
Congenital Atrophic Dermatofibrosarcoma Protuberans: A Case Report and Review of the Literature

 and
and
Abstract
1. Introduction
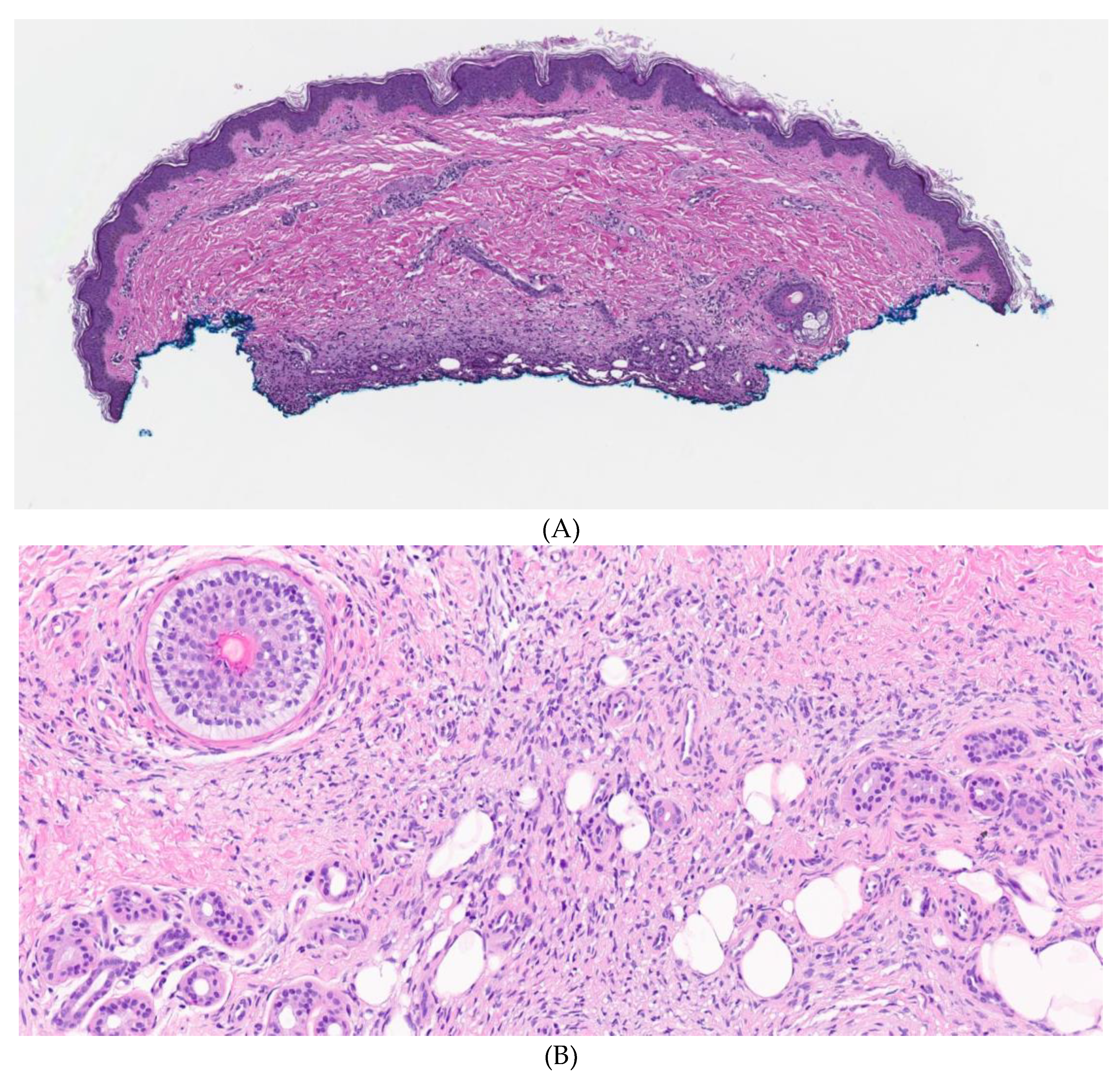
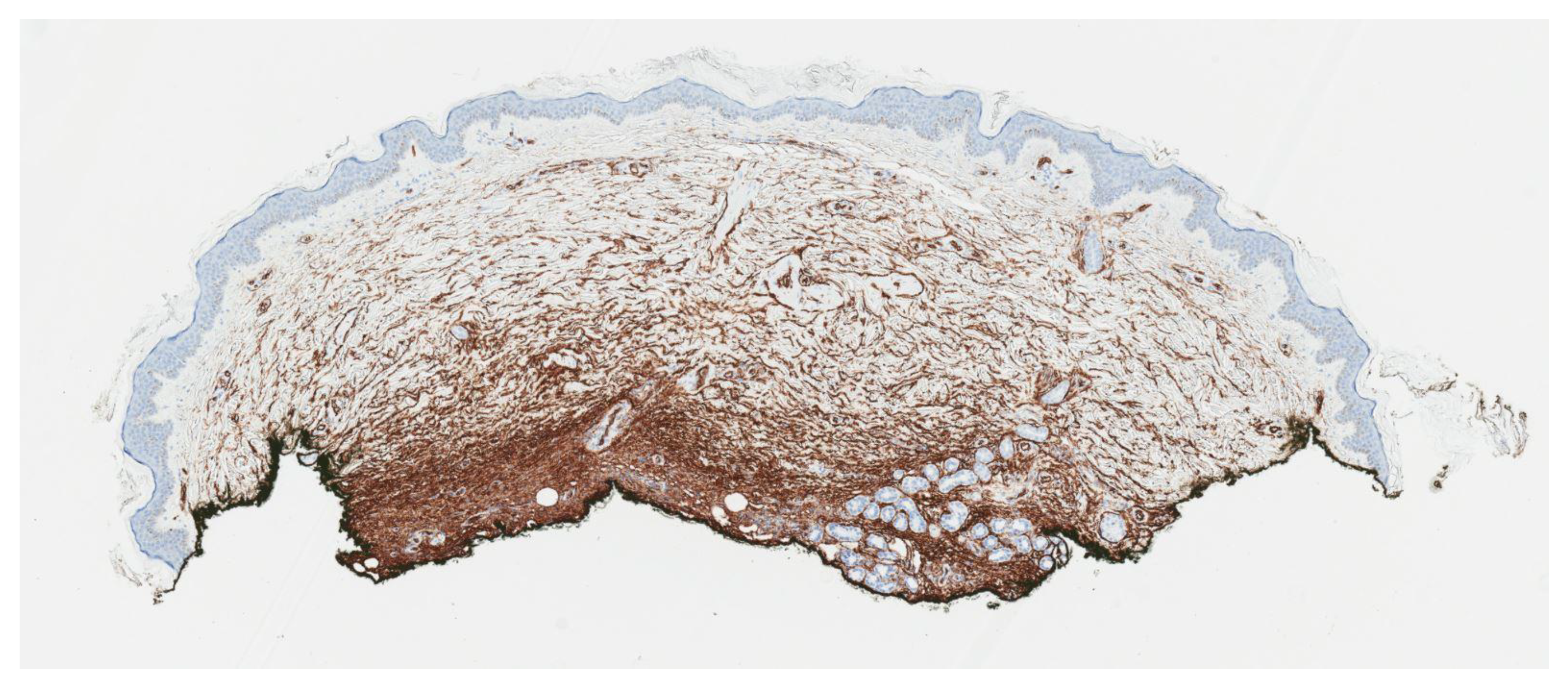
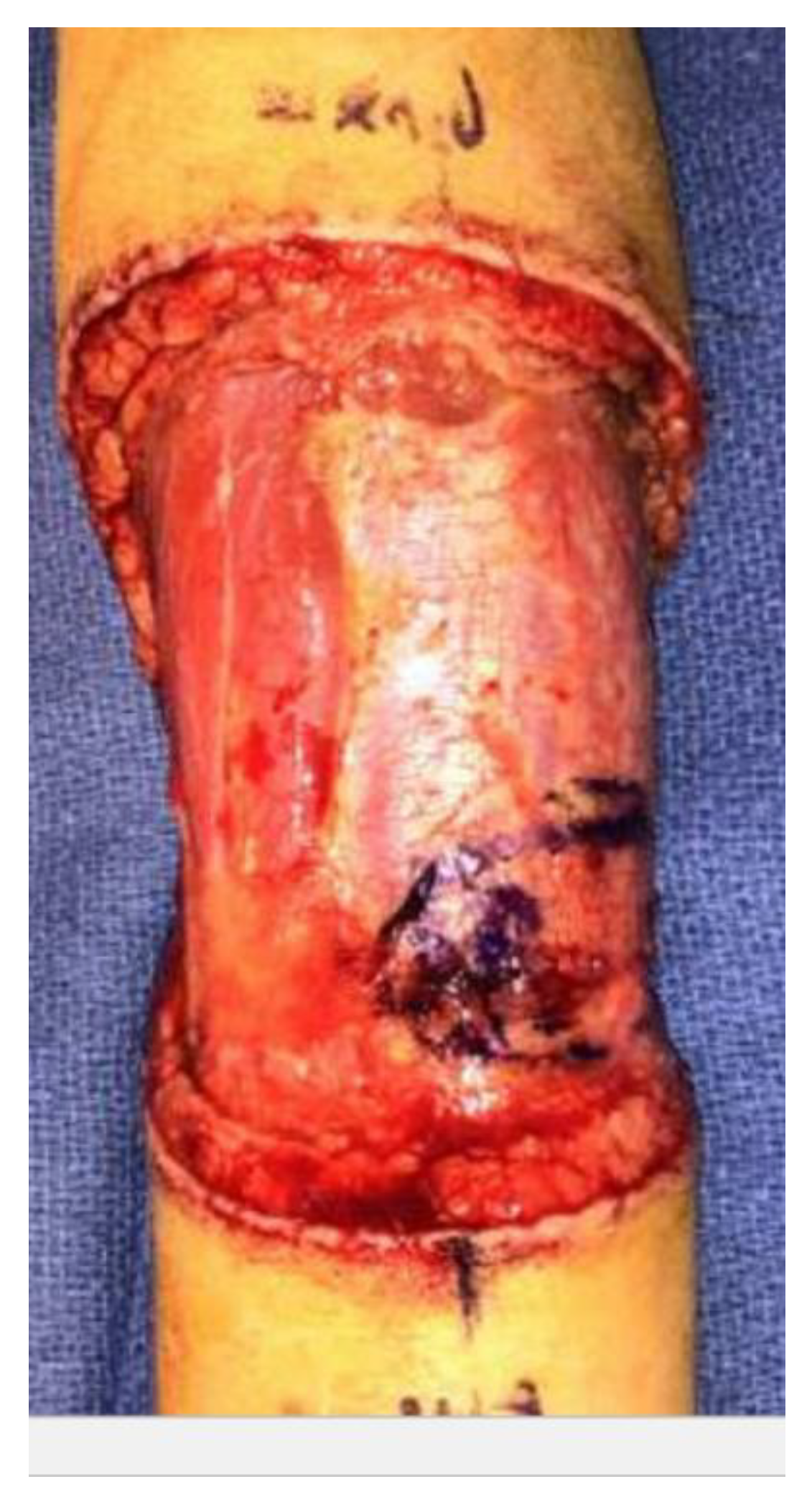
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brooks, J.; Ramsey, M.L. Dermatofibrosarcoma Protuberans. 2018. Available online: https://europepmc.org/article/NBK/nbk513305 (accessed on 19 July 2018).
- Van Lee, C.; Kan, W.C.; Gran, S.; Mooyaart, A.; Mureau, M.; Williams, H.; Matin, R.; Van Den Bos, R.; Hollestein, L. Dermatofibrosarcoma protuberans re-excision and recurrence rates in the Netherlands Between 1989 and 2016. Acta Derm.-Venereol. 2019, 99, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Badhey, A.K.; Tikhtman, R.; Tang, A.L. Management of dermatofibrosarcoma protuberans. Curr. Opin. Otolaryngol. Head Neck Surg. 2021, 29, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shi, Y.Z.; Feng, X.H.; Wang, X.T.; He, X.L.; Zhao, M. Novel TNC-PDGFD fusion in fibrosarcomatous dermatofibrosarcoma protuberans: A case report. Diagn. Pathol. 2021, 16, 1–6. [Google Scholar] [CrossRef]
- Reddy, C.; Hayward, P.; Thompson, P.; Kan, A. Dermatofibrosarcoma protuberans in children. J. Plast. Reconstr. Aesthetic Surg. 2009, 62, 819–823. [Google Scholar] [CrossRef]
- Posso-De Los Rios, C.J.; Lara-Corrales, I.; Ho, N. Dermatofibrosarcoma protuberans in pediatric patients: A report of 17 cases. J. Cutan. Med. Surg. 2014, 18, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.W.; Cleton-Jansen, A.M.; Cleven, A.H.; Ruano, D.; van Wezel, T.; Szuhai, K.; Bovée, J.V. Molecular analysis of gene fusions in bone and soft tissue tumors by anchored multiplex PCR–based targeted next-generation sequencing. J. Mol. Diagn. 2018, 20, 653–663. [Google Scholar] [CrossRef]
- Zhou, Y.; Chin, J.; Strutin, M.D.; Lomiguen, C.M. Unmasking dermatofibrosarcoma protuberans: Case report of an atypical presentation complicated by post-surgical excision. Int. J. Surg. Case Rep. 2020, 69, 101–104. [Google Scholar] [CrossRef]
- Souiki, T.; Belhaj, A.; Ait Abderrhim, A.; Alami, B.; Tahiri, L.; Chbani, L.; Ibn Majdoub, K.; Toughrai, I.; Mazaz, K. Dermatofibrosarcoma protuberans of the anterior abdominal wall: Case report and literature review. J. Surg. Case Rep. 2022, 2022, rjac272. [Google Scholar] [CrossRef]
- Bakry, O.; Attia, A. Atrophic dermatofibrosarcoma protuberans. J. Dermatol. Case Rep. 2012, 6, 14. [Google Scholar] [CrossRef]
- Feramisco, J.; Larsen, F.; Weitzul, S.; Cockerell, C.; Ghali, F. Congenital atrophic dermatofibrosarcoma protuberans in a 7-month-old boy treated with Mohs micrographic surgery. Pediatr. Dermatol. 2008, 25, 455–459. [Google Scholar] [CrossRef]
- Llombart, B.; Serra, C.; Requena, C.; Alsina, M.; Morgado-Carrasco, D.; Través, V.; Sanmartín, O. Guidelines for diagnosis and treatment of cutaneous sarcomas: Dermatofibrosarcoma protuberans. Actas Dermo-Sifiliográficas 2018, 109, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.; Shetty K, J.; Prasad, H.L.K. An uncommon malignancy of the breast; dermatofibrosarcoma protruberans—A case report. Indian J. Surg. Oncol. 2012, 3, 242–244. [Google Scholar] [CrossRef]
- Reilly, D.J.; Loo, Y.L.; Alexander, W.M.; Wilks, D.J.; MacGregor, D.; Coombs, C.J. Diagnostic and Management Considerations in Pediatric Dermatofibrosarcoma Protuberans. Ann. Plast. Surg. 2022, 88, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Valdivielso-Ramos, M.; Hernanz, J.M. Dermatofibrosarcoma protuberans en la infancia. Actas Dermo-Sifiliogr. 2012, 103, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, B.; Han, X.; Ma, L. Pediatric atrophic dermatofibrosarcoma protuberans. Pediatr. Investig. 2017, 1, 50. [Google Scholar] [CrossRef] [PubMed]
- Akay, B.N.; Unlu, E.; Erdem, C.; Heper, A.O. Dermatoscopic findings of atrophic dermatofibrosarcoma protuberans. Dermatol. Pract. Concept. 2015, 5, 71. [Google Scholar] [CrossRef] [PubMed]
- Makino, M.; Sasaoka, S.; Nakanishi, G.; Makino, E.; Fujimoto, W. Congenital atrophic dermatofibrosarcoma protuberans detected by COL1A1-PDGFB rearrangement. Diagn. Pathol. 2016, 11, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hanabusa, M.; Kamo, R.; Harada, T.; Ishii, M. Dermatofibrosarcoma protuberans with atrophic appearance at early stage of the tumor. J. Dermatol. 2007, 34, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Marini, M.; Saponaro, A.; Magariños, G.; De Baldrich, A.; Lynch, P.; Remorino, L. Congenital atrophic dermatofibrosarcoma protuberans. Int. J. Dermatol. 2001, 40, 448–450. [Google Scholar] [CrossRef]
- Han, H.H.; Lim, S.Y.; Park, Y.M.; Rhie, J.W. Congenital dermatofibrosarcoma protuberans: A case report and literature review. Ann. Dermatol. 2015, 27, 597–600. [Google Scholar] [CrossRef]
- Weinstein, J.M.; Drolet, B.A.; Esterly, N.B.; Rogers, M.; Bauer, B.S.; Wagner, A.M.; Mancini, A.J. Congenital dermatofibrosarcoma protuberans: Variability in presentation. Arch. Dermatol. 2003, 139, 207–211. [Google Scholar] [CrossRef]
- Martin, L.; Combemale, P.; Dupin, M.; Chouvet, B.; Kanitakis, J.; Bouyssou-Gauthier, M.L.; Dubreuil, G.; Claudy, A.; Grimand, P.G. The atrophic variant of dermatofibrosarcoma protuberans in childhood: A report of six cases. Br. J. Dermatol. 1998, 139, 719–725. [Google Scholar] [PubMed]
- Martin, L.; Piette, F.; Blanc, P.; Mortier, L.; Avril, M.F.; Delaunay, M.M.; Dréno, B.; Granel, F.; Mantoux, F.; Aubin, F.; et al. Clinical variants of the preprotuberant stage of dermatofibrosarcoma protuberans. Br. J. Dermatol. 2005, 153, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Maire, G.; Fraitag, S.; Galmiche, L.; Keslair, F.; Ebran, N.; Terrier-Lacombe, M.J.; de Prost, Y.; Pedeutour, F. A clinical, histologic, and molecular study of 9 cases of congenital dermatofibrosarcoma protuberans. Arch. Dermatol. 2007, 143, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhao, L.; Wang, J. Atrophic dermatofibrosarcoma protuberans: A clinicopathological study of 16 cases. Pathology 2019, 51, 615–620. [Google Scholar] [CrossRef]
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan — a web and mobile app for systematic reviews. Syst. Rev. 2016, 5, 210. [Google Scholar] [CrossRef]
- Hao, X.; Billings, S.D.; Wu, F.; Stultz, T.W.; Procop, G.W.; Mirkin, G.; Vidimos, A.T. Dermatofibrosarcoma protuberans: Update on the diagnosis and treatment. J. Clin. Med. 2020, 9, 1752. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.K.; Bandyopadhyay, D.; Ghosh, A.; Biswas, S.K.; Barma, K.D. Acquired multifocal tufted angiomas in an immunocompetent young adult. Indian J. Dermatol. 2011, 56, 412. [Google Scholar] [CrossRef]
- Jandali, S.; Kirschner, R.E. Congenital leukemia cutis presenting as multiple violaceous lesions in a newborn. Ann. Plast. Surg. 2011, 66, 310–312. [Google Scholar] [CrossRef]
- Tantcheva-Poor, I.; Marathovouniotis, N.; Kutzner, H.; Mentzel, T. Vascular congenital dermatofibrosarcoma protuberans: A new histological variant of dermatofibrosarcoma protuberans. Am. J. Dermatopathol. 2012, 34, e46–e49. [Google Scholar] [CrossRef]
- Marque, M.; Bessis, D.; Pedeutour, F.; Viseux, V.; Guillot, B.; Fraitag-Spinner, S. Medallion-like dermal dendrocyte hamartoma: The main diagnostic pitfall is congenital atrophic dermatofibrosarcoma. Br. J. Dermatol. 2008, 160, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Liu, B.; Liu, T.; Qiao, J.; Fang, H. Atrophic Pigmented Dermatofibrosarcoma Protuberans: A Case Report and Literature Review. Front. Oncol. 2021, 11, 669754. [Google Scholar] [CrossRef]
- Rodríguez-Jurado, R.; Palacios, C.; Durán-McKinster, C.; Mercadillo, P.; Orozco-Covarrubias, L.; del Mar Saez-de-Ocariz, M.; Ruiz-Maldonado, R. Medallion-like dermal dendrocyte hamartoma: A new clinically and histopathologically distinct lesion. J. Am. Acad. Dermatol. 2004, 51, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.N.; Anderson, E.; Junkins-Hopkins, J.; James, W.D. Medallion-like dermal dendrocyte hamartoma. Pediatr. Dermatol. 2007, 24, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Sirvent, N.; Maire, G.; Pedeutour, F. Genetics of dermatofibrosarcoma protuberans family of tumors: From ring chromosomes to tyrosine kinase inhibitor treatment. Genes Chromosomes Cancer 2003, 37, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kadam, S.S.; Kadam, T. Recurrent Dermatofibrosarcoma Protruberance over Shoulder: An Unresolved Problem. Int. Res. J. Oncol. 2021, 4, 11–20. [Google Scholar]
- Sleiwah, A.; Wright, T.C.; Chapman, T.; Dangoor, A.; Maggiani, F.; Clancy, R. Dermatofibrosarcoma protuberans in children. Curr. Treat. Options Oncol. 2022, 23, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Chicaud, M.; Frassati-Biaggi, A.; Kaltenbach, S.; Karanian, M.; Orbach, D.; Fraitag, S. Dermatofibrosarcoma protuberans, fibrosarcomatous variant: A rare tumor in children. Pediatr. Dermatol. 2021, 38, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Macarenco, R.S.; Zamolyi, R.; Franco, M.F.; Nascimento, A.G.; Abott, J.J.; Wang, X.; Erickson-Johnson, M.R.; Oliveira, A.M. Genomic gains of COL1A1-PDFGB occur in the histologic evolution of giant cell fibroblastoma into dermatofibrosarcoma protuberans. Genes Chromosomes Cancer 2008, 47, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Gupta, A.K.; Ansari, M.; Mehra, S.K.; Barolia, D.K. Very rare childhood tumor: Giant cell fibroblastoma. Med. J. Dr. DY Patil Univ. 2018, 11, 452. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Age of Presentation | Age of Diagnosis | Quiescent Period to Protuberant Phase | Location | Misdiagnosis | Translocation T (17; 22)/ COL1A1-PDGFB Fusion Gene | Treatment |
|---|---|---|---|---|---|---|---|
| Salem et al. | One year * | 10 years | Remained atrophic | Forearm | Bruise, morphea | Positive | Wide excision |
| Makino et al. [18] | Birth | 19 years | 10 years | Anterior chest | N/A | Positive | Wide excision |
| Marini et al. [20] | Birth | 16 years | 7 years | Anterior leg | Congenital fibroma | N/A | Mohs surgery |
| Han et al. [21] | Birth | 6 years | 4 years | Posterior neck | Dermatitis | Negative | Wide excision |
| Feramisco et al. [11] | 7 m * | 7 m | Remained atrophic | Left inguinal | N/A | Positive | Mohs surgery |
| Weinstein et al. [22] | 6 years * | 14 years | 6 years | Back | Vacular malformation, Fibrous histocytoma | N/A | Wide excision |
| Martin et al. [23,24] | Birth | 13 years | 7 years | Calf | Hematoma | Positive | Wide excision |
| Weinstein et al. [22] | 6 m * | 1 year | 6 m | Right thigh | Aplasia cutis | N/A | Wide excision |
| Martin et al. [23,24] | Birth | 3 years | 2 years | Periumbilical | Morphea | N/A | Wide excision |
| Maire et al. [25] | N/A | 3 years | N/A | Lumbar | “Difficult to Characterize” | Positive | Wide excision |
| Maire et al. [25] | N/A | 11 years | N/A | Lumbar | N/A | Positive | Wide excision |
| Maire et al. [25] | 2 years * | 7 years | N/A | Occipital | Aplasia cutis, Fibrous hamartoma, Infantile fibromatosis | Positive | Wide excision |
| Maire et al. [25] | N/A | 10 years | N/A | Foot | Difficult to characterize | Positive | Wide excision |
| Maire et al. [25] | 15 years * | 17 years | N/A | Trunk | Fibromatosis, Xanthomatous hamartoma, Aplasia cutis, Infantile fibrosarcoma | Positive | Wide excision |
| Maire et al. [25] | 2 years * | 3 years | N/A | Thorax | Fibrosarcoma, Angioma, Mastocytoma | N/A | Wide excision |
| Wide Local Excision | Mohs Excision | Staged Excision “Slow Mohs ” | |
|---|---|---|---|
| Description | Excision with 2–4 cm margins followed by closure of the defect |
|
|
| Advantage |
|
|
|
| Disadvantage |
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salem, I.; Bradley, K.; Mann, J.A.; Shin, J.H.; LeBoeuf, M.; Sriharan, A. Congenital Atrophic Dermatofibrosarcoma Protuberans: A Case Report and Review of the Literature. Dermato 2023, 3, 97-108. https://doi.org/10.3390/dermato3020008
Salem I, Bradley K, Mann JA, Shin JH, LeBoeuf M, Sriharan A. Congenital Atrophic Dermatofibrosarcoma Protuberans: A Case Report and Review of the Literature. Dermato. 2023; 3(2):97-108. https://doi.org/10.3390/dermato3020008
Chicago/Turabian StyleSalem, Iman, Katherine Bradley, Julianne A. Mann, Joseph H. Shin, Matthew LeBoeuf, and Aravindhan Sriharan. 2023. "Congenital Atrophic Dermatofibrosarcoma Protuberans: A Case Report and Review of the Literature" Dermato 3, no. 2: 97-108. https://doi.org/10.3390/dermato3020008
APA StyleSalem, I., Bradley, K., Mann, J. A., Shin, J. H., LeBoeuf, M., & Sriharan, A. (2023). Congenital Atrophic Dermatofibrosarcoma Protuberans: A Case Report and Review of the Literature. Dermato, 3(2), 97-108. https://doi.org/10.3390/dermato3020008