Abstract
Guanine-rich sequences of nucleic acids, including DNA and RNA, are known to fold into non-canonical structures named G-quadruplexes (G4s). Such arrangements of these macromolecular polymers are mainly located in telomeres and in promoter regions of oncogenes and, for this reason, they represent a potential target for compounds with therapeutic applications. In fact, the ligand-mediated stabilization of G4s inhibits telomerase and the activity of transcriptional machinery and counteracts cancer cell immortalization. Flavonoids, along with other classes of small molecules, have been previously tested for their ability to stabilize G4s, but the mechanism of their interaction has not been fully elucidated. In the current work, we report a multi-technique investigation on the binding of tosylated isoflavones obtained by the B-ring modification of compounds from Maclura pomifera to a telomeric DNA sequence. Our study demonstrates that such derivatization leads to compounds showing lower binding affinity but with an increased selectivity toward G4 with respect to double-stranded DNA. The binding mode to the macromolecular target G4 was studied by combining results from electrospray mass spectrometry binding studies, nuclear magnetic resonance experiments and computational simulations. Overall, our findings show that tosylation influences the selectivity toward the macromolecular target by affecting the interaction mode with the nucleic acid.
Keywords:
DNA; G-quadruplex; flavonoids; ESI-MS; molecular modeling; molecular dynamics; drug discovery 1. Introduction
In the context of nucleic acids, besides the canonical double-stranded DNA (dsDNA) structure, nucleic acids can fold into different arrangements, such as G-quadruplexes (G4s) [1]. In fact, guanine-rich (G-rich) sequences can be organized into planar quartets of Gs held together by Hoogsten base pairing. These systems are stabilized by monovalent cations coordinated by the carbonyl oxygen atoms pointing toward the inner channel at the center of the quartet and by the stacking of two or more of these arrangements [2]. G4s are present in telomeres, where their stabilization with small molecules inhibits telomerase activity, preventing sequence elongation and, therefore, cell immortalization [3]. G4s were also found in the promoter region of oncogenes, such as c-KIT, c-MYC, k-RAS, B-RAF, BCL-2, RET, VEGF, HIF, hTERT and HSP90.
In such cases, the stabilization causes the inhibition of the transcriptional machinery and the subsequent down-regulation of the expression of the targeted gene [4]. Small molecules targeting nucleic acids are also endowed with antiviral, antibacterial, anticancer and antiprotozoal activity.
In more detail, G4 stabilization can be achieved through the intervention of small molecules that bind this structure and counteract their conversion to other arrangements. To date, several G4 ligands have been reported to bind this macromolecular structure and several of them are being studied for their potential antiproliferative activity. These molecules include anthraquinones, porphyrins, perylenes, acridines and flavonoids [5,6,7,8]. In the literature, such binders are classified based on their chemical structure or on their preferential interaction mode with G4, which are indeed interconnected. A relevant ligand–G4 binding motif is pi-stacking, inspired by dsDNA intercalating agents. This peculiar interaction mode is allowed by the large planar aromatic core of the ligand, which can stack to the guanines of the external tetrads. A second motif consists of groove and loop binding, which is normally favored by the presence of positive charges on the ligands that may interact through an H-bond or Coulomb interaction with the phosphate groups [9,10].
Flavonoids represent a wide class of bioactive compounds that have beneficial effects in the modulation of the enzymatic activity and inhibition of cellular proliferation [11]. More specifically, isoflavones derived from Maclura pomifera, namely osajin, pomiferin, scandenone and auriculasin, possess antibacterial, anti-inflammatory, antidiabetic and antinociceptive properties [12,13], and they have been previously tested against telomeric G4 and dsDNA by our group using both blind molecular docking and electrospray ionization–mass spectrometry (ESI-MS). In this context, osajin and scandenone demonstrated higher selectivity for G4 [14,15], and the B-ring derivatization of osajin through the introduction of different aryl sulfonate groups leads to enhanced selectivity toward G4 over dsDNA with retained G4 stabilization properties [16].
Indeed, selectivity toward G4 over dsDNA is a feature of primary relevance to limit off-target reactivity and side effects [17,18]. Thus, in the current work, the binding of semi-synthetic flavonoids deriving from M. pomifera, modified with a toluenesulfonyl moiety in the hydroxyl group(s) of the B-ring, to a telomeric G4 sequence was tested using ESI-MS as a screening tool. Sequence selectivity was assessed by also considering a dsDNA sequence as reference. Furthermore, we performed a series of molecular docking experiments and carried out molecular dynamics (MD) simulations to study the binding mode of ligands, correlating computational results with MS-based fragmentation studies. The interaction motif was further investigated by means of solution nuclear magnetic resonance (NMR).
2. Materials and Methods
2.1. ESI-MS Binding Studies
DNA sequences were obtained from Sigma-Aldrich (Milan, Italy). Samples were heat-denatured and folded in 150 mM ammonium acetate before incubation with the ligands. Stock solutions of the compounds were prepared in methanol. The final concentration of the oligonucleotide was 5 μM in 150 mM ammonium acetate, with a 10:1 compound/oligo ratio. Samples were acquired after an equilibration time of 30 min and methanol was added to the samples to obtain a stable ESI signal. Mass spectra were recorded by direct infusion ESI on a Thermo Fisher Scientific (Waltham, MA, USA) LCQ Fleet ion trap mass spectrometer. The instrument was set in negative ionization mode with a 3.4 kV capillary voltage, 120 °C capillary temperature and a flow rate of 5 μL/min. Binding affinity (BA) was calculated based on signal intensities (I) as in the following: BA = IG4 bound/(IG4 bound + IG4 unbound). Collision-induced dissociation (CID) experiments were performed on the complexes by isolating the precursor ion in the trap. The fragmentation was promoted, increasing the “normalized collision energy” parameter. Exact mass for the G4 sequence (5′-AGGGTTAGGGTTAGGGTTAGGGT3′): 7270.774 Da. Exact mass for dsDNA (5′-ACTATTTACGTATAATGA-3′, 5′-TCATTATACGTAAATAGT-3′): 10987.922 Da. For data processing, Qual Browser Thermo Xcalibur 4.0.27.13 software was used.
2.2. Molecular Docking
G4 structure file was retrieved from the RCSB Protein Data Bank (PDB, www.rcsb.org, accessed on 24 May 2024) (PDB ID: 7KLP) and was prepared with the Protein Preparation Wizard included in the Schrodinger suite [19], using default settings, i.e., adding hydrogens, removing waters further than 5 Å from the ligand, adjusting charges, capping termini, optimizing hydrogen bond clusters and performing a final minimization under the OPLS3e force field [20]. The ligands were prepared for docking with Ligprep tool under OPLS3e force field using Epik ionizer, and all possible states in the 7 ± 2 pH range were generated, including the possible metal binding states, therefore allowing for the interaction with the potassium ions. The docking protocol consisted of rigid receptor/flexible ligand docking, which was performed with Glide, under the OPLS3e force field, with default settings, i.e., a 0.80 scaling factor for the ligand atom van der Waals radius and 0.15 for the partial charge cut-off, with generation of 1 pose per ligand. Different parameters, such as grid dimension, docking precision (standard precision is labelled as SP, whereas higher precision docking is marked as XP) and sampling mode were changed throughout the study to investigate their influence on the computation. Five cubic grids were prepared with Receptor Grid Generation tool included in Schrodinger suite and generated by manually selecting as center the two potassium ions in the inner channel of the G4 quartets. The dimension of the grids was varied as mentioned earlier and the third potassium ion included in the 3D structure was either left unmodified or deleted. The settings used for each calculation are summarized in Table S1.
2.3. Molecular Dynamics
The structures of the G4–ligand complexes, obtained by the molecular docking study, were prepared for the simulations with Schrodinger application System Builder as follows: the structures were solvated in a triclinic water box (10 Å buffer) composed by 4-site model TIP4Pew water molecules, the total charge was neutralized by the addition of an adequate number of K+ ions and a 0.20 M KCl concentration was set. The system initially prepared for Schrodinger default force field OPLS3e was converted through Viparr application to the AMBER OL15 force field specific for nucleic acids [21,22,23], and the parameters set used for the ions was the one developed by Joung et al. [24]. The ligands were parameterized with Antechamber application of AmberTools20 using GAFF2 parameters and AM1-BCC atomic charges, and missing parameters were generated with Parmchk2. The simulations were conducted with Schrodinger Desmond for 100 ns each, recording 1 frame every 10 ps using the NPT ensemble at a temperature and pressure of 300 K and 1.01 bar. Default settings were used for the RESPA integrator (2.0 fs for bonded interactions, 2.0 fs for near and 6.0 fs for far interactions), and the system was relaxed prior to the simulation using the default Desmond protocol. The trajectories were analyzed after the simulations with Simulation Event Analysis Schrodinger application, and, for the over-time ligand RMSD calculations, the ligand atoms positions were fitted to the guanine planes.
2.4. Nuclear Magnetic Resonance
Saturation transfer difference (STD) NMR experiments were performed on a Bruker Ascend spectrometer (frequency: 400.13 MHz for 1H) (Bruker, Billerica, MA, USA) using the pulse program stddiffesgp.3. For data processing, TopSpin 4.3.0 was used. In these experiments, the same G4 sequence used in ESI-MS binding studies was adopted (5′-AGGGTTAGGGTTAGGGTTAGGGT-3′), and a ligand/target ratio of 400:1 was used. NMR samples were prepared in a 5 mm NMR tube with a final volume of 465 µL using a 2/1 mixture of deuterated phosphate buffered saline (6.218 mM K2HPO4, 3.782 mM KH2PO4 dissolved in D2O, pH 7.1) and DMSO-d6. The final concentration of the DNA sequence and of the ligand was 10 µM and 4 mM, respectively. In all the experiments, 4096 scans were recorded, and a saturation time (d20) and relaxation delay (d1) of, respectively, 2 and 2.5 s were applied. The water 1H signal was suppressed by using an excitation sculpting with perfect echo (Bruker library: zgesgppe) pulse sequence. The STD effects of the individual protons were calculated in each sample in relation to a reference spectrum recorded with off-resonance saturation at δ = −40 ppm. The on-resonance spectra were acquired by saturating the DNA sequence in three different spectral regions, thus performing three experiments. In particular, the portions that have been irradiated are the imino, aromatic and deoxyribose/backbone spectral regions at δ = 11.0 ppm, δ = 9.5 ppm and δ = 5.8 ppm, respectively. The three sets of STD spectra were recorded with target saturation achieved with a series of 90° Gaussian-shaped 50 ms pulses repeated to reach the desired d20 time.
3. Results and Discussion
In the current work, isoosajin (5), isopomiferin (6) and their tosylated derivatives (11, 12), together with di-p-toluensulfonyl pomiferin, di-p-toluensulfonyl auriculasin- and p-toluensulfonyl scadenone (8–10), which were extracted and synthetized by our research group [16,25], were tested against a 23-mer telomeric G4 sequence and a dsDNA oligonucleotide using ESI-MS, comparing the results with our previous data on the binding of natural flavonoids (see Scheme S1 in the Supplementary Materials for chemical structures) [14,15,16].
In the 1990s, ESI-MS emerged as a useful tool for the discovery of DNA binders, considering the “softness” of the ionization technique in which minimal fragmentation occurs and, as a result, non-covalent interactions being retained during the ESI process [26]. The experiment is carried out using a negative ionization mode since the analyte is dissolved in an aqueous environment and infused at atmospheric pression, conditions in which the phosphodiester backbone of the DNA sequence is fully deprotonated (pKa < 1) [27]. Because of the similar topologies adopted by G4s that fold in the presence of ammonium and potassium ions, the ESI-MS experiment is performed in ammonium acetate (150 mM) [8,28]. Indeed, more specifically, we also observed in a previous study by means of circular dichroism that the same 23-mer G4 telomeric sequence used in the current work behaves comparably in buffer solutions containing either K+ or NH4+ [29]. In the MS spectra, the intensities of the peaks corresponding to the different species are assumed to be proportional to the concentrations of the species in the analyzed solution. While accurate Kd determination would require titration experiments at an increasing concentration of the species, BA is a semiquantitative property that efficiently describes the tendency of a molecule to interact with its target and is used to rank the binding of small molecules to G4s in ESI-MS screenings [30,31]. It is based on the fact that relative intensities of the ion peaks of comparable species within a mass spectrum are assumed to be proportional to their abundance. Since this value is calculated as the ratio of the bound over the total nucleic acid, as detailed in the Materials and Methods section, BA is generally expressed as a percentage without units of measurement [30,32,33].
The results of the ESI-MS study are resumed in Table 1, and a representative mass spectrum is shown in Figure 1. Most of the studied molecules, except for compound 8, which does not bind the nucleic acid, interact with both G4 and dsDNA. Notably, the selectivity of the molecules for G4 increased upon derivatization with the tosyl moiety for most of the ligands compared to natural isoflavones, even though lower BA values were recorded (see Figures S1–S4 in the Supplementary Materials for representative ESI-MS spectra) [16]. Only in the case of compound 7 did the interaction occur exclusively with G4. Nevertheless, it must be taken into account that, in the context of G4 ligands screenings based on ESI-MS, a selectivity ratio above 2 is considered promising [34] and that, more in general, very popular and biologically active G4 ligands such as BRACO-19 and PIPER show limited selectivity over dsDNA [34,35].

Table 1.
BA and ECOM50% values determined by ESI-MS interaction assays between compounds 1–12 and the 23-mer G4 telomeric or dsDNA sequences. Selectivity ratio is calculated according to the following equation: BA G4/BA dsDNA (* indicates data from references [14,16]).

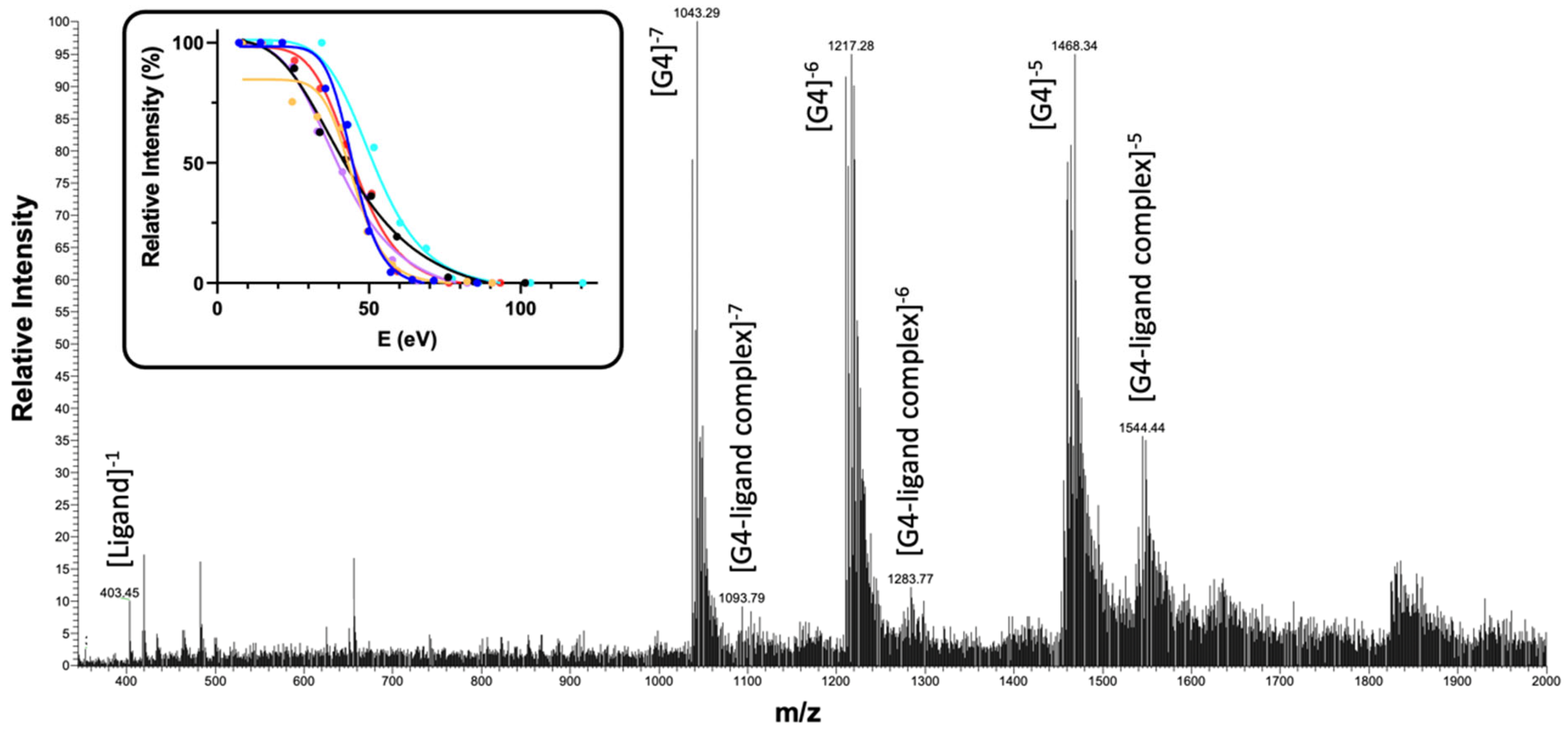
Figure 1.
Representative mass spectrum showing the interaction of a ligand (isoosajin 5) with G4 DNA. In the inset, CID curves of G4–ligand complexes for compounds 5 (yellow), 6 (pink), 9 (blue), 10 (red), 11 (black) and 12 (cyan) are reported.
The efficiency in stabilizing the macromolecular arrangement, which is a pivotal aspect for ligands targeting G4s, can be determined by using ESI-MS through collision-induced dissociation (CID) experiments in which the molecular ion of the G4–ligand complex is isolated and accelerated, applying an increasing potential, causing a collision with a gas that promotes its fragmentation. Based on the relative intensities of the adduct and of the dissociation products (i.e., the unbound nucleic acid, the ligand or fragments) in m/z spectra recorded at increasing collision energy values, it is possible to calculate the gas-phase stability in terms of ECOM50%, which is expressed in electron-volts (eV). This value, also defined as CE50, represents the collision energy needed to cause the dissociation of the complex to its relative half-intensity (see Figure 1 and Figure S8 in the Supplementary Materials for CID curves). Thus, a higher ECOM50% value indicates a more stable ligand–G4 complex [27,33,36,37], and this could be directly related to biological effects, as it has been shown that compounds promoting a higher stabilization of G4s also induce more efficient telomerase inhibition [38]. When considering this parameter, tosylated derivatives (7–12) retained the ECOM50% values measured for natural isoflavones; thus, it can be deduced that their ability to stabilize the G4 arrangement is maintained upon tosylation. In most of the recorded fragmentation spectra (see Figures S5–S7 in the Supplementary Materials for representative spectra), the first and main observed event upon CID fragmentation consists of the loss of the ligand and the appearance of the peaks corresponding to the unbound G4. This behavior suggests that interaction may occur via stacking, as observed for isopomiferin 6 (Figure S7 in the Supplementary Materials) [39]. Nevertheless, it is also worth pointing out that some tosylated isoflavones, such as p-toluenesulfonyl-scandenone 10, show the ability to alkylate the G4 sequence (Figure S5 in the Supplementary Materials). Importantly, the same effect was not observed for dsDNA, thus paving the way for the development of small molecules selectively delivering DNA damage to G4s. On the other hand, in the case of the complex generated by di-p-toluensulfonyl-isopomiferin 12, base loss upon fragmentation was noted (Figure S6 in the Supplementary Materials).
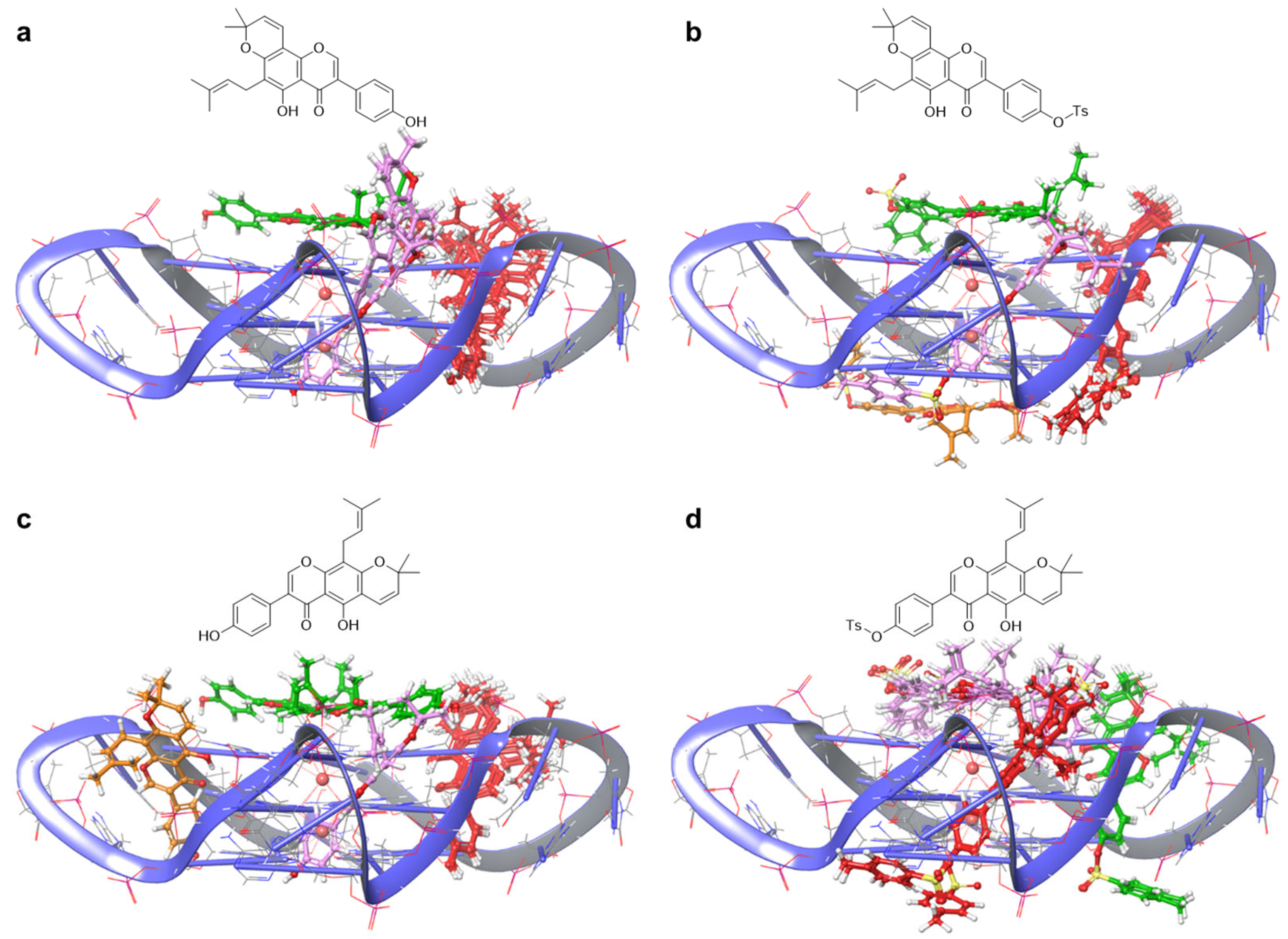
The investigation of the ligand–G4 interaction motif can be carried out by combining information from ESI-MS screening and molecular modeling [33]. Thus, a computational study was performed on the most promising compounds, i.e., those showing the highest experimental selectivity ratio values for G4 over dsDNA. Osajin, p-toluenesulfonyl-osajin, scandenone and p-toluenesulfonyl-scandenone (1, 4, 7 and 10) were considered as models for this step of the investigation. In particular, we studied the interaction pattern toward a telomeric parallel G4 structure, which is reported as the prevalent topology in the cellular overcrowded solution conditions [40]. The study was composed of two parts, consisting of molecular docking and MD. In the first part, we generated, through a multiparametric docking process, the initial structures of the complexes for the following MD runs. Precisely, the four compounds were docked to the telomeric parallel G4 structure (PDB ID: 7KLP) [41] using Glide [42]. In order to obtain a complete ensemble of interaction modes for each complex, key parameters were varied in every computation, namely grid dimension (15 to 32 Å), docking precision (SP or XP), ions presence (two or three) and exhaustiveness of the sampling of conformer generation and of poses selection. This workflow produced a multitude of possible poses for every docked compound, and we obtained the formation of clusters of poses in stacking and groove binding modes for the studied molecules. Within these clusters, we observed an overall similarity among poses, as can be deduced from Figure 2. Thus, we then selected for each compound the stacking and groove binding poses showing the best energetic score for performing the subsequent MD simulations.

Figure 2.
Poses overlap of the multiparametric molecular docking: osajin 1 (a), p-toluensulfonyl osajin 7 (b), scandenone 4 (c), p-toluensulfonyl scandenone 10 (d). The chemical structures of the compounds are reported in the upper part of every panel.
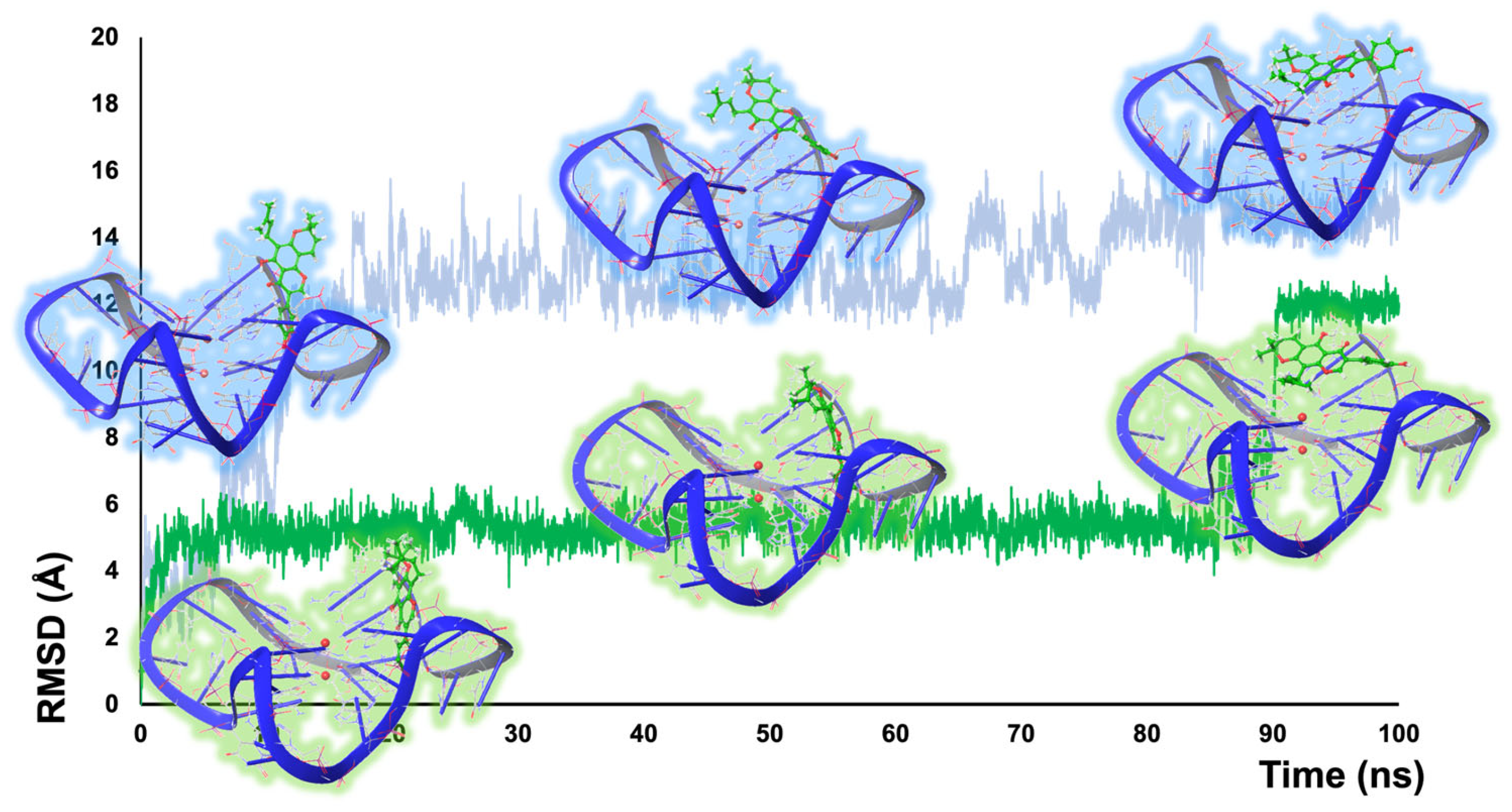
After parameterization of the ligands (Antechamber: GAFF2, AM1-BCC), the ligand–G4 complexes were enclosed in a cubic water box (TIP4Pew) with 0.20 M of KCl ions and subjected to a 100 ns simulation with Desmond [43] under a specific AMBER force field for nucleic acids (OL15) (see Supplementary Materials for the detailed procedure and Figure S9 for MD trajectories) [21,22,23]. The eight trajectories obtained were then analyzed (Schrodinger embedded app—Simulation Event Analysis) and the over-time RMSD values were calculated for the ligands and for the guanines of the G4 structures. The MD study showed a preference for a stacking mode of interaction for osajin and scandenone (1 and 4), whereas their tosylated derivatives (7 and 10) demonstrated a preference to interact with the G4 by groove binding. More specifically, the stacking pose of scandenone 4 produced a stable trajectory for all of the simulation timeframe, with only minor RMSD fluctuations, probably connected with the flexibility of the isoprene moiety (Figure S9 in the Supplementary Materials). A different behavior was otherwise observed for the same compound for what concerns the groove binding pose. Indeed, throughout MD simulation, a transition was detected, and the ligand changed its interaction mode to stacking after about 85 ns (Figure 3). In a similar fashion, the simulation starting from the groove binding mode of osajin 1 produced a transition to a stacking position after 10 ns, which was retained throughout the simulation time (Figure 3). To further confirm this observation, we measured the distance from the center of mass of the ligands and the upper tetrad targeted through stacking in the frames across the binding pose transition event. In the case of osajin 1, a change between 12.25 (groove binding, 6 ns) and 5.04 Å (stacking, 15 ns) was measured. For scandenone 4, a transition from 8.32 (groove binding, 85 ns) to 6.59 Å (stacking, 95 ns) was observed. Interestingly, in the case of the tosylated derivatives (7 and 10), an opposite behavior was observed. In fact, the stacking mode produced high RMSD fluctuations, whereas the groove binding produced stable trajectories after an initial stabilization period (Figure S9 in the Supplementary Materials). This could be explained by the steric effects of the tosylate moiety that may direct the molecules toward the grooves. Additionally, this substituent could hinder intercalation in dsDNA, thus leading to an improved selectivity.

Figure 3.
MD trajectories and snapshots showing the pose transition from groove to stacking binding mode for osajin 1 (blue) and scandenone 4 (green).
To provide further insights on the interaction motif, another analytical technique was eventually enrolled. Nuclear Overhauser effect (NOE)-based NMR experiments, such as STD, are used to study the binding of small molecules to macromolecular targets in solution. NOE causes the transfer of nuclear spin polarization from one population of spin-active nuclei to another that is located in close proximity, thus providing interaction information in conditions that closely resemble the physiological environment. STD is an established technique used for compounds targeting peculiar arrangements of nucleic acids [44].
In this experiment, p-toluensulfonyl osajin 7 was used as a model compound. STD spectra showed that, in agreement with what was observed in docking studies (Figure 2), the different portions of the molecule do not interact identically with the macromolecular target. In fact, while the experiment confirmed the interaction of the compound with G4 in solution, STD signals were only observed for the protons of the double bonds inserted in the cycles of the scaffold and for the Ph-CH3 moiety (Figure S10 in the Supplementary Materials). On the other hand, the isoprenyl chain and the aromatic rings showed weaker interactions. Importantly, this behavior was observed when irradiating in the deoxyribose/backbone spectral region (δ = 5.8 ppm), while no STD effect was observed when considering the imino and aromatic portions (δ = 11.0 ppm and δ = 9.5 ppm, respectively). This observation further suggests that the binding would preferentially occur with the loops of the G4 rather than with the tetrads.
4. Conclusions
The experimental and computational results of this study support the potential of isoflavones as promising scaffolds for developing selective G4 binders and stabilizers. Within the set of studied compounds, we observed that the derivatization of the B-ring with the tosyl moiety, despite affecting BA, led to an increase in selectivity over dsDNA. Most importantly, the stability of the complex was retained for semi-synthetic derivatives.
Concerning the structural details of the ligand–target interaction motif, combined docking and MD studies demonstrate that tosylated derivatives interact with G4 in a different manner when compared to their natural precursors. In fact, pi-stacking is not predicted as the favored binding motif for these molecules since MD trajectories for groove binding interactions showed lower RMSD fluctuations. This observation was also confirmed by NMR analysis. On the other hand, the preferred binding motif of unmodified isoflavones is pi–pi stacking, as demonstrated by docking and MD simulations. These results partially parallel experimental observations from CID fragmentation studies, from which a conventional dissociation mode was detected for non-tosylated flavonoids while more complex phenomena were observed in the case of tosylated derivatives.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/macromol4030033/s1, Scheme S1: chemical structures of the studied compounds; Figures S1–S7: mass spectra; Figure S8: plot of relative intensity of the complex dsDNA–ligand against collision energy; Table S1: molecular docking settings; Figure S9: MD trajectories; Figure S10: STD NMR experiments and representative model of the interaction motif.
Author Contributions
Conceptualization, G.R., M.M. and A.G.; software, M.A.; investigation, M.G., E.O. and A.O.; writing—original draft preparation, G.R.; writing—review and editing, G.R., M.M. and A.G.; supervision, M.M. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge funding from University of Brescia and Regione Lombardia.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, Topology and Structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Kosiol, N.; Juranek, S.; Brossart, P.; Heine, A.; Paeschke, K. G-Quadruplexes: A Promising Target for Cancer Therapy. Mol. Cancer 2021, 20, 40. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct Evidence for a G-Quadruplex in a Promoter Region and Its Targeting with a Small Molecule to Repress c-MYC Transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef]
- Chaudhuri, R.; Bhattacharya, S.; Dash, J.; Bhattacharya, S. Recent Update on Targeting c-MYC G-Quadruplexes by Small Molecules for Anticancer Therapeutics. J. Med. Chem. 2021, 64, 42–70. [Google Scholar] [CrossRef]
- Ongaro, A.; Ribaudo, G.; Zagotto, G.; Memo, M.; Gianoncelli, A. Synthesis via A3 Coupling Reaction of Anthracene-Propargylamine as a New Scaffold for the Interaction with DNA. ChemistrySelect 2019, 4, 13138–13142. [Google Scholar] [CrossRef]
- Ribaudo, G.; Scalabrin, M.; Pavan, V.; Fabris, D.; Zagotto, G. Constrained Bisantrene Derivatives as G-Quadruplex Binders. Arkivoc 2016, 2016, 145–160. [Google Scholar] [CrossRef]
- Murat, P.; Singh, Y.; Defrancq, E. Methods for Investigating G-Quadruplex DNA/Ligand Interactions. Chem. Soc. Rev. 2011, 40, 5293. [Google Scholar] [CrossRef]
- Ribaudo, G.; Ongaro, A.; Zorzan, M.; Pezzani, R.; Redaelli, M.; Zagotto, G.; Memo, M.; Gianoncelli, A. Investigation of the Molecular Reactivity of Bioactive Oxiranylmethyloxy Anthraquinones. Arch. Der Pharm. 2019, 352, 1900030. [Google Scholar] [CrossRef]
- Jucá, M.M.; Filho, F.M.S.C.; De Almeida, J.C.; Da Mesquita, D.D.; De Barriga, J.R.D.; Dias, K.C.F.; Barbosa, T.M.; Vasconcelos, L.C.; Leal, L.K.A.M.; Ribeiro, J.E.; et al. Flavonoids: Biological Activities and Therapeutic Potential. Nat. Prod. Res. 2020, 34, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Hajimahmoodi, M.; Shams-Ardakani, M.; Saniee, P.; Siavoshi, F.; Mehrabani, M.; Hosseinzadeh, H.; Foroumadi, P.; Safavi, M.; Khanavi, M.; Akbarzadeh, T.; et al. In Vitro Antibacterial Activity of Some Iranian Medicinal Plant Extracts against Helicobacter Pylori. Nat. Prod. Res. 2011, 25, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Wang, P.; Yuan, W.; Grant, G.; Li, S. Phenolics from the Fruits of Maclura pomifera. Nat. Prod. Commun. 2017, 12, 1934578X1701201. [Google Scholar] [CrossRef]
- Ribaudo, G.; Ongaro, A.; Zagotto, G.; Memo, M.; Gianoncelli, A. Evidence on Selective Binding to G-Quadruplex DNA of Isoflavones from Maclura pomifera by Mass Spectrometry and Molecular Docking. Nat. Prod. Res. 2019, 35, 2583–2587. [Google Scholar] [CrossRef]
- Ribaudo, G.; Oselladore, E.; Ongaro, A.; Zagotto, G.; Memo, M.; Gianoncelli, A. Enhanced G-Quadruplex Selectivity of Flavonoid Glycoside Rutin over Quercetin. Nat. Prod. Res. 2020, 36, 3469–3473. [Google Scholar] [CrossRef] [PubMed]
- Ribaudo, G.; Ongaro, A.; Zagotto, G.; Memo, M.; Gianoncelli, A. Photoactivated Semi-Synthetic Derivative of Osajin Selectively Interacts with G-Quadruplex DNA. Nat. Prod. Res. 2020, 36, 405–410. [Google Scholar] [CrossRef]
- Arola, A.; Vilar, R. Stabilisation of G-Quadruplex DNA by Small Molecules. Curr. Top. Med. Chem. 2008, 8, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Zuffo, M.; Guédin, A.; Leriche, E.-D.; Doria, F.; Pirota, V.; Gabelica, V.; Mergny, J.-L.; Freccero, M. More Is Not Always Better: Finding the Right Trade-off between Affinity and Selectivity of a G-Quadruplex Ligand. Nucleic Acids Res. 2018, 46, e115. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2020-1: Protein Preparation Wizard; Epik, 2016; Impact, 2016; Prime, 2020; Schrödinger, LLC.: New York, NY, USA, 2020.
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Zgarbová, M.; Šponer, J.; Otyepka, M.; Cheatham, T.E.; Galindo-Murillo, R.; Jurečka, P. Refinement of the Sugar–Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA. J. Chem. Theory Comput. 2015, 11, 5723–5736. [Google Scholar] [CrossRef]
- Pérez, A.; Marchán, I.; Svozil, D.; Sponer, J.; Cheatham, T.E.; Laughton, C.A.; Orozco, M. Refinement of the AMBER Force Field for Nucleic Acids: Improving the Description of α/γ Conformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef]
- Ribaudo, G.; Coghi, P.; Zanforlin, E.; Law, B.Y.K.; Wu, Y.Y.J.; Han, Y.; Qiu, A.C.; Qu, Y.Q.; Zagotto, G.; Wong, V.K.W. Semi-Synthetic Isoflavones as BACE-1 Inhibitors against Alzheimer’s Disease. Bioorg. Chem. 2019, 87, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Siegel, M. Early Discovery Drug Screening Using Mass Spectrometry. Curr. Top. Med. Chem. 2002, 2, 13–33. [Google Scholar] [CrossRef]
- Rosu, F.; De Pauw, E.; Gabelica, V. Electrospray Mass Spectrometry to Study Drug-Nucleic Acids Interactions. Biochimie 2008, 90, 1074–1087. [Google Scholar] [CrossRef]
- Monchaud, D.; Allain, C.; Bertrand, H.; Smargiasso, N.; Rosu, F.; Gabelica, V.; De Cian, A.; Mergny, J.-L.; Teulade-Fichou, M.-P. Ligands Playing Musical Chairs with G-Quadruplex DNA: A Rapid and Simple Displacement Assay for Identifying Selective G-Quadruplex Binders. Biochimie 2008, 90, 1207–1223. [Google Scholar] [CrossRef]
- Ongaro, A.; Desiderati, G.; Oselladore, E.; Auricchio, D.; Memo, M.; Ribaudo, G.; Sissi, C.; Gianoncelli, A. Amino-Acid-Anthraquinone Click Chemistry Conjugates Selectively Target Human Telomeric G-Quadruplexes. ChemMedChem 2022, 17, e202100665. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Zhou, J.; Yuan, G. Electrospray Ionization Mass Spectrometry Probing of Binding Affinity of Berbamine, a Flexible Cyclic Alkaloid from Traditional Chinese Medicine, with G-quadruplex DNA. Rapid Commun. Mass Spectrom. 2014, 28, 143–147. [Google Scholar] [CrossRef]
- Cubrilovic, D.; Biela, A.; Sielaff, F.; Steinmetzer, T.; Klebe, G.; Zenobi, R. Quantifying Protein-Ligand Binding Constants Using Electrospray Ionization Mass Spectrometry: A Systematic Binding Affinity Study of a Series of Hydrophobically Modified Trypsin Inhibitors. J. Am. Soc. Mass Spectrom. 2012, 23, 1768–1777. [Google Scholar] [CrossRef]
- Yuan, G.; Zhang, Q.; Zhou, J.; Li, H. Mass Spectrometry of G-quadruplex DNA: Formation, Recognition, Property, Conversion, and Conformation. Mass Spectrom. Rev. 2011, 30, 1121–1142. [Google Scholar] [CrossRef]
- Ribaudo, G.; Ongaro, A.; Oselladore, E.; Memo, M.; Gianoncelli, A. Combining Electrospray Mass Spectrometry (ESI-MS) and Computational Techniques in the Assessment of G-Quadruplex Ligands: A Hybrid Approach to Optimize Hit Discovery. J. Med. Chem. 2021, 64, 13174–13190. [Google Scholar] [CrossRef] [PubMed]
- Mazzitelli, C.L.; Brodbelt, J.S.; Kern, J.T.; Rodriguez, M.; Kerwin, S.M. Evaluation of Binding of Perylene Diimide and Benzannulated Perylene Diimide Ligands to Dna by Electrospray Ionization Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2006, 17, 593–604. [Google Scholar] [CrossRef]
- Machireddy, B.; Sullivan, H.-J.; Wu, C. Binding of BRACO19 to a Telomeric G-Quadruplex DNA Probed by All-Atom Molecular Dynamics Simulations with Explicit Solvent. Molecules 2019, 24, 1010. [Google Scholar] [CrossRef] [PubMed]
- Rosu, F.; Nguyen, C.-H.; De Pauw, E.; Gabelica, V. Ligand Binding Mode to Duplex and Triplex Dna Assessed by Combining Electrospray Tandem Mass Spectrometry and Molecular Modeling. J. Am. Soc. Mass Spectrom. 2007, 18, 1052–1062. [Google Scholar] [CrossRef]
- Torvinen, M.; Kalenius, E.; Sansone, F.; Casnati, A.; Jänis, J. Noncovalent Complexation of Monoamine Neurotransmitters and Related Ammonium Ions by Tetramethoxy Tetraglucosylcalix[4]Arene. J. Am. Soc. Mass Spectrom. 2012, 23, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Lin, Z.; Zhao, S.; Wang, G.; Shen, Z.; Liu, W.; Cai, Y.; Wang, K.; Wan, C.C.; Yan, T. Research Progress on G-Quadruplexes in Human Telomeres and Human Telomerase Reverse Transcriptase (hTERT) Promoter. Oxid. Med. Cell. Longev. 2022, 2022, 2905663. [Google Scholar] [CrossRef]
- Xu, N.; Yang, H.; Cui, M.; Song, F.; Liu, Z.; Liu, S. Evaluation of Alkaloids Binding to the Parallel Quadruplex Structure [d(TGGGGT)]4 by Electrospray Ionization Mass Spectrometry: ESI-MS of Alkaloids/G-Quadruplex DNA Complexes. J. Mass Spectrom. 2012, 47, 694–700. [Google Scholar] [CrossRef]
- Pagano, B.; Amato, J.; Iaccarino, N.; Cingolani, C.; Zizza, P.; Biroccio, A.; Novellino, E.; Randazzo, A. Looking for Efficient G-Quadruplex Ligands: Evidence for Selective Stabilizing Properties and Telomere Damage by Drug-Like Molecules. ChemMedChem 2015, 10, 640–649. [Google Scholar] [CrossRef]
- Li, K.; Yatsunyk, L.; Neidle, S. Water Spines and Networks in G-Quadruplex Structures. Nucleic Acids Res. 2021, 49, 519–528. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-1: Glide; Schrödinger, LLC.: New York, NY, USA, 2020.
- Schrödinger Release 2020-1: Desmond Molecular Dynamics System; D. E. Shaw Research: New York, NY, USA; Maestro-Desmond Interoperability Tools, Schrödinger: New York, NY, USA, 2020.
- Martino, L.; Virno, A.; Pagano, B.; Virgilio, A.; Di Micco, S.; Galeone, A.; Giancola, C.; Bifulco, G.; Mayol, L.; Randazzo, A. Structural and Thermodynamic Studies of the Interaction of Distamycin A with the Parallel Quadruplex Structure [d(TGGGGT)]4. J. Am. Chem. Soc. 2007, 129, 16048–16056. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).