1. Introduction
In recent times, mAbs-based therapeutics products are rapidly moving up in biosimilar industries [
1]. It is well-known that mAbs have great clinical applications for the treatment of immunological disorders, malignancy and other critical infections. To obtain approval for clinical use as a biosimilar product, bio similarity data must be generated with an innovator molecule with various analytical tools. The isoelectric point of a protein is the pH, at which a protein carries no net charge and hence is considered neutral. The isoelectric point (pI), where the net charge of the protein is neutral, is pH-dependent. The pI of a protein is crucial to understanding its biochemical function [
2]. The pI of mAbs is mostly dependent on the dissociation constant of the ionizable groups. Amino acids containing acidic or basic functionalities form the peptide bounds, which ultimately give rise to the polypeptides or the proteins. These functionalities influence the overall surface charge, which, in turn, can be regulated by monitoring the pH of buffer system surrounding the protein.
Quality by design (QbD) is a next generation step in developing and producing biopharmaceutical products in the biopharma industry. The best quality and most consistent product can be generated using the QbD approach for the characterization of therapeutic proteins. [
3]. mAbs and related products have modifications such as aggregation, deamidation, disulfide bond scrambling, fragmentation, glycosylation, lysine truncation, oxidation, isomerization and pyro glutamate formation. These modifications may impact the critical quality attributes (CQAs) of the drug with relation to its efficacy and safety. The composition and physiochemical parameters of the cell culture media at the time of up-stream fermentation can be impacted by critical process parameters [
3,
4].
Therapeutic mAbs usually display heterogeneity when they are produced using cell culture methods. These heterogeneities or variations are caused by deamidation, glycosylation, mutation, N-terminal glutamine cyclization, C-terminal lysine truncation, and oxidation, resulting in isoforms of the peptides. Due to the aforementioned causes, mAbs tend to display hurdles with respect to their characterization, of which charge variants are an important attribute. Charge deviations are natural features, not impurities of this type of therapeutic mAbs, and they have critical impacts on protein stability and bioactivity.
mAbs and other therapeutic proteins are routinely characterized, processed and analyzed for charge heterogeneity by ion exchange chromatography (IEX), as this is a gold standard technique for assessing the quality of proteins and to allow for separate control of individual proteoforms. To assess the charge heterogeneity, IEX relies on electrostatic interactions between the resin functional groups (cation/anion) and mAbs. The purification and characterization of charge sensitive mAbs can be carried out using cation exchange chromatography (CEX) which is based on the pI of the mAbs. However, due to the minute differences in the binding properties charge variant mAbs, separation sometimes becomes difficult using CEX. Tiny particle analytical stationary phases called column and mobile phase gradient chromatography are used for separation.
In biopharmaceutical industry, charge variant analysis is a critical attribute used for direct comparison of biosimilar product lots with reference products to certify constant product quality, which is very important for biosimilar product profile.
Currently, in the biopharmaceutical industry, the tool of choice for quality control, optimization and product retrieval from upstream and downstream processing is analytical technology [
5]. However, for identification of the unknown peaks in charge variants analysis which are out-of-specification for product manufacturing, it requires identification and characterization, which is a time-consuming process, involving initial sample preparation procedures and follow-up experiments. These experimental procedures generally involve peak collection followed by buffer exchange to carry out further analysis [
6,
7,
8]. The entire identification and characterization process leads to more time and more cost. Sometimes, these experimental protocols lead to forceful generation of modifications in the protein product that are actually not present in its native form [
9,
10].
mAbs are traditionally eluted in IEX using salt gradients of low-to-high concentration. This eluent, however, is not suitable for MS analysis, due to its high salt concentration. Furthermore, due to denaturation of tertiary structure and exposure of charged residues, highly charged mAb ions are produced, which results in the generation of complex MS [
11].
Traditional ion exchange chromatography uses salt gradients of low-to-high concentration to elute proteins/mAbs, but when connecting to mass, the high concentrations of salt can be problematic, which is not recommended. Another limitation of this approach is denaturation of the protein/mAbs, resulting in the generation of complex mass spectra containing highly charged mAb ions due to the loss of tertiary structure and exposure of charged residues [
12,
13,
14,
15].
The ICH Q2 (R1) guideline, which is directly related to the validation of analytical methods for conventional medications but can also be utilized with biopharmaceutical products, should be followed when qualifying analytical processes employed in the characterization of biopharmaceuticals products. The method qualification of a stability-indicating parameter requires the analysis of stressed or force-deteriorated samples to show that the method is suitable for active ingredient analysis in the presence of the degradation conditions. The method can be submitted to the appropriate regulatory agency for approval as part of the product license application if the qualification findings are satisfactory and within the limitations of ICH recommendations. The goal of this research is to create and validate a simple, precise, and robust pH gradient-based IEX approach for determining charge variations in mAb samples that can be replicated in any lab. This method can be used in routine testing for quality control analysis, giving manufacturer’s confidence in the quality of their products. Furthermore, the approach can be used to track the stability of mAb in biopharmaceutical formulations under a variety of force-degraded circumstances. For regulatory data submission of biosimilar products, the analytical method must be qualified and validated for analysis. Thereby, in this study, the method is qualified as per ICH guidelines for quality control batch release use and regulated data submission.
Additionally, the aim of this work is to develop and qualify a mass spectrometry-based analytical method for charge variant analysis based on volatile pH-based buffers with low ionic strength. This allows for on-line mass spectrometric detection of charge variants in different mAbs/Proteins. Low ionic strength eluents, which are employed for MS detection and have a low buffering capacity, are a feature and quality of this approach. A sufficient amount of data can be acquired from a single chromatographic injection without the need for any sample preparation operations, which would ordinarily necessitate many phases of analysis and various sample preparation procedures [
13,
14]. As a result, the charge variant analysis-MS technique will enable the monitoring of many quality parameters at the intact protein level, which will aid in process optimization both upstream and downstream. It is also possible to investigate new information on protein heterogeneity and breakdown ability, which may have implications for the launch of prospective biosimilar products in the future.
2. Materials and Methods
2.1. Materials
Ammonium bicarbonate, water (mass grade, JT Baker, Phillipsburg, NJ, USA), ammonium hydroxide solution, and acetic acid (ACS mass grade) were used for analysis. MAbs (Bevacizumab) was kindly provided from the university. It has a molecular weight of about 149 kDa and pI around 8.3. Bevacizumab is a humanized monoclonal IgG1 antibody, and inhibits angiogenesis by binding and neutralizing VEGF-A. The concentration of IgG1 is claimed as 25 mg/mL.
2.2. Method
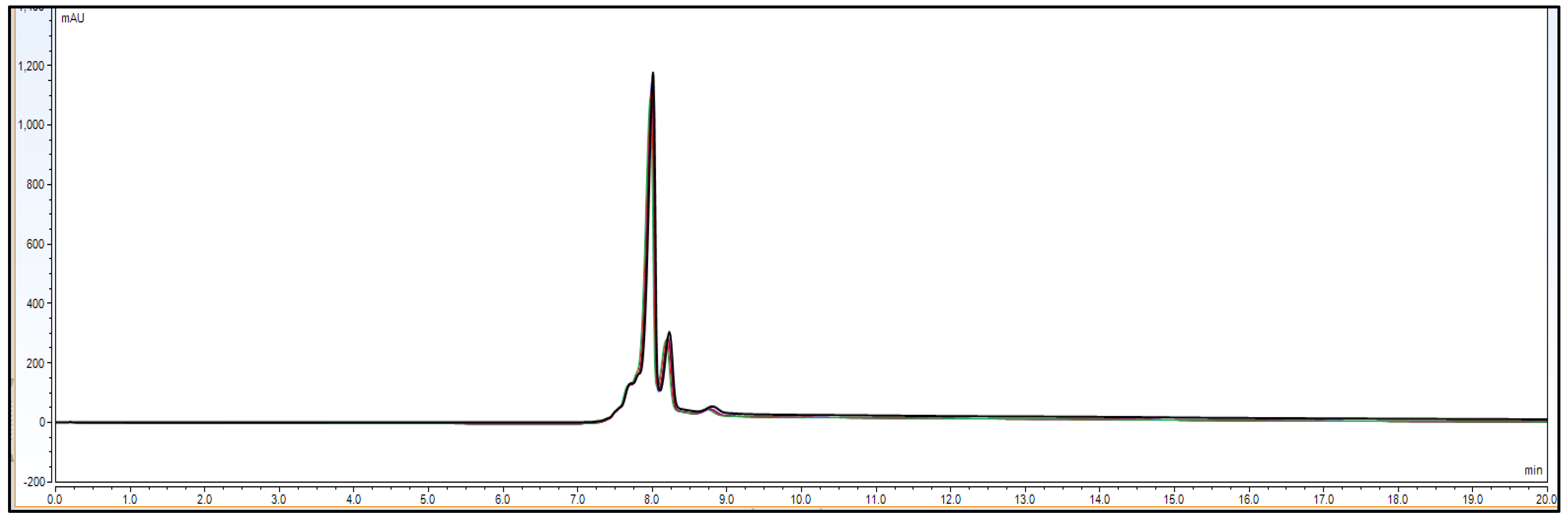
CEX-UPLC analyses of untreated Bevacizumab was carried out on an UPLC system (Binary, Waters). Separation was performed on a MAbPac SCX-10 RS 5 µm, 2.1 × 50 mm column (Thermo Fisher Scientific, Sunnyvale, CA, USA) using a mobile phase A comprising 25 mmol·L−1 ammonium bicarbonate and 30 mmol·L−1 acetic acid in water (pH 5.3) and a mobile phase B comprising 10 mmol·L−1 ammonium hydroxide in water (pH 10.9). A pH gradient was applied consisting of 35 to 60% mobile-phase B in 7 min followed by a flushing step with 90% B and a re-equilibration step with 35% B for 5 min, flowrate 400 µL min−1, T 25 °C, and UV detection at 280 nm. Per run, 30 µg of IgG1 antibody was injected.
Result: Based on
Table 1, the system parameters program, CEX UPLC run was performed using Bevacizumab IgG1 antibody sample with a 30 microgram injection volume on the column. Total run time was 20 min. The representative chromatogram is shown as below.
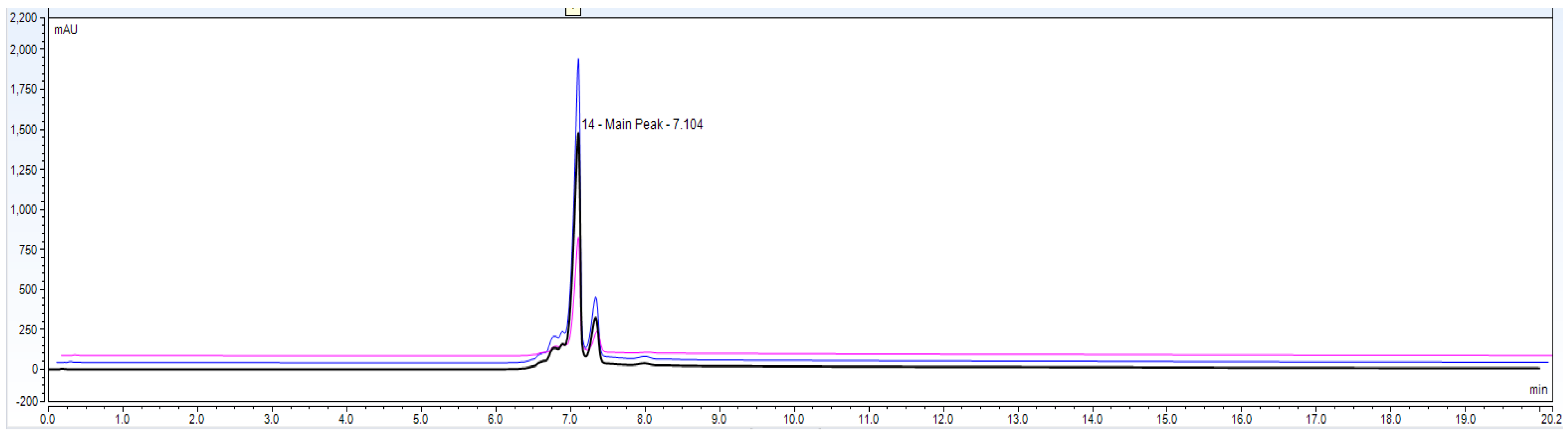
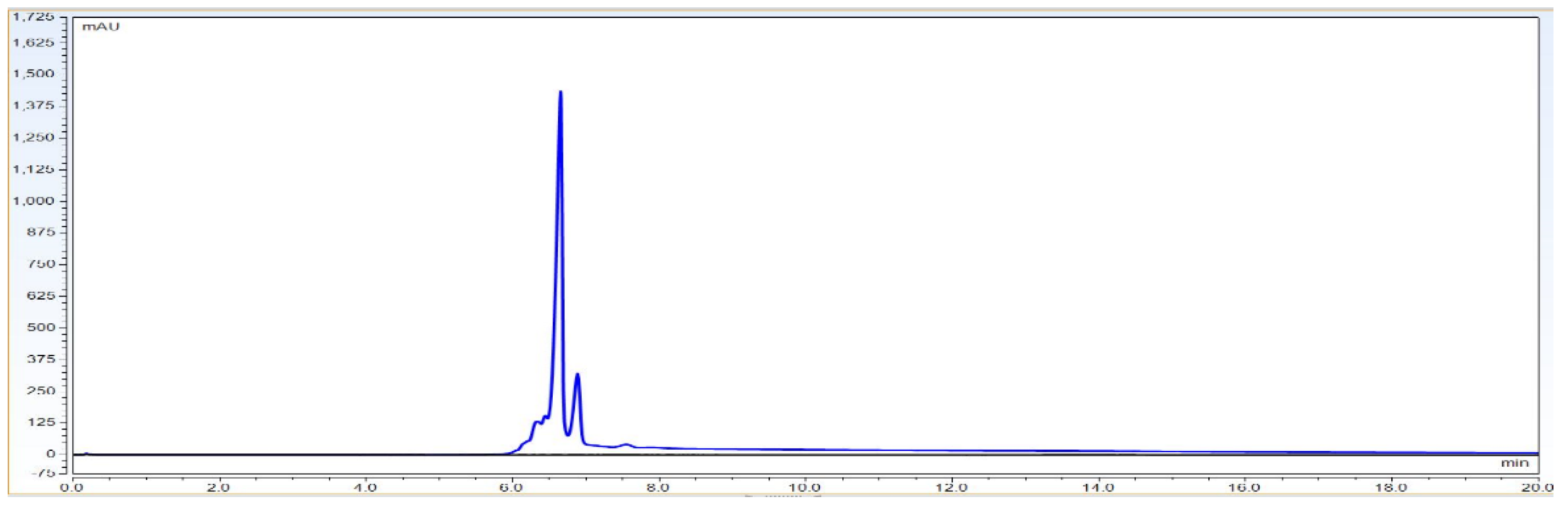
As per
Figure 1 chromatogram, main peak which is obtained around 7 min represent IgG1 purity having charge variants as pre peak (acidic variants) and post peak (basic variants). Those charge variants are present due to PTMs. From this it is very clear that the method is producing quite good resolution in acidic and basic variants separation using volatile mobile phase gradient. Additionally, the method run time is short, around 20 min, which shows that the method is compact and provides a platform to characterize charge variant at intact level within a short time period.
2.3. Method Qualification
Transfer of analytical qualified method to the quality control lab of mAbs/proteins is a crucial final step in the acceptance of this method, which has quality attributes parameters that are under a good scan. Method qualification is a systematic approach that states with assurance that a specific process will meet its pre-defined acceptance criteria. Additionally, an effective analytical method qualification will improve the precision and accuracy of the previously developed chromatographic methods and decrease their errors. Development of a qualified ion exchange method will help to avoid time-consuming experiments and product developmental and manufacturing cost. In analytical approach, before a developed chromatographic method can be implemented into quality system, it must first be qualified and validated and shown that it is a suitable method for the reporting critical quality data generation. If the qualified analytical method data are acceptable, the method can be send to the appropriate regulatory authority for approval of product license application. Validation of analytical methods used in the characterization of biopharmaceuticals industries should be performed in compliance with the ICH Q2 (R1) guideline, which outlines the validation of analytical procedures. The qualification of a stability-indicating method requires analysis of stressed samples to show that the method is capable of analyzing pure protein in the presence of the degradation of products.
3. Qualification Parameters Evaluation
3.1. Linearity
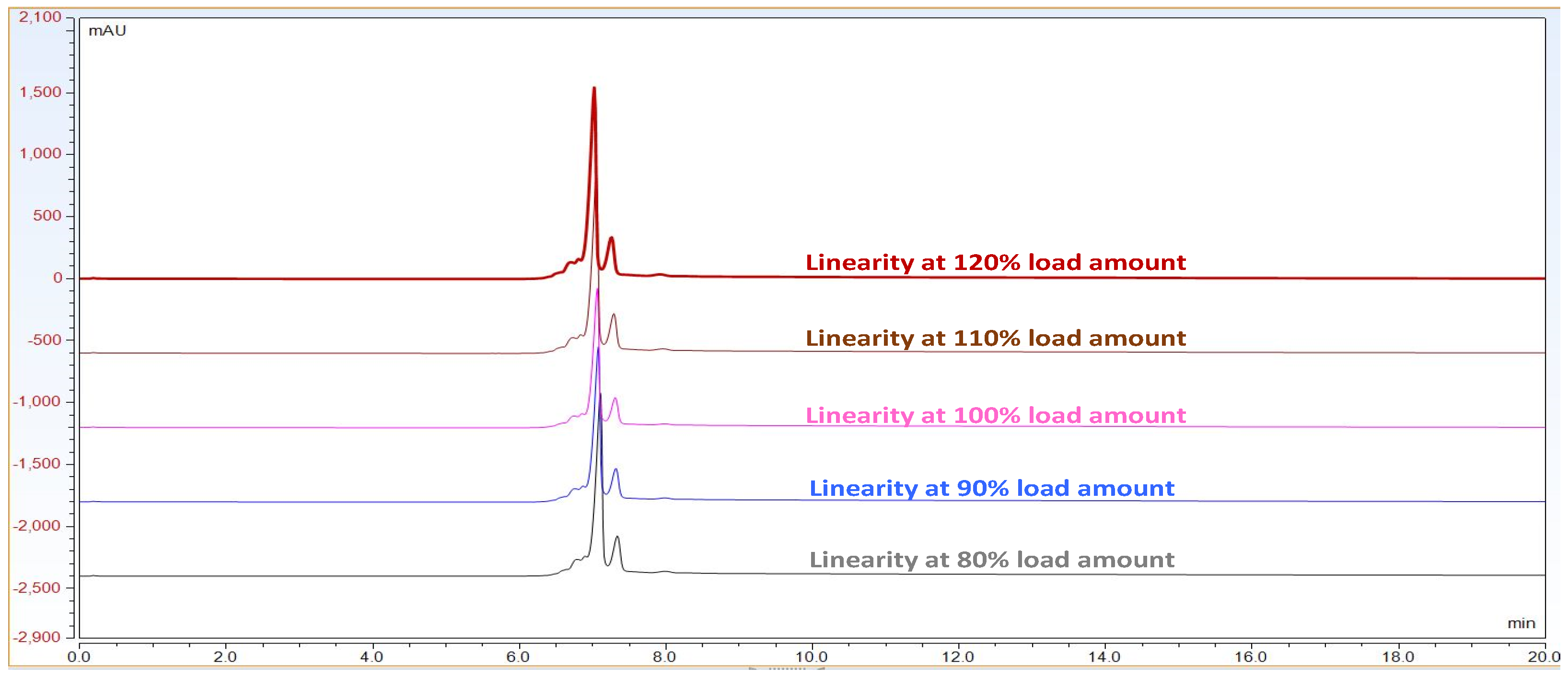
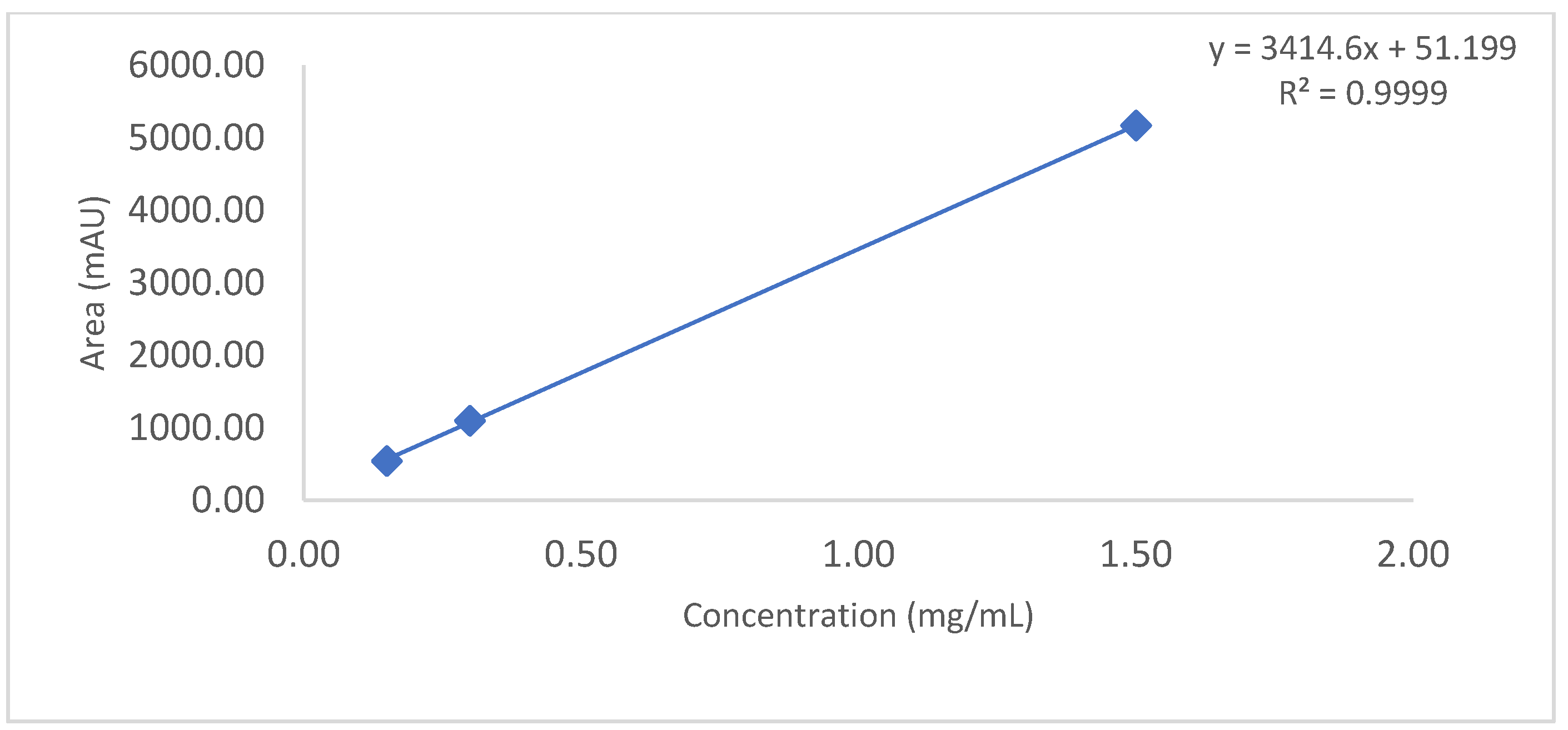
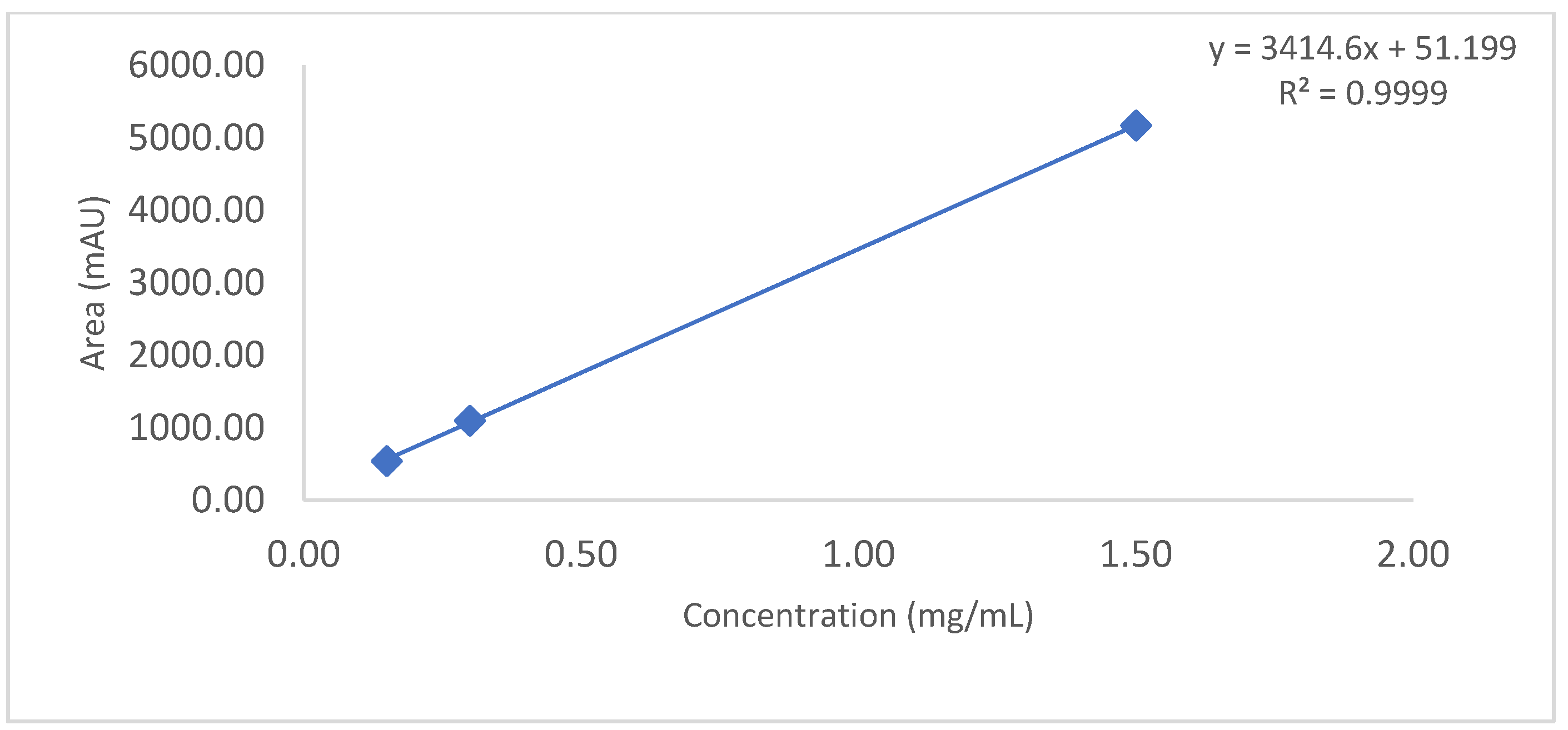
The linearity of an analytical method is its ability to obtain test results which are directly proportional to the amount/concentration of analyte in the protein sample within a specified range. Linearity of the CEX method was performed up to 3.6 mg/mL. For this study, mAb RMP Bevacizumab working standard solutions of 2.4, 2.7, 3.0, 3.3 and 3.6 mg/mL were prepared in duplicate from 25 mg/mL and injected into the Waters UPLC instrument. A calibration curve was plotted for main peak area vs. drug concentration/amount and a coefficient of determination (R2) is calculated by linear least-square regression mathematical analysis.
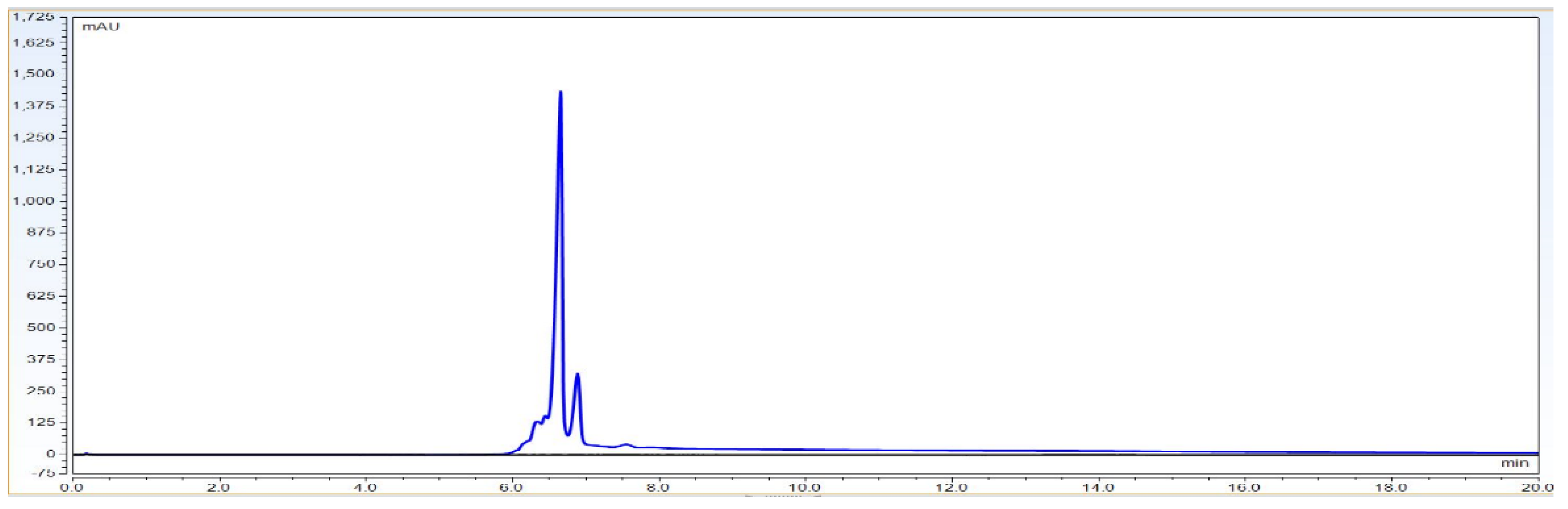
Observation: As per chromatogram in
Figure 2, calibration curve obtained from
Figure 3 and compiled data obtained from
Table 2, it can be concluded that CEX UPLC method has load amount linearity from 80 to 120%, with respect to main peak area. It also shows R
2 0.99 with % recovery between 95 and 105%, suggesting that the method is linear within 10% of recovery.
3.2. Limit of Detection (LOD) and Limit of Quantification (LOQ)
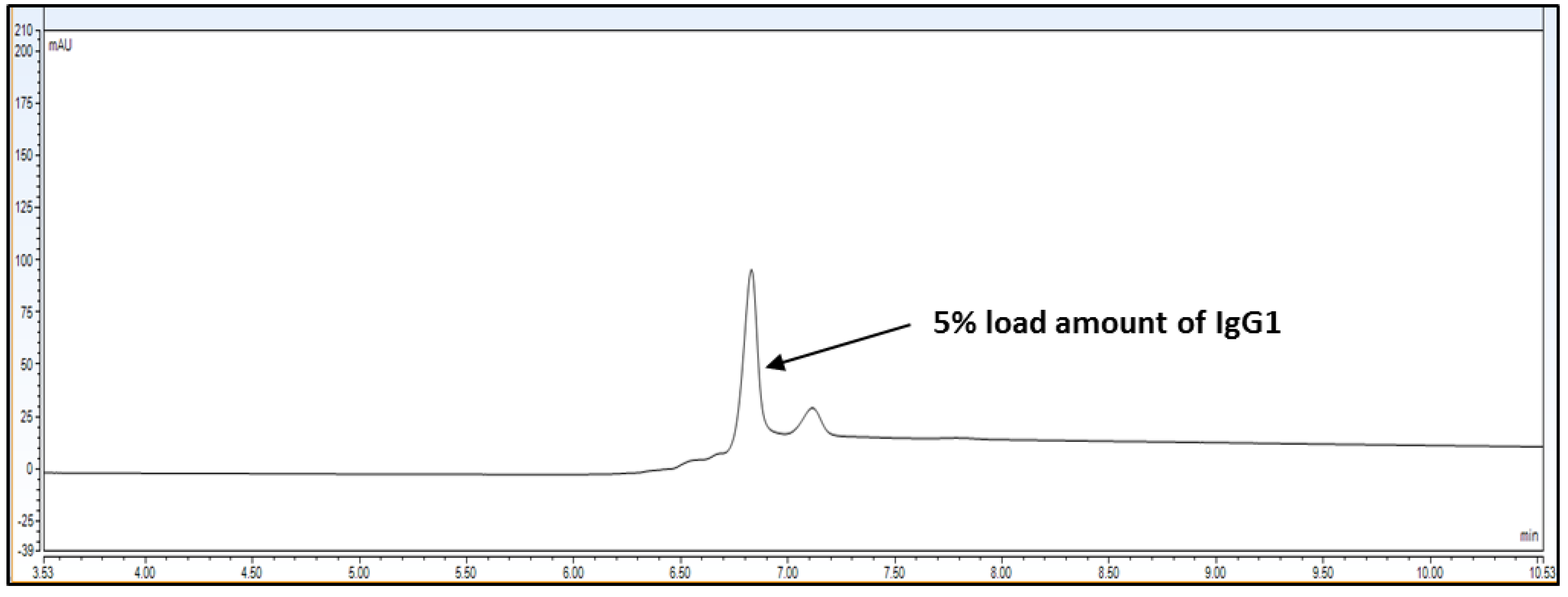
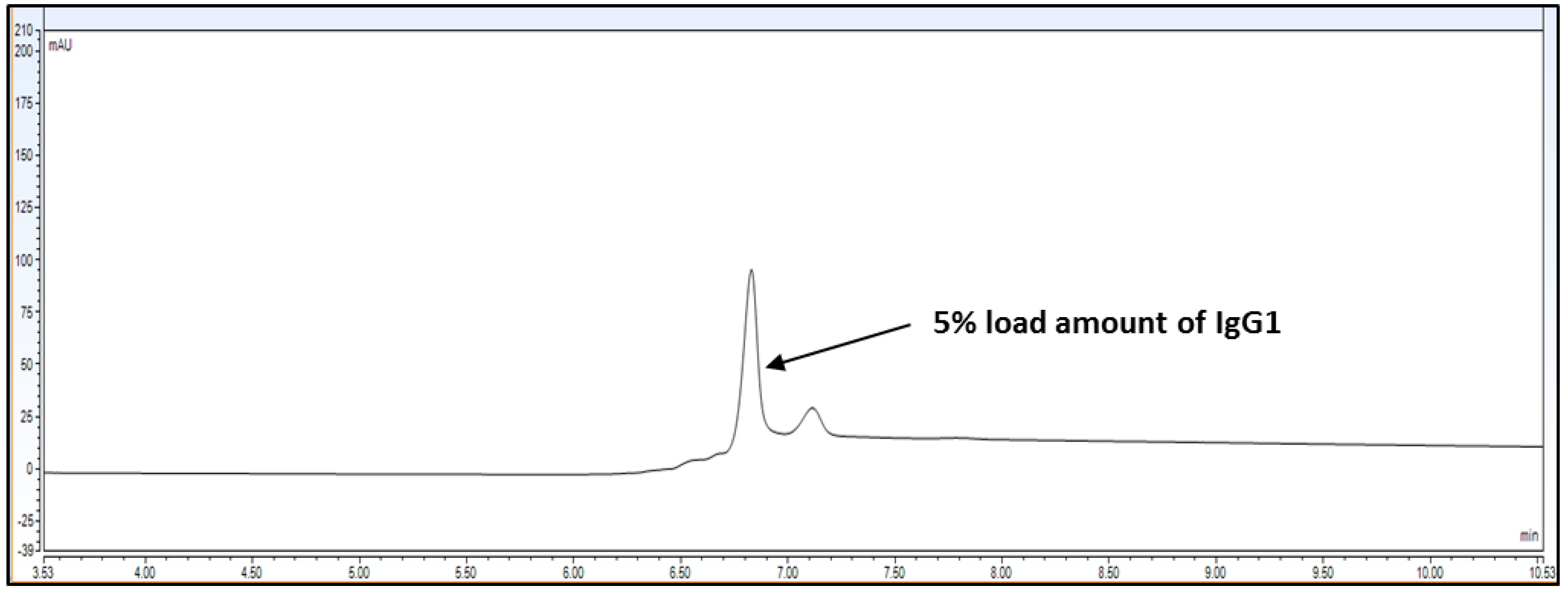
The limit of detection for analytical method is the lowest amount of analyte/protein in a sample which can be detected but not exactly quantitated as a reporting value. The limit of quantitation for analytical procedure is the lowest amount of analyte/protein in a sample which can be quantify with accuracy and precision. The LOQ limit is a parameter of quantitative value for low levels of compounds/impurities in sample matrices. It is used for the determination of impurities for force degradation products. The LOQ or LOD is reported by either signal-to-noise ration or impurity level area determination. For LOD and LOQ determination, Bevacizumab RMP was injected at 5% loading amount, which is 0.15 mg/mL on the column. Back-calculated recovery was determined using slope and intercept.
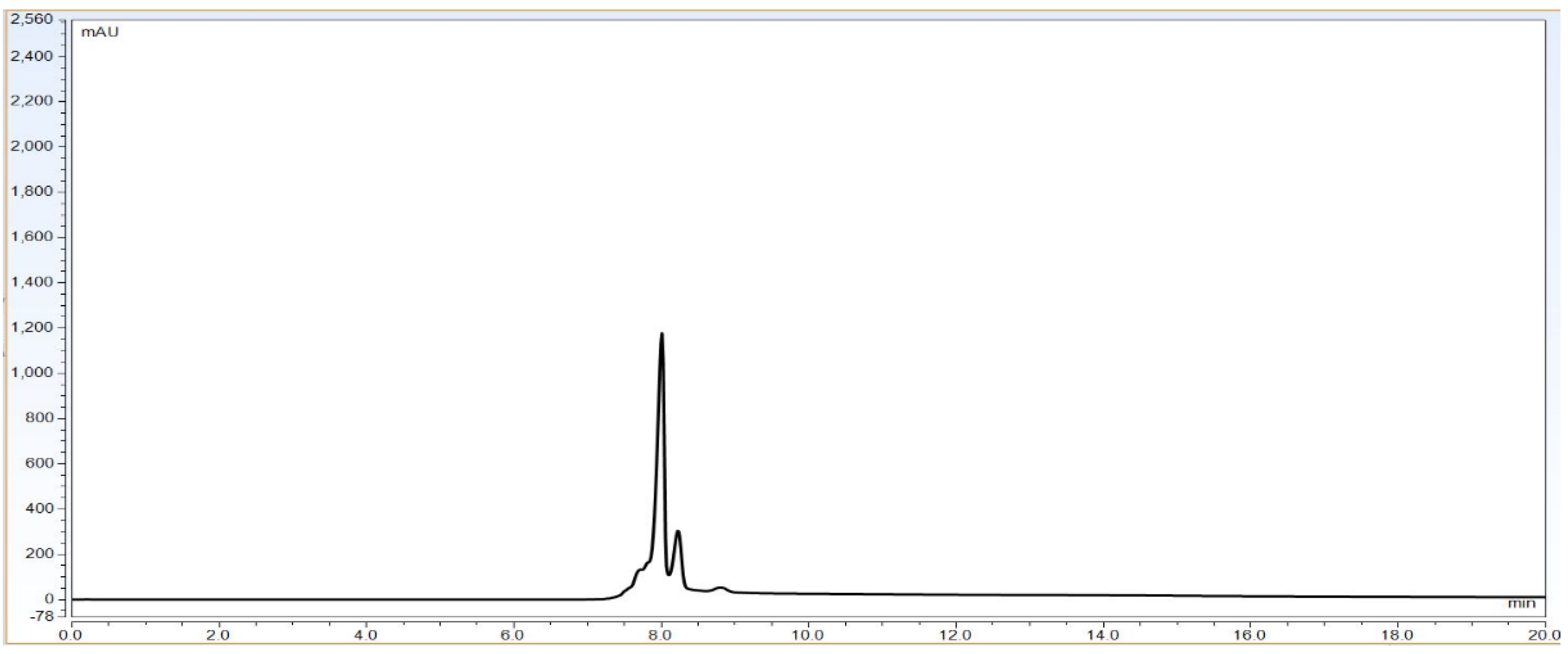
Observation: Based on above chromatography data obtained from
Figure 4,
Figure 5 and
Figure 6 and data
Table 3, it is concluded that LOQ of the method is 5% of loading sample amount out of 100% which is 0.15 mg/mL. This method can detect LOD <5% of loading sample amount but this cannot be quantitated.
3.3. Precision and Accuracy
The precision of an analytical method is based on the closeness of agreement (degree of scatter) between a linear series of measurements taken from multiple concentrations of the same sample under the defined conditions. Precision can be determined at three levels: repeatability, reproducibility and intermediate precision.
Repeatability: Repeatability provides information for the precision under the same operating conditions with a short time interval. Repeatability can also be named as intra-assay precision.
Reproducibility: Reproducibility provides the precision between labs.
Intermediate precision: Intermediate precision provides data regarding within-laboratories variations: different days, different analysts, different equipment, different column and different mobile phase preparation variation.
The accuracy of an analytical method is closeness of agreement between the value which is accepted as a true value or an accepted reference value and the value obtained. Accuracy is defined as the closeness of the measured value to the actual value. The determination of accuracy was carried out with calculation of the average recovery value by analyzing three standard solutions triplicates at three concentration levels high (2.00 mg/mL), the medium (0.50 mg/mL), and the low (0.12 mg/mL) of the linear range of the calibration curve.
The precision of the method was performed as the intra-day and inter-day precision, which represent repeatability and intermediate precision, respectively. It is defined with given as relative standard deviations (RSDs). Intra-day precision was determined using 12 samples with same concentration levels of 3.0 mg/mL injected on UPLC system. These samples were prepared and injected into the chromatography system on different days with different system, different column, and different analyst.
Observation: Based on above
Figure 7 and
Figure 8 and
Table 4,
Table 5,
Table 6 and
Table 7 data, it is found that repeatability data % RSD of six different injections for analyst 1, analyst 2 and intermediate precision data for a combined 12 injections with respect to main peak and total peak area is found to be RSD < 5.0%. This data shows that the method can be precise for the purpose of reporting charge variants percentage with precision of 95–105%.
Table 7 accuracy experiment data show that it is falling 50 to 150% of analyte loading amount with <±10% recovery.
3.4. Specificity and Stability Indicating Property
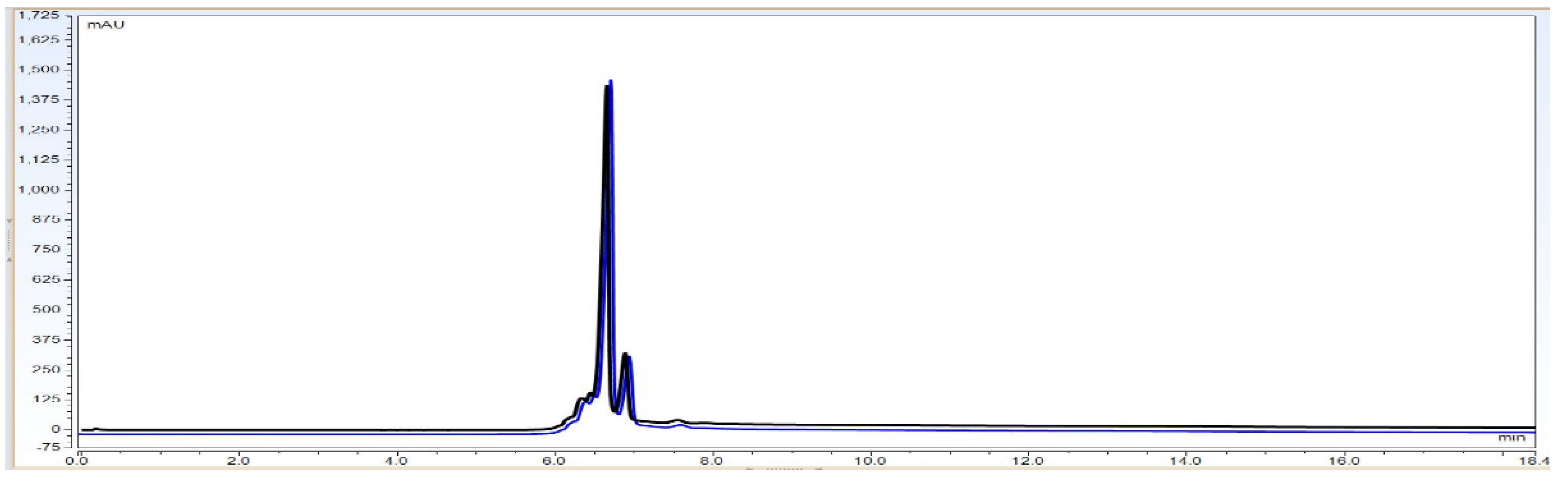
Specificity is defined as two major qualification parameters evaluation as Matrix interference and selectivity. It is the ability to assess and measure accurate and specific analyte of interest in the presence of other additives, including impurities, degradants, matrix, etc. It must be demonstrated that our analytical method is not affected by any impurities or excipients. For the specificity experiment, an IgG1 antibody concentration of 3.0 mg/mL injected with buffer blank was used to check whether any buffer peak has interference with the elution time of the Bevacizumab peak.
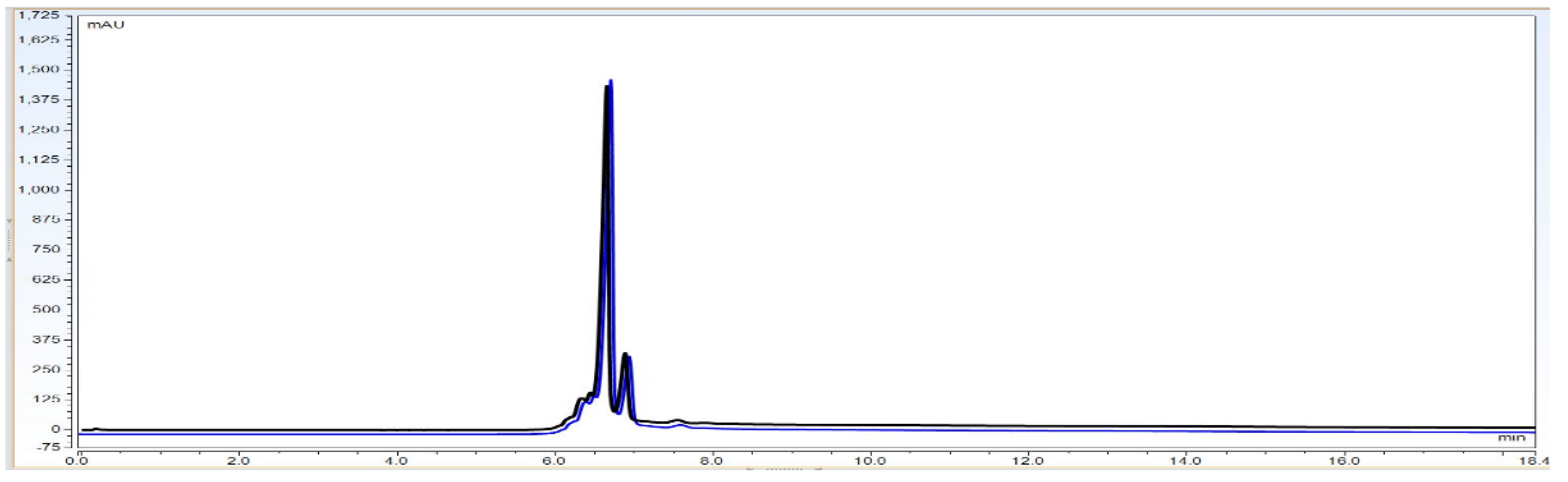
Observation: As per
Figure 9 chromatography overlay data the matrix components did not interfere with the signal obtained for the analyte and thus there was no matrix interference. The method is therefore specific for evaluating the identity of the BevacizumAb sample.
3.5. Robustness
The robustness is defined as a measure of a method’s capacity to remain unaffected by small but deliberate changes in procedural parameters. Examples of typical variations are stability of analytical solutions, variations in pH of solvents, column temperature, auto sampler temperature, and flow rate variation. For this study, mAb/protein at a concentration of 2.00 mg/mL was analyzed to evaluate the impact of each modified condition on the data results. Bevacizumab sample was run on UPLC system at initial time point and after 48 h kept in an auto sampler (2–8 °C) to check the sample stability.
Observation: With the
Figure 10 robustness data it can be concluded that the mAb sample which is kept in an auto sampler and the same mobile phase can be used for up to 48 h after preparation.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}