Cell-Free Protein Synthesis by Diversifying Bacterial Transcription Machinery



Abstract
1. Introduction
2. Materials and Methods
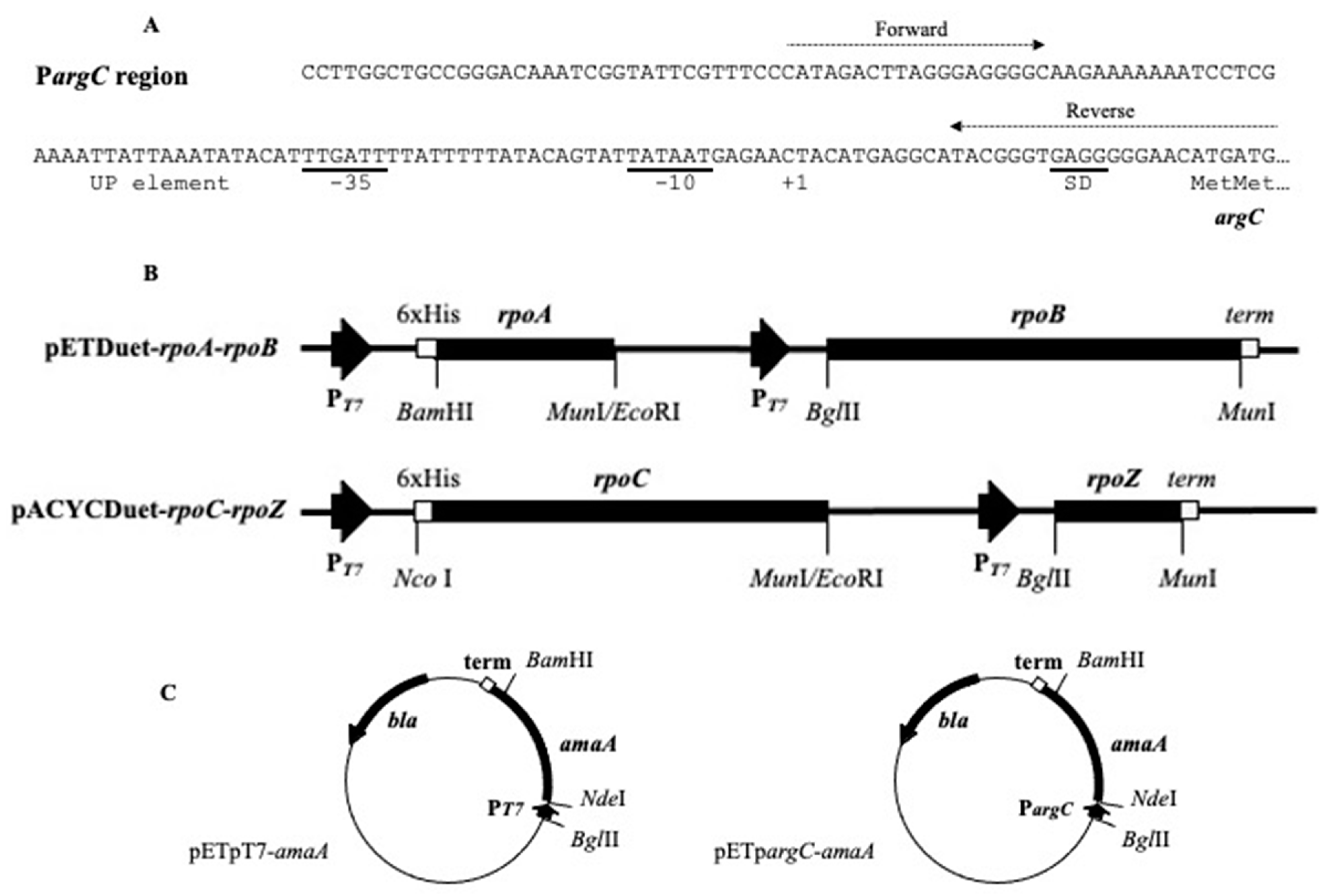
2.1. DNA Cloning
2.2. Construction of Linear DNA Templates
2.3. P1 Transduction
2.4. Ribonuclease Assay
2.5. Purification of Proteins and Western Blotting
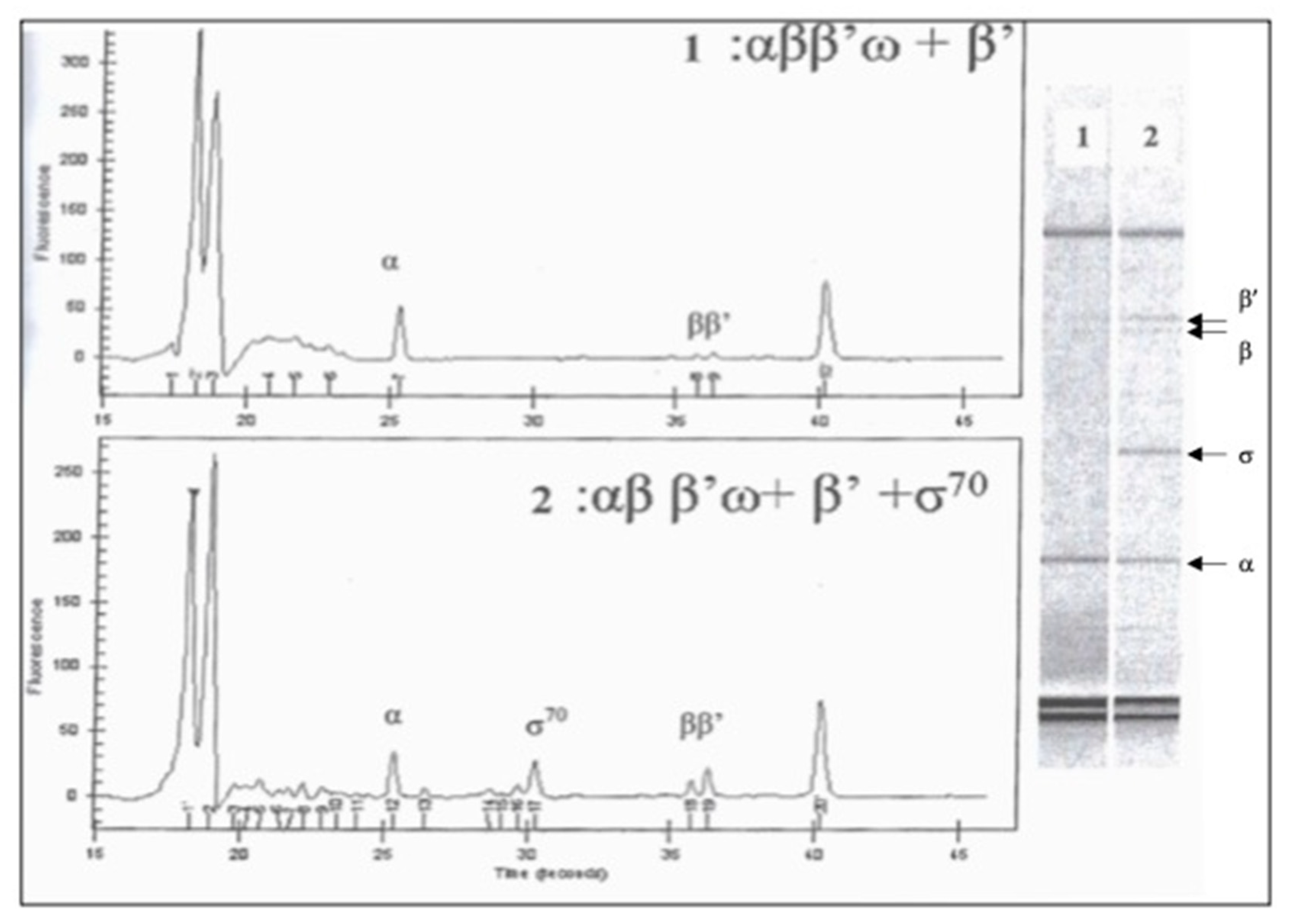
2.6. Purification of E. coli RNA Polymerase Subunits from Living Cells
2.7. Preparation of S30 Cell-Free Extracts
2.8. Cell-Free Protein Synthesis
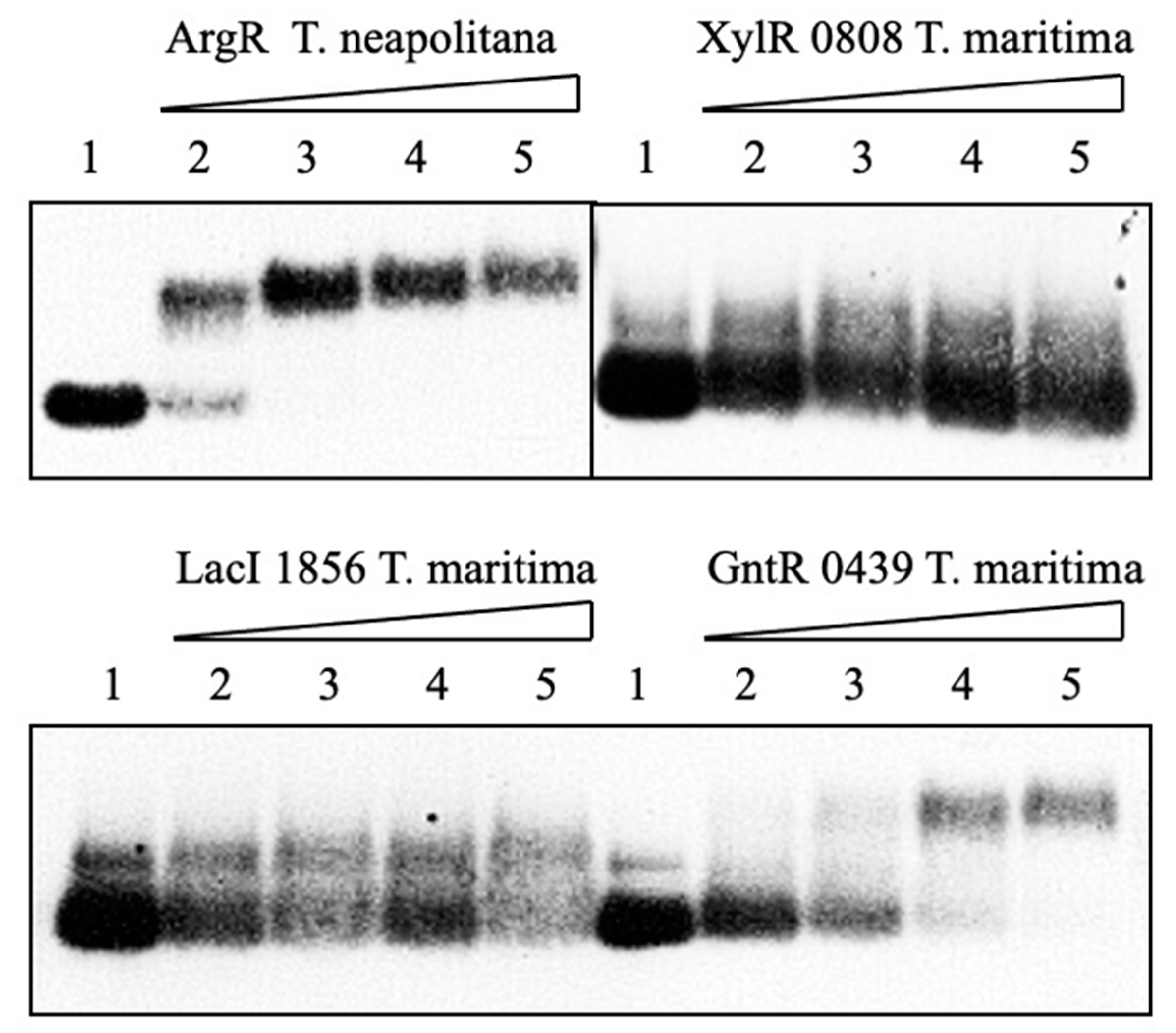
2.9. Mobility-Shift Assay
3. Results
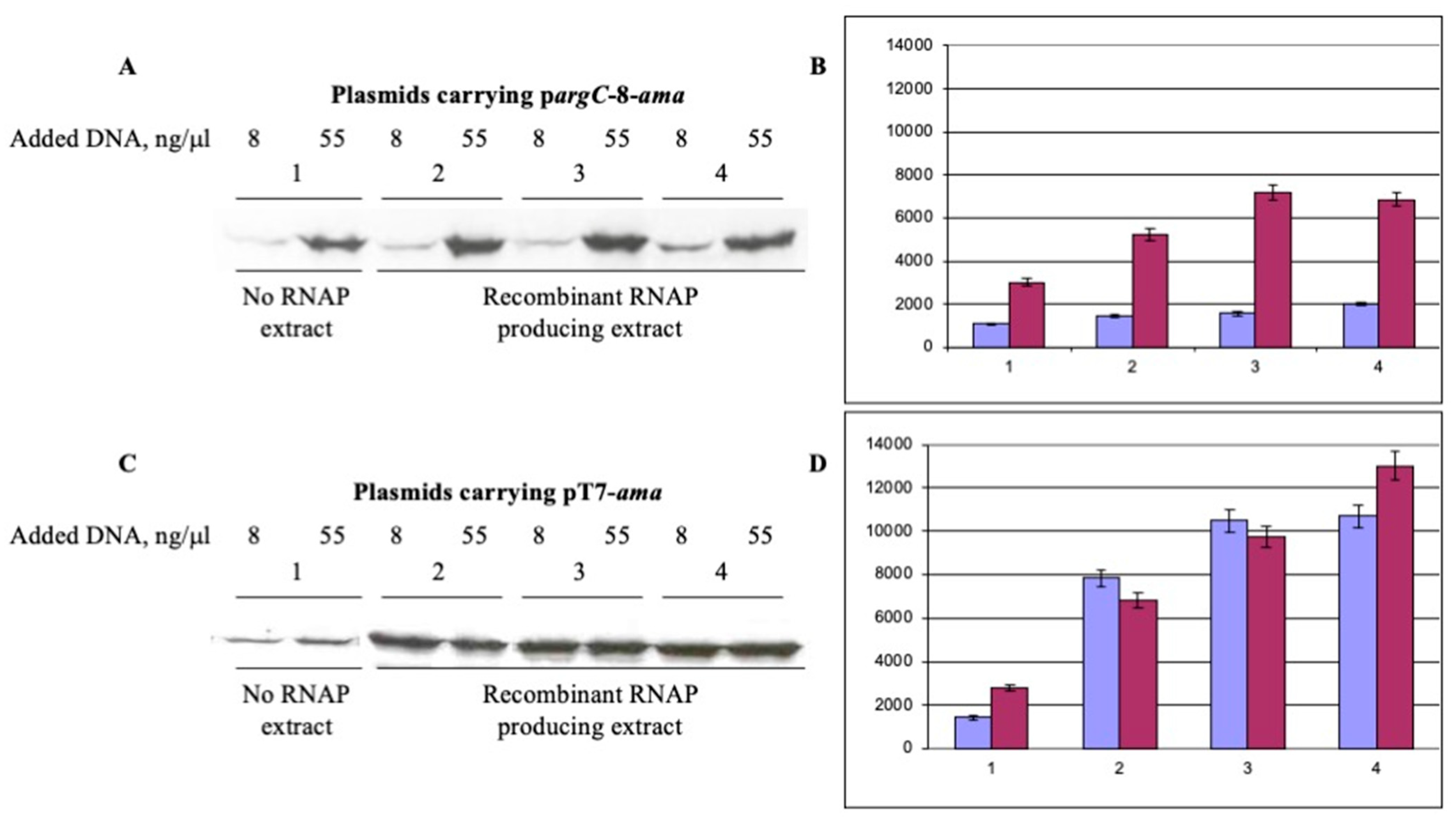
3.1. Protein Synthesis in S30 Extracts Prepared from Cells Overexpressing RNA Polymerase Subunits

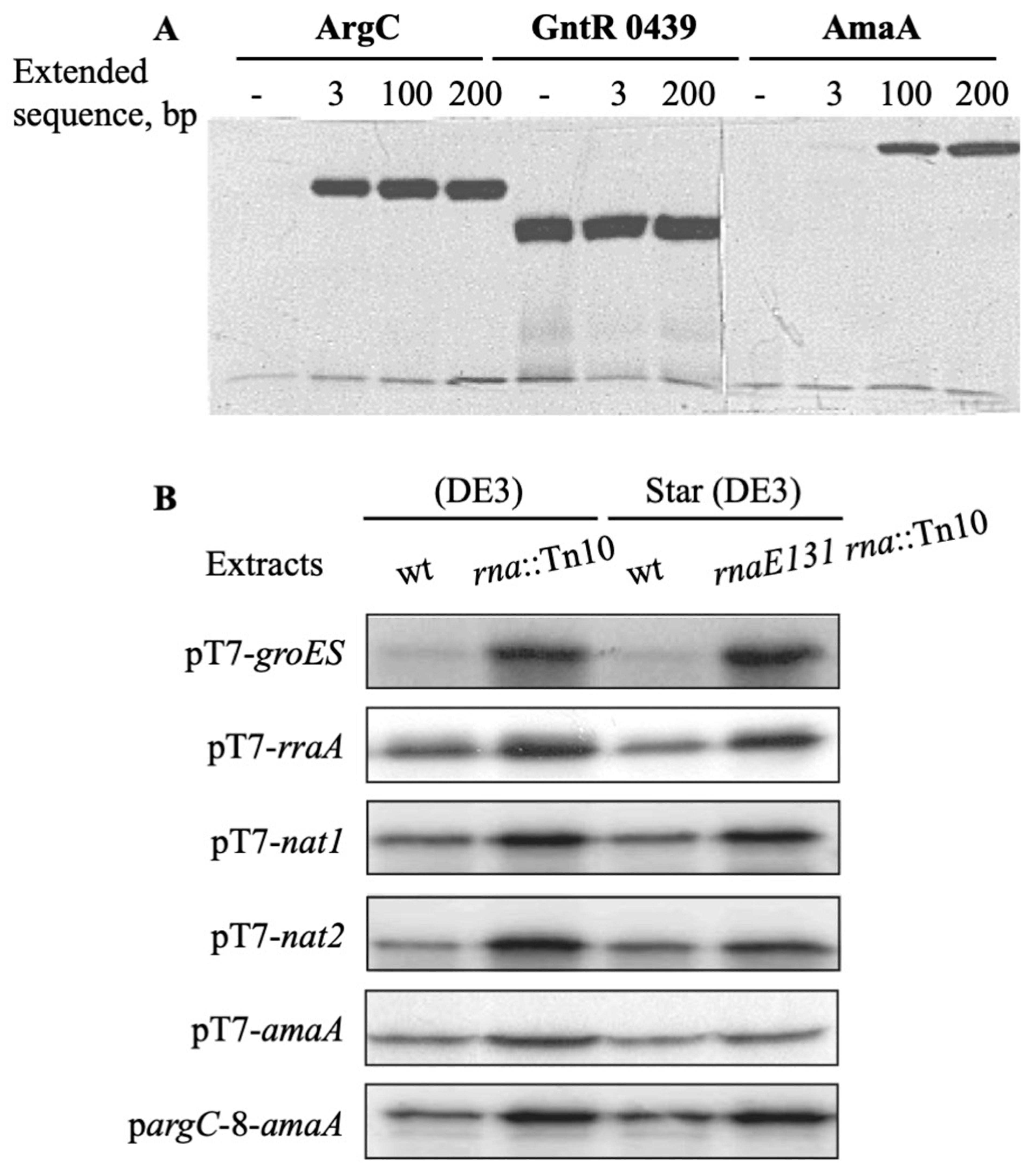
3.2. Extension of a 3′ Extremity of mRNA Templates and T7 Transcription Terminator Increase CFPS

3.3. Protein Synthesis Is Higher in RNase-I-Deficient Cell-Free Extracts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNase Status | Relative Endoribonuclease Activity (%) | |
|---|---|---|
| Crude Extracts | S30 Extracts | |
| BL21 (DE3) | 100 | 100 |
| BL21 (DE3) rna::Tn10 | 42 | 51 |
| BL21 Star (DE3) (it is rnaE131 strain) | 113 | 92 |
| BL21 Star (DE3) rnaE131 rna::Tn10 | 38 | 48 |
3.4. DNA–Protein Interaction in Cell-Free System
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nirenberg, M.W.; Matthaei, J.H. The dependence of cell-free protein synthesis in E. coli upon naturally occurring or synthetic polyribonucleotides. Proc. Natl. Acad. Sci. USA 1961, 47, 1588–1602. [Google Scholar] [CrossRef]
- Golomb, M.; Chamberlin, M. Characterization of T7-specific ribonucleic acid polymerase. IV. Resolution of the major in vitro transcripts by gel electrophoresis. J. Biol. Chem. 1974, 249, 2858–2863. [Google Scholar]
- Hui, M.P.; Foley, P.L.; Belasco, J.G. Messenger RNA degradation in bacterial cells. Annu. Rev. Genet. 2014, 48, 537–559. [Google Scholar] [CrossRef]
- Bechhofer, D.H.; Deutscher, M.P. Bacterial ribonucleases and their roles in RNA metabolism. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 242–300. [Google Scholar] [CrossRef] [PubMed]
- Catherine, C.; Lee, K.H.; Oh, S.J.; Kim, D.M. Cell-free platforms for flexible expression and screening of enzymes. Biotechnol. Adv. 2013, 31, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.D.; Miklos, A.E.; Chiao, A.C.; Sun, Z.Z.; Lux, M.W. Methodologies for preparation of prokaryotic extracts for cell-free expression systems. Synth. Syst. Biotechnol. 2020, 5, 252–267. [Google Scholar] [CrossRef]
- Kido, M.; Yamanaka, K.; Mitani, T.; Niki, H.; Ogura, T.; Hiraga, S. RNase E polypeptides lacking a carboxyl-terminal half suppress a mukB mutation in Escherichia coli. J. Bacteriol. 1996, 178, 3917–3925. [Google Scholar] [CrossRef] [PubMed]
- Subbarayan, P.R.; Deutscher, M.P. Escherichia coli RNase M is a multiply altered form of RNase, I. RNA 2001, 7, 1702–1707. [Google Scholar]
- Kennell, D. Processing endoribonucleases and mRNA degradation in bacteria. J. Bacteriol. 2002, 184, 4645–4657. [Google Scholar] [CrossRef][Green Version]
- Zhu, L.; Gangopadhyay, T.; Padmanabha, K.P.; Deutscher, M.P. Escherichia coli RNA gene encoding RNase I: Cloning, overexpression, subcellular distribution of the enzyme, and use of a rna deletion to identify additional RNases. J. Bacteriol. 1990, 172, 3146–3151. [Google Scholar] [CrossRef] [PubMed]
- Snapyan, M.; Lecocq, M.; Guével, L.; Arnaud, M.C.; Ghochikyan, A.; Sakanyan, V. Dissecting DNA-protein and protein-protein interactions involved in bacterial transcriptional regulation by a sensitive protein array method combining a near-infrared fluorescence detection. Proteomics 2003, 3, 647–657. [Google Scholar] [CrossRef]
- Yeretssian, G.; Lecocq, M.; Lebon, G.; Hurst, H.C.; Sakanyan, V. Competition on nitrocellulose-immobilized antibody arrays: From bacterial protein binding assay to protein profiling in breast cancer cells. Mol. Cell. Proteom. 2005, 4, 605–617. [Google Scholar] [CrossRef]
- Dimova, D.; Weigel, P.; Takahashi, M.; Marc, F.; Van Duyne, G.D.; Sakanyan, V. Thermostability, oligomerization and DNA-binding properties of the regulatory protein ArgR from the hyperthermophilic bacterium Thermotoga Neapolitana. Mol. Gen. Genet. 2000, 263, 119–130. [Google Scholar] [CrossRef]
- Nelson, K.E.; Clayton, R.A.; Gill, S.R.; Gwinn, M.L.; Dodson, R.J.; Haft, D.H.; Hickey, E.K.; Peterson, J.D.; Nelson, W.C.; Ketchum, K.A.; et al. Evidence for lateral gene transfer between Archaea and Bacteria from genome sequence of Thermotoga maritima. Nature 1999, 399, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Zhan, X.; Gao, J.; Qiu, J.; Feng, Y.; Meganathan, R.; Cohen, S.N.; Georgiou, G. RraA, a protein inhibitor of RNase E activity that globally modulates RNA abundance in E. coli. Cell 2003, 114, 623–634. [Google Scholar] [CrossRef]
- Sim, E.; Abuhammad, A.; Ryan, A. Arylamine N-acetyltransferases: From drug metabolism and pharmacogenetics to drug discovery. Br. J. Pharmacol. 2014, 171, 2705–2725. [Google Scholar] [CrossRef] [PubMed]
- Sakanyan, V.; Charlier, D.; Legrain, C.; Kochikyan, A.; Mett, I.; Pierard, A.; Glansdorff, N. Primary structure, partial purification and regulation of key enzymes of the acetyl cycle of arginine biosynthesis in Bacillus stearothermophilus: Dual function of ornithine acetyltransferase. J. Gen. Microbiol. 1993, 139, 393–402. [Google Scholar] [CrossRef][Green Version]
- Sakanyan, V.; Desmarez, L.; Legrain, C.; Charlier, D.; Mett, I.; Kochikyan, A.; Savchenko, A.; Boyen, A.; Falmagne, P.; Piérard, A.; et al. Gene cloning, sequence analysis, purification, and characterization of a thermostable aminoacylase from Bacillus stearothermophilus. Appl. Environ. Microbiol. 1993, 59, 3878–3888. [Google Scholar] [CrossRef]
- Zheng, M.; Cooper, D.R.; Grossoehme, N.E.; Yu, M.; Hung, L.W.; Cieslik, M.; Derewenda, U.; Lesley, S.A.; Wilson, I.; Giedroc, D.P.; et al. Structure of Thermotoga maritima TM0439: Implications for the mechanism of bacterial GntR transcription regulators with Zn2+-binding FCD domains. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Karaivanova, I.M.; Weigel, P.; Takahashi, M.; Fort, C.; Versavaud, A.; Van Duyne, G.; Charlier, D.; Hallet, J.N.; Glansdorff, N.; Sakanyan, V. Mutational analysis of the thermostable arginine repressor from Bacillus stearothermophilus: Dissecting residues involved in DNA binding properties. J. Mol. Biol. 1999, 291, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Rosner, J.L. Formation, induction and curing of bacteriophage P1 lysogens. Virology 1972, 48, 679–689. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Cannistraro, V.J.; Kennell, D. Broad-specificity endoribonucleases and mRNA degradation in Escherichia coli. J. Bacteriol. 1992, 174, 56–62. [Google Scholar] [CrossRef][Green Version]
- Borukhov, S.; Goldfarb, A. Recombinant Escherichia coli RNA polymerase: Purification of individually overexpressed subunits and in vitro assembly. Protein Expr. Purif. 1993, 4, 503–511. [Google Scholar] [CrossRef]
- Zubay, G. In vitro synthesis of protein in microbial systems. Annu. Rev. Genet. 1973, 7, 267–287. [Google Scholar] [CrossRef]
- Nevin, D.E.; Pratt, J.M. A coupled in vitro transcription-translation system for the exclusive synthesis of polypeptides expressed from the T7 promoter. FEBS Lett. 1991, 291, 259–263. [Google Scholar] [CrossRef]
- Kim, D.-M.; Swartz, J.R. Prolonging cell-free protein synthesis with a novel ATP regeneration system. Biotechnol. Bioeng. 1999, 66, 180–188. [Google Scholar] [CrossRef]
- Batisse, N. Étude Génétique d’enzymes Responsables de la Résolution des Acides Aminés de la Série L à Partir de Dérivés N-Substitués chez la Bactérie Thermophile Bacillus stearothermophilus. Ph.D. Thesis, University of Nantes, Nantes, France, 1997. [Google Scholar]
- Ghochikyan, A.; Karaivanova, I.M.; Lecocq, M.; Vusio, P.; Arnaud, M.C.; Snapyan, M.; Weigel, P.; Guével, L.; Buckle, M.; Sakanyan, V. Arginine operator binding by heterologous and chimeric ArgR repressors from Escherichia coli and Bacillus stearothermophilus. J. Bacteriol. 2002, 184, 6602–6614. [Google Scholar] [CrossRef]
- Savchenko, A.; Weigel, P.; Dimova, D.; Lecocq, M.; Sakanyan, V. The Bacillus stearothermophilus argCJBD operon harbours a strong promoter as evaluated in Escherichia coli cells. Gene 1998, 212, 167–177. [Google Scholar] [CrossRef]
- Higgins, C.F.; McLaren, R.S.; Newbury, S.F. Repetitive extragenic palindromic sequences, mRNA stability and gene expression: Evolution by gene conversion? Gene 1988, 72, 3–14. [Google Scholar] [CrossRef]
- Régnier, P.; Hajnsdorf, E. Decay of mRNA encoding ribosomal protein S15 of Escherichia coli is initiated by an RNase E-dependent endonucleolytic cleavage that removes the 3’ stabilizing stem and loop structure. J. Mol. Biol. 1991, 217, 282–292. [Google Scholar] [CrossRef]
- Studier, F.W.; Rosenberg, A.H.; Dunn, J.J.; Dubendorff, J.W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990, 185, 60–89. [Google Scholar]
- Köhrer, C.; Mayer, C.; Grobner, P.; Piendl, W. Use of T7 RNA polymerase in an optimized Escherichia coli coupled in vitro transcription-translation system. Application in regulatory studies and expression of long transcription units. Eur. J. Biochem. 1996, 236, 234–239. [Google Scholar]
- Lopez, P.J.; Marchand, I.; Joyce, S.A.; Dreyfus, M. The C-terminal half of RNase E, which organizes the Escherichia coli degradosome, participates in mRNA degradation but not rRNA processing in vivo. Mol. Microbiol. 1999, 33, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Porco, A.; Isturiz, T.; Conway, T. Cloning and molecular genetic characterization of the Escherichia coli gntR, gntK, and gntU genes of GntI, the main system for gluconate metabolism. J. Bacteriol. 1996, 178, 3260–3269. [Google Scholar] [CrossRef]
- Song, S.; Park, C. Utilization of D-ribose through D-xylose transporter. FEMS Microb. Lett. 1998, 163, 255–261. [Google Scholar]
- Oehler, S. Feedback regulation of Lac repressor expression in Escherichia coli. J. Bacteriol. 2009, 191, 5301–5303. [Google Scholar] [CrossRef] [PubMed]
- Dondapati, S.K.; Stech, M.; Zemella, A.; Kubick, S. Cell-Free Protein Synthesis: A Promising Option for Future Drug Development. BioDrugs 2020, 34, 327–348. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Chu, H.S.; Kim, T.W.; Oh, I.S.; Choi, C.Y.; Hahn, G.H.; Park, C.G.; Kim, D.M. Cell-free synthesis of recombinant proteins from PCR-amplified genes at a comparable productivity to that of plasmid-based reactions. Biochem. Biophys. Res. Commun. 2005, 338, 1346–1352. [Google Scholar] [CrossRef]
- Michel-Reydellet, N.; Woodrow, K.; Swartz, J. Increasing PCR fragment stability and protein yields in a cell-free system with genetically modified Escherichia coli extracts. J. Mol. Microbiol. Biotechnol. 2005, 9, 26–34. [Google Scholar] [CrossRef]
- Hahn, G.H.; Kim, D.M. Production of milligram quantities of recombinant proteins from PCR-amplified DNAs in a continuous-exchange cell-free protein synthesis system. Anal. Biochem. 2006, 335, 151–153. [Google Scholar] [CrossRef]
- Liou, G.-G.; Jane, W.-N.; Cohen, S.N.; Lin, N.-S.; Lin-Chao, S. RNA degradosomes exist in vivo in Escherichia coli as multicomponent complexes associated with the cytoplasmic membrane via the N-terminal region of ribonuclease E. Proc. Natl. Acad. Sci. USA 2001, 98, 63–68. [Google Scholar] [CrossRef]
- Hibi, K.; Amikura, K.; Sugiura, N.; Masuda, K.; Ohno, S.; Yokogawa, T.; Ueda, T.; Shimizu, Y. Reconstituted cell-free protein synthesis using in vitro transcribed tRNAs. Commun. Biol. 2020, 3, 350. [Google Scholar] [CrossRef]
- Dekhtyar, M.; Morin, A.; Sakanyan, V. Triad pattern algorithm for predicting strong promoter candidates in bacterial genomes. BMC Bioinform. 2008, 9, 233. [Google Scholar] [CrossRef] [PubMed]
- Failmezger, J.; Rauter, M.; Nitschel, R.; Kraml, M.; Siemann-Herzberg, M. Cell-free protein synthesis from non-growing, stressed Escherichia coli. Sci. Rep. 2017, 7, 16524. [Google Scholar] [CrossRef] [PubMed]
- Chiba, C.H.; Knirsch, M.C.; Azzoni, A.R.; Moreira, A.R.; Stephano, M.A. Cell-free protein synthesis: Advances on production process for biopharmaceuticals and immunobiological products. Biotechniques 2020, 70, 2. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Snapyan, M.; Robin, S.; Yeretssian, G.; Lecocq, M.; Marc, F.; Sakanyan, V. Cell-Free Protein Synthesis by Diversifying Bacterial Transcription Machinery. BioTech 2021, 10, 24. https://doi.org/10.3390/biotech10040024
Snapyan M, Robin S, Yeretssian G, Lecocq M, Marc F, Sakanyan V. Cell-Free Protein Synthesis by Diversifying Bacterial Transcription Machinery. BioTech. 2021; 10(4):24. https://doi.org/10.3390/biotech10040024
Chicago/Turabian StyleSnapyan, Marina, Sylvain Robin, Garabet Yeretssian, Michèle Lecocq, Frédéric Marc, and Vehary Sakanyan. 2021. "Cell-Free Protein Synthesis by Diversifying Bacterial Transcription Machinery" BioTech 10, no. 4: 24. https://doi.org/10.3390/biotech10040024
APA StyleSnapyan, M., Robin, S., Yeretssian, G., Lecocq, M., Marc, F., & Sakanyan, V. (2021). Cell-Free Protein Synthesis by Diversifying Bacterial Transcription Machinery. BioTech, 10(4), 24. https://doi.org/10.3390/biotech10040024