
Pathway Analysis Interpretation in the Multi-Omic Era

 , , ,
, , , 

Abstract

1. Introduction
2. Key Challenges to Pathway Analysis
2.1. Pathway Annotation
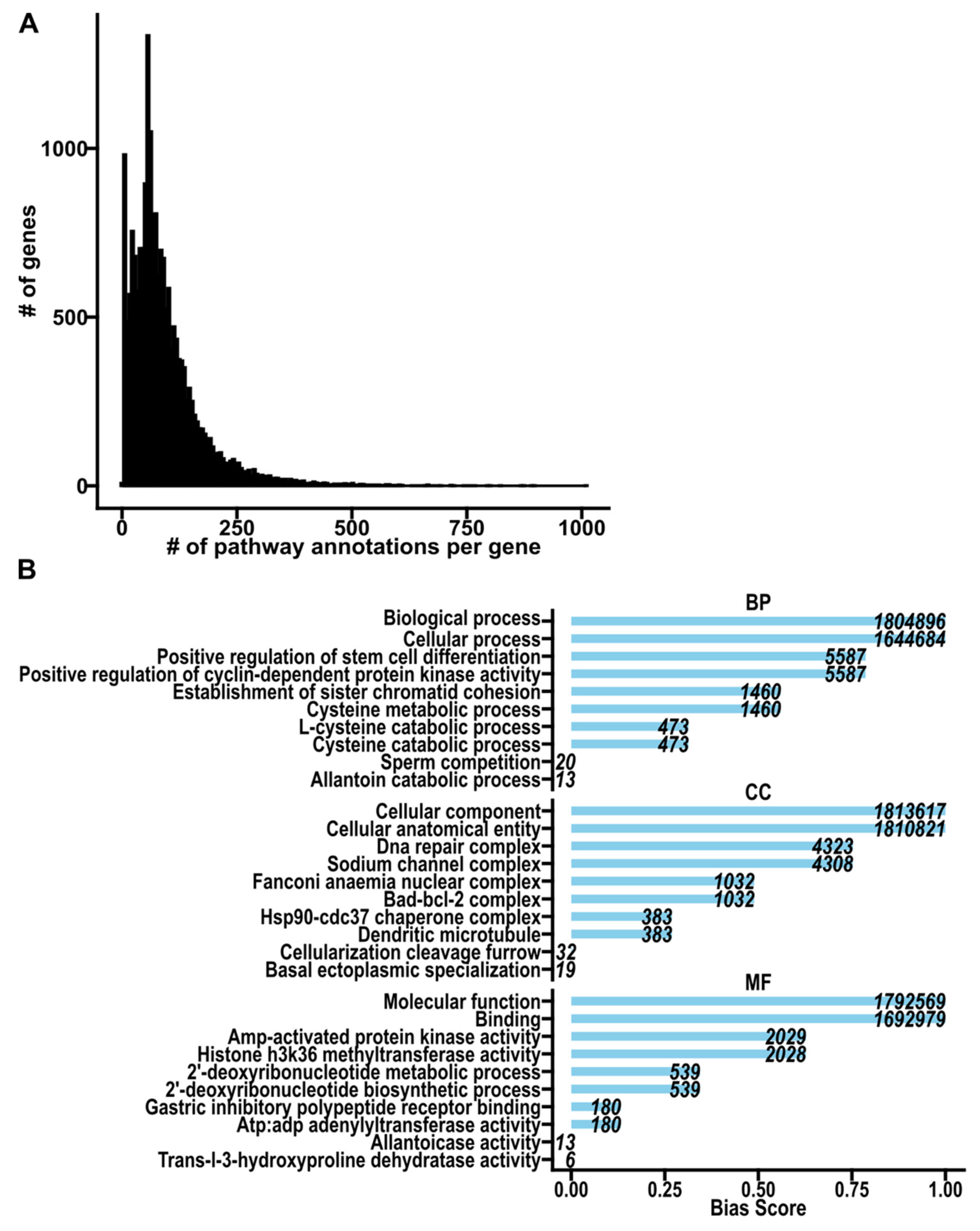

{kind=link}
{kind=link}
| Locus Type | Count |
|---|---|
| pseudogene | 13,940 |
| RNA, long non-coding | 5640 |
| RNA, micro | 1912 |
| gene with protein product | 611 |
| RNA, transfer | 591 |
| RNA, small nucleolar | 568 |
| immunoglobulin pseudogene | 202 |
| readthrough | 143 |
| RNA, cluster | 119 |
| fragile site | 116 |
| endogenous retrovirus | 92 |
| T cell receptor gene | 67 |
| RNA, ribosomal | 58 |
| immunoglobulin gene | 55 |
| RNA, small nuclear | 51 |
| region | 46 |
| unknown | 46 |
| T cell receptor pseudogene | 38 |
| RNA, misc | 29 |
| virus integration site | 8 |
| complex locus constituent | 6 |
| RNA, vault | 4 |
| RNA, Y | 4 |
| Tool | Year | Method | Access | Database | Visualization | Description |
|---|---|---|---|---|---|---|
| REVIGO [57] | 2011 | Semantic | Web | GO | Scatterplots, Interactive graph, tree maps | Summarizes GO term list using Semantic Similarity and clustering |
| clusterProfiler [58] | 2013 | Semantic | R package | GO, KEGG, DO | Dot plot | Enrichment analysisfor GO/KEGG terms and visualization |
| ReCiPa [52] | 2018 | Semantic | R package | KEGG, Reactome | Data tables | Controls redundancy in pathway databases |
| GOGO [59] | 2018 | Semantic | Web, Perl | GO | Data tables | Calculates semantic similarity of GO terms using improved algorithms |
| FunSet [60] | 2019 | Semantic | Web, Standalone | GO | 2D plots | Performs GO enrichment analysis with interactive visualizations |
| GeneSetCluster [61] | 2020 | Semantic | R package | Any | Network graph, dendrogram, heatmap | Groups gene sets post analysis based on shared genes |
| GOMCL [62] | 2020 | Semantic | Python | GO | Heatmap, Network graph | Clusters GO terms using Markov clustering algorithm |
| GoSemSim [63] | 2020 | Semantic | R package | GO | Data tables | Computes semantic similarity among GO terms for comparison |
| GO-FIGURE! [15] | 2021 | Semantic | Python | GO | Scatterplot | Visualizes GO term similarity with custom scatterplots |
| Simplify Enrichment [64] | 2022 | Semantic | R package | GO | Heatmap | Clusters with a unique binary cut algorithm. |
| iDEP [65] | 2018 | Semantic | Web, R package | GO, KEGG, Reactome | Tree, Heatmap, Network Graph | Web app for transcriptomics And pathway exploration |
| DAVID [66] | 2009 | Semantic | Web, REST API | KEGG, Any | Tabular, Barchart, Network graph | Enrichment analysis with functional annotation clustering |
| g:Profiler [67] | 2007 | Semantic | Web, R package | KEGG, Reactome, WikiPathways, Any | Dot plot, Tabular, Network graph | Orthology-aware enrichment analysis and clustering |
| RICHNET [68] | 2019 | Network | R protocol | MSigDB | Network graph | Automated gene-set network creation |
| EnrichmentMap [69] | 2019 | Network | Cytoscape | Any | Interactive network | Detailed enrichment mapping |
| Gscluster [70] | 2019 | Network | Web, R Package | MSigDB | Interactive network | Network-weighted gene-set clustering integrating PPI data |
| aPEAR [71] | 2019 | Network | R package | Any | Network graph | Clustering with automated naming |
| GeneFEAST [72] | 2023 | Network | Web, Python | Any | Heatmap, Dot plot, Upset plot | Highlights multi-enrichment genes |
| vissE [73,74] | 2023 | Network | R package | MSigDB, Any | Network graph | Visualizes higher-order interactions |
| pathlinkR [75] | 2024 | Network | R package | Reactome, MSigDB, InnateDB | Network graph, Volcano plot, Dot plot | Integrated PPI network construction |
| Pathview [76] | 2017 | Network | Web, R Package, REST API | KEGG | Network Graph | Visualizes and maps data onto KEGG pathways |
| PAVER [77] | 2024 | Embedding | Web, R package | Any | UMAP, Heatmap, Dot plot | Embedding-based clustering with UMAP for clear pathway visualization |
| Mondrian- Map [78] | 2024 | Embedding | Python | WikiPathways | Mondrian Map | Embedding visualizations highlighting pathway interactions and crosstalk |
| GOsummaries [79] | 2015 | Word Cloud | R package | GO | PCA, Boxplot | Visualizes GO analyses as word clouds and overlays results |
| genesetSV [80] | 2023 | Game Theory | Python | KEGG, MSigDB | Scatterplot | Uses Shapley values for ranking and reducing pathway sets |
| Archetype- Discovery [81] | 2024 | Non-negative Matrix Factorization | MATLAB | MSigDB, Any | Radar, Scatter- and Boxplot, Heatmap | Derives compact archetypal gene-set patterns and their pathway associations |
2.2. Visualizing Pathway Findings
2.3. Limitations to Pathway Analysis Utility
2.4. Discrepancies in Molecular Biology Mislead Validation
2.5. Research Data Mismanagement
3. Methods for Pathway Analysis Interpretation
3.1. Semantic Similarity Based Methods
3.2. Network-Based Methods
3.3. Embedding Based Methods
3.4. Applications of Tools for Pathway Interpretation
3.5. Choosing the Right Tool for Your Research
4. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Glossary
| Pathway Analysis | A method for identifying biological pathways enriched with differentially expressed genes or proteins in datasets. |
| Dimensionality Reduction | Techniques used to visualize high-dimensional data in simpler forms for clearer pathway analysis. |
| Pathway Redundancy | The occurrence of overlapping or repeated pathways in analysis, which can complicate interpretation and reduce clarity. |
| Embedding-Based Methods | Computational approaches that represent biological pathways as high-dimensional numerical vectors for analysis. |
| Semantic Similarity | A metric that quantifies the functional similarity between different biological terms or pathways. |
| Network-Based Analysis | A method that visualizes relationships between pathways as interconnected networks, highlighting shared functions or genes. |
References
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2016, 19, 286–302. [Google Scholar] [CrossRef]
- Veenstra, T.D. Omics in Systems Biology: Current Progress and Future Outlook. Proteomics 2021, 21, e2000235. [Google Scholar] [CrossRef] [PubMed]
- Herr, T.M.; Bielinski, S.J.; Bottinger, E.; Brautbar, A.; Brilliant, M.; Chute, C.G.; Denny, J.; Freimuth, R.R.; Hartzler, A.; Kannry, J.; et al. A conceptual model for translating omic data into clinical action. J. Pathol. Inform. 2015, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- García-Campos, M.A.; Espinal-Enríquez, J.; Hernández-Lemus, E. Pathway Analysis: State of the Art. Front. Physiol. 2015, 6, 383. [Google Scholar] [CrossRef] [PubMed]
- Wegman-Points, L.; Alganem, K.; Imami, A.S.; Mathis, V.; Creeden, J.F.; McCullumsmith, R.; Yuan, L.-L. Subcellular partitioning of protein kinase activity revealed by functional kinome profiling. Sci. Rep. 2022, 12, 17300. [Google Scholar] [CrossRef]
- Ramanan, V.K.; Shen, L.; Moore, J.H.; Saykin, A.J. Pathway analysis of genomic data: Concepts, methods, and prospects for future development. Trends Genet. 2012, 28, 323–332. [Google Scholar] [CrossRef]
- Krassowski, M.; Das, V.; Sahu, S.K.; Misra, B.B. State of the Field in Multi-Omics Research: From Computational Needs to Data Mining and Sharing. Front. Genet. 2020, 11, 610798. [Google Scholar] [CrossRef]
- Sboner, A.; Mu, X.J.; Greenbaum, D.; Auerbach, R.K.; Gerstein, M.B. The real cost of sequencing: Higher than you think! Genome Biol. 2011, 12, 125. [Google Scholar] [CrossRef]
- D’Adamo, G.L.; Widdop, J.T.; Giles, E.M. The future is now? Clinical and translational aspects of “Omics” technologies. Immunol. Cell Biol. 2021, 99, 168–176. [Google Scholar] [CrossRef]
- Denecker, T.; Lelandais, G. Omics Analyses: How to Navigate Through a Constant DataData Deluge, in Yeast Functional Genomics: Methods and Protocols; Devaux, F., Ed.; Springer: New York, NY, USA, 2022; pp. 457–471. [Google Scholar]
- Bell, G.; Hey, T.; Szalay, A. Computer science. Beyond the data deluge. Science 2009, 323, 1297–1298. [Google Scholar] [CrossRef]
- Stead, W.W.; Searle, J.R.; Fessler, H.E.; Smith, J.W.; Shortliffe, E.H. Biomedical informatics: Changing what physicians need to know and how they learn. Acad. Med. 2011, 86, 429–434. [Google Scholar] [CrossRef]
- Pita-Juárez, Y.; Altschuler, G.; Kariotis, S.; Wei, W.; Koler, K.; Green, C.; Tanzi, R.E.; Hide, W. The Pathway Coexpression Network: Revealing pathway relationships. PLoS Comput. Biol. 2018, 14, e1006042. [Google Scholar] [CrossRef] [PubMed]
- Chicco, D.; Agapito, G. Nine quick tips for pathway enrichment analysis. PLoS Comput. Biol. 2022, 18, e1010348. [Google Scholar] [CrossRef] [PubMed]
- Reijnders, M.J.; Waterhouse, R.M. Summary visualizations of gene ontology terms with GO-Figure! Front. Bioinform. 2021, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Woo, H.J.; Yu, X.; Oyama, T.; Wallqvist, A.; Reifman, J. A strategy for evaluating pathway analysis methods. BMC Bioinform. 2017, 18, 453. [Google Scholar] [CrossRef]
- Searson, P.C. The Cancer Moonshot, the role of in vitro models, model accuracy, and the need for validation. Nat. Nanotechnol. 2023, 18, 1121–1123. [Google Scholar] [CrossRef]
- Durinikova, E.; Buzo, K.; Arena, S. Preclinical models as patients’ avatars for precision medicine in colorectal cancer: Past and future challenges. J. Exp. Clin. Cancer Res. 2021, 40, 185. [Google Scholar] [CrossRef]
- Diaz-Uriarte, R.; de Lope, E.G.; Giugno, R.; Fröhlich, H.; Nazarov, P.V.; Nepomuceno-Chamorro, I.A.; Rauschenberger, A.; Glaab, E. Ten quick tips for biomarker discovery and validation analyses using machine learning. PLoS Comput. Biol. 2022, 18, e1010357. [Google Scholar] [CrossRef]
- Grabowski, T.; Tomczyk, A.; Wolc, A.; Gad, S.C. Between Biological Relevancy and Statistical Significance—Step for Assessment Harmonization. Am. J. Biomed. Sci. Res. 2021, 13. [Google Scholar] [CrossRef]
- Committee, E.S.; Hardy, A.; Benford, D.; Halldorsson, T.; Jeger, M.J.; Knutsen, H.K.; More, S.; Naegeli, H.; Noteborn, H.; Ockleford, C.; et al. Guidance on the assessment of the biological relevance of data in scientific assessments. EFSA J. 2017, 15, e04970. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Zorin, A.; Dass, G.; Vu, M.T.; Xu, P.; Glont, M.; Vizcaino, J.A.; Jarnuczak, A.F.; Petryszak, R.; Ping, P.; et al. Quantifying the impact of public omics data. Nat. Commun. 2019, 10, 3512. [Google Scholar] [CrossRef] [PubMed]
- Misra, B.B.; Langefeld, C.; Olivier, M.; Cox, L.A. Integrated omics: Tools, advances and future approaches. J. Mol. Endocrinol. 2019, 62, R21–R45. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Fernández, D.; Mubeen, S.; Marín-Llaó, J.; Hoyt, C.T.; Hofmann-Apitius, M. PathMe: Merging and exploring mechanistic pathway knowledge. BMC Bioinform. 2019, 20, 243. [Google Scholar] [CrossRef] [PubMed]
- Wieder, C.; Cooke, J.; Frainay, C.; Poupin, N.; Bowler, R.; Jourdan, F.; Kechris, K.J.; Lai, R.P.J.; Ebbels, T. PathIntegrate: Multivariate modelling approaches for pathway-based multi-omics data integration. PLoS Comput. Biol. 2024, 20, e1011814. [Google Scholar] [CrossRef]
- Canzler, S.; Hackermüller, J. multiGSEA: A GSEA-based pathway enrichment analysis for multi-omics data. BMC Bioinform. 2020, 21, 561. [Google Scholar] [CrossRef]
- Ivanisevic, T.; Sewduth, R.N. Multi-Omics Integration for the Design of Novel Therapies and the Identification of Novel Biomarkers. Proteomes 2023, 11, 34. [Google Scholar] [CrossRef]
- Mohr, A.E.; Ortega-Santos, C.P.; Whisner, C.M.; Klein-Seetharaman, J.; Jasbi, P. Navigating Challenges and Opportunities in Multi-Omics Integration for Personalized Healthcare. Biomedicines 2024, 12, 1496. [Google Scholar] [CrossRef]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten years of pathway analysis: Current approaches and outstanding challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef]
- Nguyen, T.-M.; Shafi, A.; Nguyen, T.; Draghici, S. Identifying significantly impacted pathways: A comprehensive review and assessment. Genome Biol. 2019, 20, 203. [Google Scholar] [CrossRef]
- Nam, D.; Kim, S.Y. Gene-set approach for expression pattern analysis. Brief. Bioinform. 2008, 9, 189–197. [Google Scholar] [CrossRef]
- Maghsoudi, Z.; Nguyen, H.; Tavakkoli, A.; Nguyen, T. A comprehensive survey of the approaches for pathway analysis using multi-omics data integration. Brief. Bioinform. 2022, 23, bbac435. [Google Scholar] [CrossRef]
- Winston, J.E. Twenty-First Century Biological Nomenclature—The Enduring Power of Names. Integr. Comp. Biol. 2018, 58, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Vassalli, P. The pathophysiology of tumor necrosis factors. Annu. Rev. Immunol. 1992, 10, 411–452. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.D.; Vucic, D. The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front. Cell Dev. Biol. 2020, 8, 365. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yang, Q.; Abdul, A.; Lim, B.Y. Designing Theory-Driven User-Centric Explainable AI. In Proceedings of the 2019 CHI Conference on Human Factors in Computing Systems, Glasgow, UK, 4–9 May 2019; Association for Computing Machinery: Glasgow, UK, 2019; p. 601. [Google Scholar]
- Jara, J.H.; Singh, B.B.; Floden, A.M.; Combs, C.K. Tumor necrosis factor alpha stimulates NMDA receptor activity in mouse cortical neurons resulting in ERK-dependent death. J. Neurochem. 2007, 100, 1407–1420. [Google Scholar] [CrossRef]
- Sebastian-Leon, P.; Vidal, E.; Minguez, P.; Conesa, A.; Tarazona, S.; Amadoz, A.; Armero, C.; Salavert, F.; Vidal-Puig, A.; Montaner, D.; et al. Understanding disease mechanisms with models of signaling pathway activities. BMC Syst. Biol. 2014, 8, 121. [Google Scholar] [CrossRef]
- Lee, J.; Jo, K.; Lee, S.; Kang, J.; Kim, S. Prioritizing biological pathways by recognizing context in time-series gene expression data. BMC Bioinform. 2016, 17, 477. [Google Scholar] [CrossRef]
- Sjöström, J.; Bergh, J. How apoptosis is regulated, and what goes wrong in cancer. BMJ 2001, 322, 1538–1539. [Google Scholar] [CrossRef]
- Nguyen, T.T.M.; Gillet, G.; Popgeorgiev, N. Caspases in the Developing Central Nervous System: Apoptosis and Beyond. Front. Cell Dev. Biol. 2021, 9, 702404. [Google Scholar] [CrossRef]
- Ryu, J.R.; Hong, C.J.; Kim, J.Y.; Kim, E.-K.; Sun, W.; Yu, S.-W. Control of adult neurogenesis by programmed cell death in the mammalian brain. Mol. Brain 2016, 9, 43. [Google Scholar] [CrossRef]
- Anosike, N.L.; Adejuwon, J.F.; Emmanuel, G.E.; Adebayo, O.S.; Etti-Balogun, H.; Nathaniel, J.N.; Omotosho, O.I.; Aschner, M.; Ijomone, O.M. Necroptosis in the developing brain: Role in neurodevelopmental disorders. Metab. Brain Dis. 2023, 38, 831–837. [Google Scholar] [CrossRef]
- Saini, S.; Kakati, P.; Singh, K. Role of Inflammation in Tissue Regeneration and Repair. In Inflammation Resolution and Chronic Diseases; Tripathi, A., Dwivedi, A., Gupta, S., Poojan, S., Eds.; Springer Nature: Singapore, 2024; pp. 103–127. [Google Scholar]
- Choi, B.; Lee, C.; Yu, J.-W. Distinctive role of inflammation in tissue repair and regeneration. Arch. Pharmacal Res. 2023, 46, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease--a double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Gasque, P.; Neal, J.W.; Singhrao, S.K.; McGreal, E.P.; Dean, Y.D.; Van, B.J.; Morgan, B.P. Roles of the complement system in human neurodegenerative disorders: Pro-inflammatory and tissue remodeling activities. Mol. Neurobiol. 2002, 25, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Shih, R.-H.; Wang, C.-Y.; Yang, C.-M. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef]
- Sun, S.-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Adriaens, M.E.; Jaillard, M.; Waagmeester, A.; Coort, S.L.M.; Pico, A.R.; Evelo, C.T.A. The public road to high-quality curated biological pathways. Drug Discov. Today 2008, 13, 856–862. [Google Scholar] [CrossRef]
- Shin, M.-G.; Pico, A.R. Using published pathway figures in enrichment analysis and machine learning. BMC Genom. 2023, 24, 713. [Google Scholar] [CrossRef]
- Vivar, J.C.; Pemu, P.; McPherson, R.; Ghosh, S. Redundancy control in pathway databases (ReCiPa): An application for improving gene-set enrichment analysis in Omics studies and “Big data” biology. Omics 2013, 17, 414–422. [Google Scholar] [CrossRef]
- Pastrello, C.; Niu, Y.; Jurisica, I. Pathway Enrichment Analysis of Microarray Data. Methods Mol. Biol. 2022, 2401, 147–159. [Google Scholar]
- Gable, A.L.; Szklarczyk, D.; Lyon, D.; Rodrigues, J.F.M.; von Mering, C. Systematic assessment of pathway databases, based on a diverse collection of user-submitted experiments. Brief. Bioinform. 2022, 23, bbac355. [Google Scholar] [CrossRef] [PubMed]
- Maertens, A.; Tran, V.P.; Maertens, M.; Kleensang, A.; Luechtefeld, T.H.; Hartung, T.; Paller, C.J. Functionally Enigmatic Genes in Cancer: Using TCGA Data to Map the Limitations of Annotations. Sci. Rep. 2020, 10, 4106. [Google Scholar] [CrossRef] [PubMed]
- Stoney, R.A.; Schwartz, J.-M.; Robertson, D.L.; Nenadic, G. Using set theory to reduce redundancy in pathway sets. BMC Bioinform. 2018, 19, 386. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, Z. GOGO: An improved algorithm to measure the semantic similarity between gene ontology terms. Sci. Rep. 2018, 8, 15107. [Google Scholar] [CrossRef]
- Hale, M.L.; Thapa, I.; Ghersi, D. FunSet: An open-source software and web server for performing and displaying Gene Ontology enrichment analysis. BMC Bioinform. 2019, 20, 359. [Google Scholar] [CrossRef]
- Ewing, E.; Planell-Picola, N.; Jagodic, M.; Gomez-Cabrero, D. GeneSetCluster: A tool for summarizing and integrating gene-set analysis results. BMC Bioinform. 2020, 21, 443. [Google Scholar] [CrossRef]
- Wang, G.; Oh, D.H.; Dassanayake, M. GOMCL: A toolkit to cluster, evaluate, and extract non-redundant associations of Gene Ontology-based functions. BMC Bioinform. 2020, 21, 139. [Google Scholar] [CrossRef]
- Yu, G. Gene Ontology Semantic Similarity Analysis Using GOSemSim. Methods Mol. Biol. 2020, 2117, 207–215. [Google Scholar]
- Gu, Z.; Hübschmann, D. SimplifyEnrichment: A Bioconductor Package for Clustering and Visualizing Functional Enrichment Results. Genom. Proteom. Bioinform. 2022, 21, 190–202. [Google Scholar] [CrossRef]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. g:Profiler—Interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef]
- Prummer, M. Enhancing gene set enrichment using networks. F1000Research 2019, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Kim, J.; Kim, S.-K.; Baik, B.; Chi, S.-M.; Kim, S.-Y.; Nam, D. GScluster: Network-weighted gene-set clustering analysis. BMC Genom. 2019, 20, 352. [Google Scholar] [CrossRef] [PubMed]
- Kerseviciute, I.; Gordevicius, J. aPEAR: An R package for autonomous visualization of pathway enrichment networks. Bioinformatics 2023, 39, btad672. [Google Scholar] [CrossRef]
- Taylor, A.; Macaulay, V.M.; Maurya, A.K.; Miossec, M.J.; Buffa, F.M. GeneFEAST: The pivotal, gene-centric step in functional enrichment analysis interpretation. arXiv 2023, arXiv:2309.00061. [Google Scholar] [CrossRef]
- Bhuva, D.D.; Tan, C.W.; Liu, N.; Whitfield, H.J.; Papachristos, N.; Lee, S.C.; Kharbanda, M.; Mohamed, A.; Davis, M.J. vissE: A versatile tool to identify and visualise higher-order molecular phenotypes from functional enrichment analysis. BMC Bioinform. 2024, 25, 64. [Google Scholar] [CrossRef]
- Mohamed, A.; Bhuva, D.D.; Lee, S.; Liu, N.; Tan, C.W.; Davis, M.J. vissE.cloud: A webserver to visualise higher order molecular phenotypes from enrichment analysis. Nucleic Acids Res. 2023, 51, W593–W600. [Google Scholar] [CrossRef] [PubMed]
- Blimkie, T.M.; An, A.; Hancock, R.E.W. Facilitating pathway and network based analysis of RNA-Seq data with pathlinkR. PLoS Comput. Biol. 2024, 20, e1012422. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Pant, G.; Bhavnasi, Y.K.; Blanchard, S.G.; Brouwer, C. Pathview Web: User friendly pathway visualization and data integration. Nucleic Acids Res. 2017, 45, W501–W508. [Google Scholar] [CrossRef]
- Ryan, V.W.; Imami, A.S.; Sajid, H.A.; Vergis, J.; Zhang, X.; Meller, J.; Shukla, R.; McCullumsmith, R. Interpreting and visualizing pathway analyses using embedding representations with PAVER. Bioinformation 2024, 20, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Al Abir, F.; Chen, J.Y. Mondrian Abstraction and Language Model Embeddings for Differential Pathway Analysis. bioRxiv 2024. [Google Scholar] [CrossRef]
- Kolde, R.; Vilo, J. GOsummaries: An R Package for Visual Functional Annotation of Experimental Data. F1000Research 2015, 4, 574. [Google Scholar] [CrossRef]
- Balestra, C.; Maj, C.; Muller, E.; Mayr, A. Redundancy-aware unsupervised ranking based on game theory: Ranking pathways in collections of gene sets. PLoS ONE 2023, 18, e0282699. [Google Scholar] [CrossRef]
- Weistuch, C.; Murgas, K.A.; Zhu, J.; Norton, L.; Dill, K.A.; Tannenbaum, A.R.; Deasy, J.O. Normal tissue transcriptional signatures for tumor-type-agnostic phenotype prediction. Sci. Rep. 2024, 14, 27230. [Google Scholar] [CrossRef]
- Hanspers, K.; Kutmon, M.; Coort, S.L.; Digles, D.; Dupuis, L.J.; Ehrhart, F.; Hu, F.; Lopes, E.N.; Martens, M.; Pham, N.; et al. Ten simple rules for creating reusable pathway models for computational analysis and visualization. PLoS Comput. Biol. 2021, 17, e1009226. [Google Scholar] [CrossRef]
- He, C.; Micallef, L.; Serim, B.; Vuong, T.; Ruotsalo, T.; Jacucci, G. Interactive visual facets to support fluid exploratory search. J. Vis. 2023, 26, 211–230. [Google Scholar] [CrossRef]
- Ovchinnikova, S.; Anders, S. Exploring dimension-reduced embeddings with Sleepwalk. Genome Res. 2020, 30, 749–756. [Google Scholar] [CrossRef]
- Li, Y.; Ge, X.; Peng, F.; Li, W.; Li, J.J. Exaggerated false positives by popular differential expression methods when analyzing human population samples. Genome Biol. 2022, 23, 79. [Google Scholar] [CrossRef]
- Radulescu, E.; Jaffe, A.E.; Straub, R.E.; Chen, Q.; Shin, J.H.; Hyde, T.M.; Kleinman, J.E.; Weinberger, D.R. Identification and prioritization of gene sets associated with schizophrenia risk by co-expression network analysis in human brain. Mol. Psychiatry 2020, 25, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Sapienza, J.; Spangaro, M.; Guillemin, G.J.; Comai, S.; Bosia, M. Importance of the dysregulation of the kynurenine pathway on cognition in schizophrenia: A systematic review of clinical studies. Eur. Arch. Psychiatry Clin. Neurosci. 2023, 273, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Rusina, P.V.; Falaguera, M.J.; Romero, J.M.R.; McDonagh, E.M.; Dunham, I.; Ochoa, D. Genetic support for FDA-approved drugs over the past decade. Nat. Rev. Drug Discov. 2023, 22, 864. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, D.; Karim, M.; Ghoussaini, M.; Hulcoop, D.G.; McDonagh, E.M.; Dunham, I. Human genetics evidence supports two-thirds of the 2021 FDA-approved drugs. Nat. Rev. Drug Discov. 2022, 21, 551. [Google Scholar] [CrossRef]
- Diogo, D.; Bastarache, L.; Liao, K.P.; Graham, R.R.; Fulton, R.S.; Greenberg, J.D.; Eyre, S.; Bowes, J.; Cui, J.; Lee, A.; et al. TYK2 protein-coding variants protect against rheumatoid arthritis and autoimmunity, with no evidence of major pleiotropic effects on non-autoimmune complex traits. PLoS ONE 2015, 10, e0122271. [Google Scholar] [CrossRef]
- MacNamara, A.; Nakic, N.; Al Olama, A.A.; Guo, C.; Sieber, K.B.; Hurle, M.R.; Gutteridge, A. Network and pathway expansion of genetic disease associations identifies successful drug targets. Sci. Rep. 2020, 10, 20970. [Google Scholar] [CrossRef]
- de la Fuente van Bentem, S.; Mentzen, W.I.; de la Fuente, A.; Hirt, H. Towards functional phosphoproteomics by mapping differential phosphorylation events in signaling networks. Proteomics 2008, 8, 4453–4465. [Google Scholar] [CrossRef]
- Ponomarenko, E.A.; Krasnov, G.S.; Kiseleva, O.I.; Kryukova, P.A.; Arzumanian, V.A.; Dolgalev, G.V.; Ilgisonis, E.V.; Lisitsa, A.V.; Poverennaya, E.V. Workability of mRNA Sequencing for Predicting Protein Abundance. Genes 2023, 14, 2065. [Google Scholar] [CrossRef]
- Prabahar, A.; Zamora, R.; Barclay, D.; Yin, J.; Ramamoorthy, M.; Bagheri, A.; Johnson, S.A.; Badylak, S.; Vodovotz, Y.; Jiang, P. Unraveling the complex relationship between mRNA and protein abundances: A machine learning-based approach for imputing protein levels from RNA-seq data. NAR Genom. Bioinform. 2024, 6, lqae019. [Google Scholar] [CrossRef] [PubMed]
- de Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef] [PubMed]
- Upadhya, S.R.; Ryan, C.J. Experimental reproducibility limits the correlation between mRNA and protein abundances in tumor proteomic profiles. Cell Rep. Methods 2022, 2, 100288. [Google Scholar] [CrossRef] [PubMed]
- Arshad, O.A.; Danna, V.; Petyuk, V.A.; Piehowski, P.D.; Liu, T.; Rodland, K.D.; McDermott, J.E. An Integrative Analysis of Tumor Proteomic and Phosphoproteomic Profiles to Examine the Relationships Between Kinase Activity and Phosphorylation. Mol. Cell. Proteom. 2019, 18 (Suppl. S1), S26–S36. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- Handly, L.N.; Yao, J.; Wollman, R. Signal Transduction at the Single-Cell Level: Approaches to Study the Dynamic Nature of Signaling Networks. J. Mol. Biol. 2016, 428, 3669–3682. [Google Scholar] [CrossRef]
- Creeden, J.F.; Alganem, K.; Imami, A.S.; Brunicardi, F.C.; Liu, S.H.; Shukla, R.; Tomar, T.; Naji, F.; McCullumsmith, R.E. Kinome Array Profiling of Patient-Derived Pancreatic Ductal Adenocarcinoma Identifies Differentially Active Protein Tyrosine Kinases. Int. J. Mol. Sci. 2020, 21, 8679. [Google Scholar] [CrossRef]
- Litichevskiy, L.; Peckner, R.; Abelin, J.G.; Asiedu, J.K.; Creech, A.L.; Davis, J.F.; Davison, D.; Dunning, C.M.; Egertson, J.D.; Egri, S.; et al. A Library of Phosphoproteomic and Chromatin Signatures for Characterizing Cellular Responses to Drug Perturbations. Cell Syst. 2018, 6, 424–443.e7. [Google Scholar] [CrossRef]
- Reinecke, M.; Heinzlmeir, S.; Wilhelm, M.; Médard, G.; Klaeger, S.; Kuster, B. Kinobeads: A Chemical Proteomic Approach for Kinase Inhibitor Selectivity Profiling and Target Discovery. In Target Discovery and Validation; Wiley: Hoboken, NJ, USA, 2019; pp. 97–130. [Google Scholar]
- Patricelli, M.P.; Nomanbhoy, T.K.; Wu, J.; Brown, H.; Zhou, D.; Zhang, J.; Jagannathan, S.; Aban, A.; Okerberg, E.; Herring, C.; et al. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol. 2011, 18, 699–710. [Google Scholar] [CrossRef]
- Alganem, K.; Hamoud, A.R.; Creeden, J.F.; Henkel, N.D.; Imami, A.S.; Joyce, A.W.; Ryan, V.W.; Rethman, J.B.; Shukla, R.; O’Donovan, S.M.; et al. The active kinome: The modern view of how active protein kinase networks fit in biological research. Curr. Opin. Pharmacol. 2022, 62, 117–129. [Google Scholar] [CrossRef]
- Cowen, L.; Ideker, T.; Raphael, B.J.; Sharan, R. Network propagation: A universal amplifier of genetic associations. Nat. Rev. Genet. 2017, 18, 551–562. [Google Scholar] [CrossRef]
- Qiu, P. Embracing the dropouts in single-cell RNA-seq analysis. Nat. Commun. 2020, 11, 1169. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Khoo, W.H.; Moran, I.; Croucher, P.I.; Phan, T.G. Single Cell RNA Sequencing of Rare Immune Cell Populations. Front. Immunol. 2018, 9, 1553. [Google Scholar] [CrossRef] [PubMed]
- Franchini, M.; Pellecchia, S.; Viscido, G.; Gambardella, G. Single-cell gene set enrichment analysis and transfer learning for functional annotation of scRNA-seq data. NAR Genom. Bioinform. 2023, 5, lqad024. [Google Scholar] [CrossRef] [PubMed]
- Ronen, J.; Akalin, A. netSmooth: Network-smoothing based imputation for single cell RNA-seq. F1000Research 2018, 7, 8. [Google Scholar] [CrossRef]
- Pavel, A.; Grønberg, M.G.; Clemmensen, L.H. The impact of dropouts in scRNAseq dense neighborhood analysis. Comput. Struct. Biotechnol. J. 2025, 27, 1278–1285. [Google Scholar] [CrossRef]
- Bouland, G.A.; Mahfouz, A.; Reinders, M.J.T. Consequences and opportunities arising due to sparser single-cell RNA-seq datasets. Genome Biol. 2023, 24, 86. [Google Scholar] [CrossRef]
- Davis, D.; Wizel, A.; Drier, Y. Accurate estimation of pathway activity in single cells for clustering and differential analysis. Genome Res. 2024, 34, 925–936. [Google Scholar] [CrossRef]
- Livne, D.; Efroni, S. Pathway metrics accurately stratify T cells to their cells states. BioData Min. 2024, 17, 60. [Google Scholar] [CrossRef]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef]
- Conroy, G. Retractions caused by honest mistakes are extremely stressful, say researchers. Nature, 2025; online ahead of print. [Google Scholar]
- Kovacs, M.; Varga, M.A.; Dianovics, D.; Poldrack, R.A.; Aczel, B. Opening the black box of article retractions: Exploring the causes and consequences of data management errors. R. Soc. Open Sci. 2024, 11, 240844. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [PubMed]
- Doniparthi, G.; Mühlhaus, T.; Deßloch, S. Integrating FAIR Experimental Metadata for Multi-omics Data Analysis. Datenbank-Spektrum 2024, 24, 107–115. [Google Scholar] [CrossRef]
- Jan, M.; Gobet, N.; Diessler, S.; Franken, P.; Xenarios, I. A multi-omics digital research object for the genetics of sleep regulation. Sci. Data 2019, 6, 258. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Skunca, N. Visualizing GO Annotations. Methods Mol. Biol. 2017, 1446, 207–220. [Google Scholar]
- Gan, M.; Dou, X.; Jiang, R. From ontology to semantic similarity: Calculation of ontology-based semantic similarity. Sci. World J. 2013, 2013, 793091. [Google Scholar] [CrossRef]
- Pesquita, C.; Faria, D.; Falcao, A.O.; Lord, P.; Couto, F.M. Semantic similarity in biomedical ontologies. PLoS Comput. Biol. 2009, 5, e1000443. [Google Scholar] [CrossRef]
- Galeota, E.; Kishore, K.; Pelizzola, M. Ontology-driven integrative analysis of omics data through Onassis. Sci. Rep. 2020, 10, 703. [Google Scholar] [CrossRef]
- Duong, D.; Ahmad, W.U.; Eskin, E.; Chang, K.W.; Li, J.J. Word and Sentence Embedding Tools to Measure Semantic Similarity of Gene Ontology Terms by Their Definitions. J. Comput. Biol. 2019, 26, 38–52. [Google Scholar] [CrossRef]
- Major, V.; Surkis, A.; Aphinyanaphongs, Y. Utility of General and Specific Word Embeddings for Classifying Translational Stages of Research. AMIA Annu. Symp. Proc. 2018, 2018, 1405–1414. [Google Scholar]
- Chiu, B.; Crichton, G.; Korhonen, A.; Pyysalo, S. How to train good word embeddings for biomedical NLP. In Proceedings of the 15th Workshop on Biomedical Natural Language Processing, Berlin, Germany, 12 August 2016. [Google Scholar]
- Mikolov, T.; Chen, K.; Corrado, G.; Dean, J. Efficient estimation of word representations in vector space. arXiv 2013, arXiv:1301.3781. [Google Scholar] [CrossRef]
- Ofer, D.; Brandes, N.; Linial, M. The language of proteins: NLP, machine learning & protein sequences. Comput. Struct. Biotechnol. J. 2021, 19, 1750–1758. [Google Scholar] [CrossRef]
- Xenos, A.; Malod-Dognin, N.; Milinkovic, S.; Przulj, N. Linear functional organization of the omic embedding space. Bioinformatics 2021, 37, 3839–3847. [Google Scholar] [CrossRef] [PubMed]
- Asgari, E.; Mofrad, M.R. Continuous Distributed Representation of Biological Sequences for Deep Proteomics and Genomics. PLoS ONE 2015, 10, e0141287. [Google Scholar] [CrossRef]
- Kulmanov, M.; Smaili, F.Z.; Gao, X.; Hoehndorf, R. Semantic similarity and machine learning with ontologies. Brief. Bioinform. 2021, 22, bbaa199. [Google Scholar] [CrossRef] [PubMed]
- Lerman, G.; Shakhnovich, B.E. Defining functional distance using manifold embeddings of gene ontology annotations. Proc. Natl. Acad. Sci. USA 2007, 104, 11334–11339. [Google Scholar] [CrossRef]
- Chen, D.-Q.; Kong, X.-S.; Shen, X.-B.; Huang, M.-Z.; Zheng, J.-P.; Sun, J.; Xu, S.-H. Identification of Differentially Expressed Genes and Signaling Pathways in Acute Myocardial Infarction Based on Integrated Bioinformatics Analysis. Cardiovasc. Ther. 2019, 2019, 8490707. [Google Scholar] [CrossRef]
- Jia, R.; Li, Z.; Liang, W.; Ji, Y.; Weng, Y.; Liang, Y.; Ning, P. Identification of key genes unique to the luminal a and basal-like breast cancer subtypes via bioinformatic analysis. World J. Surg. Oncol. 2020, 18, 268. [Google Scholar] [CrossRef]
- Niu, Y.; Zhang, Y.; Zha, Q.; Shi, J.; Weng, Q. Bioinformatics to analyze the differentially expressed genes in different degrees of Alzheimer’s disease and their roles in progress of the disease. J. Appl. Genet. 2024, 66, 73–85. [Google Scholar] [CrossRef]
- Gamazon, E.R.; Zwinderman, A.H.; Cox, N.J.; Denys, D.; Derks, E.M. Multi-tissue transcriptome analyses identify genetic mechanisms underlying neuropsychiatric traits. Nat. Genet. 2019, 51, 933–940. [Google Scholar] [CrossRef]
- Kulasinghe, A.; Liu, N.; Tan, C.W.; Monkman, J.; Sinclair, J.E.; Bhuva, D.D.; Godbolt, D.; Pan, L.; Nam, A.; Sadeghirad, H.; et al. Transcriptomic profiling of cardiac tissues from SARS-CoV-2 patients identifies DNA damage. Immunology 2023, 168, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Dalit, L.; Tan, C.W.; Sheikh, A.A.; Munnings, R.; Alvarado, C.; Hussain, T.; Zaini, A.; Cooper, L.; Kirn, A.; Hailes, L.; et al. Divergent cytokine and transcriptional signatures control functional T follicular helper cell heterogeneity. bioRxiv 2024. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Park, S.; Park, E.J.; Pagire, H.S.; Pagire, S.H.; Choi, B.-W.; Park, M.; Fang, S.; Ahn, J.H.; Oh, C.-M. Inhibition of HTR2B-mediated serotonin signaling in colorectal cancer suppresses tumor growth through ERK signaling. Biomed. Pharmacother. 2024, 179, 117428. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.H.; Curtis, M.A.; Imami, A.S.; Ryan, W.G.; Alganem, K.; Neifer, K.L.; Saferin, N.; Nawor, C.N.; Kistler, B.P.; Miller, G.W.; et al. Developmental pyrethroid exposure disrupts molecular pathways for MAP kinase and circadian rhythms in mouse brain. bioRxiv 2024. [Google Scholar] [CrossRef]
- Curtis, M.A.; Saferin, N.; Nguyen, J.H.; Imami, A.S.; Ryan, W.G.; Neifer, K.L.; Miller, G.W.; Burkett, J.P. Developmental pyrethroid exposure in mouse leads to disrupted brain metabolism in adulthood. Neurotoxicology 2024, 103, 87–95. [Google Scholar] [CrossRef]
- O’Donovan, S.; Ali, S.; Deng, W.; Patti, G.; Wang, J.; Eladawi, M.; Imami, A. Shared and unique transcriptional changes in the orbitofrontal cortex in psychiatric disorders and suicide. Transl. Univ. Toledo J. Med. Sci. 2024, 12. [Google Scholar] [CrossRef]
- Hu, Y.; Sun, I.; Tang, E.; Ryan, W.; Shrestha, U.; Gautam, J.; Lad, A.; Huntley, J.F.; Haller, S.; Kennedy, D. Probiotic Protects Kidneys Exposed to Microcystin-LR. Transl. Univ. Toledo J. Med. Sci. 2024, 12. [Google Scholar] [CrossRef]
- Cuevas-Diaz Duran, R.; Wei, H.; Wu, J. Data normalization for addressing the challenges in the analysis of single-cell transcriptomic datasets. BMC Genom. 2024, 25, 444. [Google Scholar] [CrossRef]
- Andrews, T.S.; Hemberg, M. False signals induced by single-cell imputation. F1000Research 2018, 7, 1740. [Google Scholar] [CrossRef]
- Yu, Y.; Mai, Y.; Zheng, Y.; Shi, L. Assessing and mitigating batch effects in large-scale omics studies. Genome Biol. 2024, 25, 254. [Google Scholar] [CrossRef]
- Hodgman, C.; French, A.; Westhead, D. BIOS Instant Notes in Bioinformatics; Taylor & Francis: Abingdon, UK, 2009. [Google Scholar]
- Karp, P.D.; Midford, P.E.; Caspi, R.; Khodursky, A. Pathway size matters: The influence of pathway granularity on over-representation (enrichment analysis) statistics. BMC Genom. 2021, 22, 191. [Google Scholar] [CrossRef] [PubMed]
- Wijesooriya, K.; Jadaan, S.A.; Perera, K.L.; Kaur, T.; Ziemann, M. Urgent need for consistent standards in functional enrichment analysis. PLoS Comput. Biol. 2022, 18, e1009935. [Google Scholar] [CrossRef]
- Ziemann, M.; Schroeter, B.; Bora, A. Two subtle problems with over-representation analysis. Bioinform. Adv. 2024, 4, vbae159. [Google Scholar] [CrossRef]
- Zhao, H.; Ma, C.; Xu, F.; Kong, L.; Deng, Z.-H. BioMaze: Benchmarking and Enhancing Large Language Models for Biological Pathway Reasoning. arXiv 2025, arXiv:2502.16660. [Google Scholar]
- Wong, C.-K.; Choo, A.; Cheng, E.C.; San, W.-C.; Cheng, K.C.-K.; Lau, Y.-M.; Lin, M.; Li, F.; Liang, W.-H.; Liao, S.-Y. Lomics: Generation of pathways and gene sets using large language models for transcriptomic analysis. arXiv 2024, arXiv:2407.09089. [Google Scholar] [CrossRef]
- Kamya, P.; Ozerov, I.V.; Pun, F.W.; Tretina, K.; Fokina, T.; Chen, S.; Naumov, V.; Long, X.; Lin, S.; Korzinkin, M.; et al. PandaOmics: An AI-Driven Platform for Therapeutic Target and Biomarker Discovery. J. Chem. Inf. Model. 2024, 64, 3961–3969. [Google Scholar] [CrossRef] [PubMed]
| Gene | # of Pathways |
|---|---|
| TGFB1 transforming growth factor beta 1 | 1010 |
| CTNNB1 catenin beta 1 | 894 |
| ACADL acyl-CoA dehydrogenase long chain | 120 |
| ACTBL2 actin beta like 2 | 120 |
| ABCA6 ATP binding cassette subfamily A member 6 | 72 |
| ACKR1 atypical chemokine receptor 1 (Duffy blood group) | 72 |
| ABCF3 ATP binding cassette subfamily F member 3 | 44 |
| ADISSP adipose secreted signaling protein | 44 |
| C6orf62 chromosome 6 open reading frame 62 | 2 |
| CTAGE3P CTAGE family member 3, pseudogene | 2 |
| Category | Representative Tools | Typical Strength | Typical Limitation |
|---|---|---|---|
| Semantic similarity-based | REVIGO, clusterProfiler, ReCiPa | Fast redundancy reduction for GO terms | Tied to GO; limited cross-database scope |
| Network-based | EnrichmentMap, vissE, GScluster | Visualizes pathway crosstalk as network modules | Computationally heavy for large networks |
| Embedding-based | PAVER, MondrianMap | Data-driven clustering with intuitive plots | Relies on text descriptions; may miss context |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryan V., W.G.; Sahay, S.; Vergis, J.; Weistuch, C.; Meller, J.; McCullumsmith, R.E. Pathway Analysis Interpretation in the Multi-Omic Era. BioTech 2025, 14, 58. https://doi.org/10.3390/biotech14030058
Ryan V. WG, Sahay S, Vergis J, Weistuch C, Meller J, McCullumsmith RE. Pathway Analysis Interpretation in the Multi-Omic Era. BioTech. 2025; 14(3):58. https://doi.org/10.3390/biotech14030058
Chicago/Turabian StyleRyan V., William G., Smita Sahay, John Vergis, Corey Weistuch, Jarek Meller, and Robert E. McCullumsmith. 2025. "Pathway Analysis Interpretation in the Multi-Omic Era" BioTech 14, no. 3: 58. https://doi.org/10.3390/biotech14030058
APA StyleRyan V., W. G., Sahay, S., Vergis, J., Weistuch, C., Meller, J., & McCullumsmith, R. E. (2025). Pathway Analysis Interpretation in the Multi-Omic Era. BioTech, 14(3), 58. https://doi.org/10.3390/biotech14030058