemostasis Research (GTH e.V.). Ann. Hematol. 2014, 93, 1953–1963. [Google Scholar]
- Marchioli, R.; Finazzi, G.; Specchia, G.; Masciulli, A.; Mennitto, M.R.; Barbui, T. The CYTO-PV: A Large-Scale Trial Testing the Intensity of CYTOreductive Therapy to Prevent Cardiovascular Events in Patients with Polycythemia Vera. Thrombosis 2011, 2011, 794240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, R.T.; Iafrate, A.J.; Amrein, P.C.; Sahani, D.V.; Misdraji, J. Case records of the Massachusetts General Hospital. Case 15-2006. A 46-year-old woman with sudden onset of abdominal distention. N. Engl. J. Med. 2006, 354, 2166–2175. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, V.; Rossi, E.; Carobbio, A.; Ghirardi, A.; Betti, S.; Finazzi, G.; Vannucchi, A.M.; Barbui, T. Hydroxyurea prevents arterial and late venous thrombotic recurrences in patients with myeloproliferative neoplasms but fails in the splanchnic venous district. Pooled analysis of 1500 cases. Blood Cancer J. 2018, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Gugliotta, L.; Bagnara, G.P.; Catani, L.; Gaggioli, L.; Guarini, A.; Zauli, G.; Belmonte, M.M.; Lauria, F.; Macchi, S.; Tura, S. In vivo and in vitro inhibitory effect of α-interferon on megakaryocyte colony growth in essential thrombocythaemia. Br. J. Haematol. 1989, 71, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Kosiorek, H.; Prchal, J.; Yacoub, A.; Berenzon, D.; Baer, M.R.; Ritchie, E.; Silver, R.T.; Kessler, C.; Winton, E.; et al. A prospective evaluation of pegylated interferon alfa-2a therapy in patients with polycythemia vera and essential thrombocythemia with a prior splanchnic vein thrombosis. Leukemia 2019, 33, 2974–2978. [Google Scholar] [CrossRef] [PubMed]
- Pieri, L.; Paoli, C.; Arena, U.; Marra, F.; Mori, F.; Zucchini, M.; Colagrande, S.; Castellani, A.; Masciulli, A.; Rosti, V.; et al. Safety and efficacy of ruxolitinib in splanchnic vein thrombosis associated with myeloproliferative neoplasms. Am. J. Hematol. 2016, 92, 187–195. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sedhom, W.G.; Stein, B.L. Anticoagulation for Splanchnic Vein Thrombosis in Myeloproliferative Neoplasms: The Drug and the Duration. Hemato 2021, 2, 255-263. https://doi.org/10.3390/hemato2020015
Sedhom WG, Stein BL. Anticoagulation for Splanchnic Vein Thrombosis in Myeloproliferative Neoplasms: The Drug and the Duration. Hemato. 2021; 2(2):255-263. https://doi.org/10.3390/hemato2020015
Chicago/Turabian StyleSedhom, Wafik G., and Brady Lee Stein. 2021. "Anticoagulation for Splanchnic Vein Thrombosis in Myeloproliferative Neoplasms: The Drug and the Duration" Hemato 2, no. 2: 255-263. https://doi.org/10.3390/hemato2020015
APA StyleSedhom, W. G., & Stein, B. L. (2021). Anticoagulation for Splanchnic Vein Thrombosis in Myeloproliferative Neoplasms: The Drug and the Duration. Hemato, 2(2), 255-263. https://doi.org/10.3390/hemato2020015