Abstract
Cardiomyopathies affect over 3 million individuals globally, with conventional treatments exhibiting up to 60% resistance and 25% 30-day readmission rates. This review synthesizes the current evidence on the role of neuro-immune interactions in the pathogenesis of cardiomyopathy and evaluates emerging therapies targeting this axis. We systematically examined clinical trials and mechanistic and multi-omics data across cardiomyopathy phenotypes, focusing on autonomic-immune dysregulation. Sympathetic overactivation, present in approximately 85% of patients, correlates with elevated pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) and contributes significantly to therapeutic non-response. Concurrent parasympathetic withdrawal impairs cholinergic anti-inflammatory pathways, as reflected by reduced heart rate variability and baroreflex sensitivity. At the molecular level, shared mechanisms include inflammasome activation, neuroimmune synaptic signaling, and neurogenic inflammation. Emerging therapies targeting this axis are promising. Vagus nerve stimulation, as demonstrated in the INOVATE-HF trial, improves functional outcomes, whereas IL-1β antagonists reduce cardiovascular events by 15–20% in the context of inflammatory diseases. Bioelectronic interventions, such as transcutaneous vagal nerve stimulation and baroreflex activation therapy, offer noninvasive dual-modulatory strategies that address both neural and immune pathways, positioning the neuroimmune axis as a central driver of cardiomyopathy, regardless of etiology. The integration of genetic and metabolomic profiling may enable precision therapies targeting neuroimmune circuits, thereby overcoming the limitations of hemodynamic-focused care. This mechanistic framework shifts the therapeutic paradigm from symptomatic relief to targeted modulation of pathogenic pathways, with implications for millions of patients with cardiomyopathy and broader inflammatory cardiovascular disorders.
1. Introduction
Cardiomyopathies are a heterogeneous group of cardiac disorders characterized by structural and functional abnormalities of the ventricular myocardium that significantly affect global cardiovascular health. They constitute the primary etiology of heart failure, a condition affecting over 64 million people worldwide and imposing substantial clinical and economic burdens [1,2,3]. Despite decades of therapeutic advances aimed at hemodynamic optimization and neurohormonal blockade, patients with cardiomyopathy continue to exhibit high morbidity and mortality, with mortality rates exceeding 50% within five years of diagnosis, resulting in more than 1 million hospitalizations annually [2,3]. Approximately 60% of patients demonstrate inadequate therapeutic responses, as reflected by persistent readmission rates as high as 25% within 30 days [4]. These poor clinical outcomes underscore the critical limitations of current therapeutic paradigms, necessitating a fundamental reevaluation of the pathophysiological processes that drive disease progression.
Historically, cardiomyopathy has been viewed predominantly as a structural and genetic entity. However, recent insights have shifted our understanding toward a more comprehensive conceptual framework, highlighting the critical role of neuroimmune interactions in disease progression [5]. The neuroimmune axis comprises a complex regulatory network characterized by constant bidirectional communication between the nervous and immune systems [6,7]. In cardiomyopathies, this axis orchestrates complex signaling between neurogenic pathways and inflammatory cascades, establishing neuro-immune regulatory feedback circuits [8]. In cardiomyopathies, these interactions orchestrate intricate signaling pathways between neurogenic and inflammatory responses, establishing regulatory feedback circuits essential for cardiac homeostasis [9]. Disruption of these neuroimmune mechanisms, evidenced by elevated norepinephrine and inflammatory biomarkers documented in up to 85% of cardiomyopathy patients, contributes significantly to the therapeutic resistance observed in current clinical management strategies [10]. This highlights the urgent need for novel precision-driven approaches that simultaneously target neural and immune pathways.
Despite recent advances, substantial knowledge gaps persist regarding the variability of neuroimmune interactions in diverse patient populations and disease contexts. Autonomic dysregulation and its differential impact on pediatric versus adult cardiomyopathies remain poorly characterized, sex-specific variations in immune-autonomic interactions are largely unexplored, and the role of metabolic dysfunction, notably insulin resistance, in neuroimmune-driven cardiac remodelling requires further elucidation [11]. The recent COVID-19 pandemic, exemplifying virus-induced myocarditis, has underscored critical gaps in our understanding of the mechanisms underlying virus-associated neuroimmune perturbations and their long-term cardiovascular implications.
This review synthesizes the current evidence on neuroimmune interactions in cardiomyopathy by examining its molecular mechanisms, clinical manifestations, and emerging therapeutic opportunities. Our central aim was to provide an integrative analysis of how distinct cardiomyopathy phenotypes, including dilated, hypertrophic, restrictive, and stress-induced variants, exhibit unique neuroimmune signatures. Furthermore, we critically evaluate novel therapeutic paradigms targeting neuroimmune pathways and explore their integration within precision medicine frameworks using multi-omics approaches. Underexplored domains, such as pediatric cardiomyopathy, sex-dependent immune-autonomic heterogeneity, metabolic dysfunction-driven remodelling, and post-viral myocarditis, are specifically addressed to outline future research directions and therapeutic potential.
2. Review Methodology
We conducted a systematic search using PubMed with established systematic review guidelines to evaluate the role of the neuroimmune axis in cardiomyopathy pathogenesis and therapeutic interventions, with particular focus on autonomic dysfunction, inflammatory mechanisms, and bioelectronic medicine applications.
2.1. Search Strategy
A comprehensive literature search was performed across multiple databases, including PubMed, covering studies published between January 2010 and August 2025, yielding a total of 3511 records. The search strategy employed both Medical Subject Headings (MeSH) terms and free-text keywords to ensure broad coverage of relevant studies. The primary search terms included:
- “CARDIOMYOPATHY AND NEUROIMMUNE AXIS” (n = 14)
- “CARDIOMYOPATHY, NEUROIMMUNE AXIS AND AUTONOMIC NERVOUS SYSTEM” (n = 4)
- “CARDIOMYOPATHY, NEUROIMMUNE AXIS, AUTONOMIC NERVOUS SYSTEM AND INFLAMMATION” (n = 4)
- “CARDIOMYOPATHY, NEUROIMMUNE AXIS AND INFLAMMATION” (n = 10)
- “CARDIOMYOPATHY, NEUROIMMUNE AXIS AND HEART FAILURE” (n = 4)
- “CARDIOMYOPATHY AND IMMUNE SYSTEM” (n = 2524)
- “CARDIOMYOPATHY AND VAGUS NERVE” (n = 96)
- “CARDIOMYOPATHY AND HEART RATE VARIABILITY” (n = 851)
- “CARDIOMYOPATHY AND BIOELECTRONIC MEDICINE” (n = 2)
- “CARDIOMYOPATHY, BIOELECTRONIC MEDICINE AND HEART FAILURE” (n = 2)
2.2. Study Selection
The search was refined to prioritize studies published between 1 January 2010, and 31 August 2025, focusing exclusively on peer-reviewed articles in indexed journals, while also including landmark foundational studies from earlier periods. After removing duplicates, 42 studies were identified as linking cardiomyopathy and neuroimmune mechanisms. Screening was conducted based on titles and abstracts, leading to the exclusion of studies irrelevant to cardiac neuroimmunology. A full-text review of the remaining articles was then performed, resulting in the exclusion of additional studies for the following reasons:
Studies focusing solely on cardiomyopathy development (n = 26)
Reviews without original data contribution (n = 62)
Irrelevant information (n = 3381)
Ultimately, 42 studies met the inclusion criteria and were selected for the analysis.
2.3. Inclusion Criteria
Studies were included if they provided experimental evidence on the role of neuro-immune mechanisms in cardiomyopathy pathogenesis, progression, or therapeutic intervention. Additionally, research exploring molecular mechanisms such as autonomic dysfunction, inflammatory cytokine signaling, vagal nerve modulation, and bioelectronic medicine applications were considered. Clinical studies and observational investigations evaluating autonomic dysfunction and inflammatory markers in cardiac disease were also included to ensure a comprehensive analysis from both mechanistic and clinical perspectives.
2.4. Exclusion Criteria
Non-peer-reviewed sources, conceptual frameworks lacking experimental validation, studies without full-text access (to maintain methodological rigor), and research unrelated to cardiomyopathy were excluded. These criteria ensured that only studies with direct relevance and high methodological quality were included in the final analyses. These criteria ensured that only studies with direct relevance to cardiac neuroimmunology and high methodological quality were included in the final analysis.
In addition to the primary database search, we conducted a comprehensive review of published literature examining the role of autonomic nervous system dysfunction, vagal nerve stimulation, inflammatory cascades, and bioelectronic medicine in cardiomyopathy. Studies were included based on detailed descriptions of neuroimmune mechanisms contributing to cardiac pathology, including cytokine-mediated inflammation, autonomic imbalance, and novel therapeutic approaches targeting the cardiac neuroimmune axis. We also evaluated clinical trials assessing neuromodulation interventions and inflammatory biomarkers in cardiomyopathy patients to provide comprehensive coverage of both mechanistic insights and therapeutic applications.
3. Fundamental Mechanisms of the Immuno-Neural Axis in Cardiac Disease
Under physiological conditions, the autonomic nervous system maintains cardiac function and immune system homeostasis. In cardiomyopathy, the disruption of this balance initiates pathological cascades that accelerate disease progression.
3.1. Autonomic Nervous System Dysregulation and Inflammatory Cascades
The pathophysiology of cardiomyopathy extends beyond structural cardiac abnormalities and encompasses a complex interplay between autonomic dysfunction and inflammatory activation [12]. This neuroimmune crosstalk operates as a bidirectional signaling network in which sympathetic overactivity and parasympathetic withdrawal exacerbate the inflammatory signaling. In cardiomyopathy, this delicate balance is profoundly disrupted, leading to sustained autonomic imbalance that perpetuates inflammation and accelerates cardiac deterioration.
Sympathetic nervous system overactivity elevates catecholamine levels, actively driving inflammation via oxidative stress and inflammatory mediator release [13,14], and chronic sympathetic activation further modulates immune cell phenotypes toward a pro-inflammatory state [15]. Excessive reactive oxygen species activate the NLRP3 inflammasome, inducing cytokine release and inflammatory cell death (pyroptosis), and perpetuating tissue damage [8].
Parasympathetic withdrawal simultaneously compromises the anti-inflammatory mechanisms [16,17]. These autonomic-inflammatory interactions influence clinical outcomes, as heart rate variability correlates directly with inflammatory markers and prognosis [18]. The recognition of autonomic-driven inflammation has prompted the development of novel therapeutic approaches [19].
3.1.1. Catecholamine-Mediated Inflammatory Activation
The mechanistic convergence between adrenergic signaling and cytokine induction marks catecholamines as the central mediators of immune activation in cardiomyopathy. Norepinephrine and epinephrine binding triggers inflammatory signaling pathways, producing cytokines (TNF-α, IL-1β, IL-6) associated with cardiotoxicity [5,20]. This creates a self-amplifying cycle that explains the relentless disease progression and therapeutic resistance [21].
3.1.2. Metabolic Reprogramming and Immune Activation
Metabolic shifts not only compromise energy production but also act as immunologic amplifiers, sustaining a pro-inflammatory milieu in cardiomyopathy [6]. This metabolic transformation has consequences that extend far beyond energy production, directly contributing to inflammatory activation. The byproducts of this altered metabolism serve as danger signals that activate immune cells and perpetuate inflammation [7]. Specific metabolic waste products, including lactate and succinate, bind directly to receptors on immune cells, triggering inflammatory signaling pathways [8].
Macrophage polarization is central to this metabolic-immune crosstalk, wherein metabolic dysfunction drives a decisive shift toward the M1 (classically activated) phenotype. M1 macrophages are characterized by enhanced glycolytic metabolism, elevated production of pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6), increased inducible nitric oxide synthase activity, and promotion of Th1-mediated responses that amplify tissue damage. This starkly contrasts with the cardioprotective M2 (alternatively activated) phenotype, which relies on oxidative metabolism, produces anti-inflammatory mediators, including IL-10 and arginase-1, supports tissue repair through growth factor secretion, and promotes Th2 responses conducive to healing. In cardiomyopathy, the metabolic milieu systematically favors M1 over M2 polarization, creating a self-perpetuating cycle of inflammation that impairs cardiac repair mechanisms and accelerates disease progression (Figure 1).
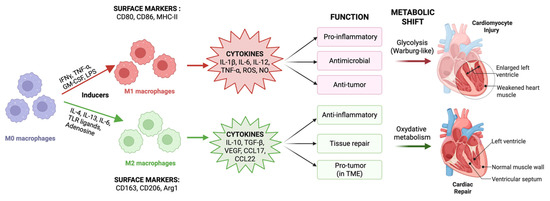
Figure 1.
Macrophage polarization and its impact on cardiomyopathy. Naïve M0 macrophages differentiate into M1 (classically activated) or M2 (alternatively activated) states depending on the specific stimuli. M1 macrophages induced by IFN-γ, TNF-α, GM-CSF, and LPS express CD80, CD86, and MHC-II, secrete pro-inflammatory mediators (TNF-α, IL-1β, IL-6, IL-12, ROS, and NO), rely on glycolytic metabolism, and contribute to cardiomyocyte injury. M2 macrophages are induced by IL-4, IL-13, TLR ligands, and adenosine, express CD163, CD206, and Arg1, secrete IL-10, TGF-β, VEGF, and chemokines (CCL17, CCL22), utilize oxidative metabolism, and promote anti-inflammation and cardiac repair. (Created with BioRender.com, Toronto, ON, Canada). Abbreviations: ROS, reactive oxygen species; NO, nitric oxide; TME, tumor microenvironment.
This metabolic-immune interaction creates another self-reinforcing pathological cycle that perpetuates disease progression. Inflammation-induced metabolic alterations perpetuate sympathetic overdrive, resulting in additional metabolic dysfunction and inflammation. A compromised cardiac energetic state increases the heart’s vulnerability to additional stressors, creating more opportunities for an inflammatory response. [22]. This metabolic dimension explains why conventional heart failure treatments that target hemodynamic parameters often fail to halt disease progression. Persistent metabolic abnormalities and their inflammatory sequelae drive disease progression, despite improvements in cardiac output and filling pressure [9]. Figure 2 provides a system-level overview of the autonomic and immune circuits involved in the pathogenesis of cardiomyopathy and highlights the key systemic triggers that dysregulate this immuno-neural balance. Targeting these metabolic-immune loops may be essential for halting cardiomyopathy progression, particularly in metabolically vulnerable patient subgroups.
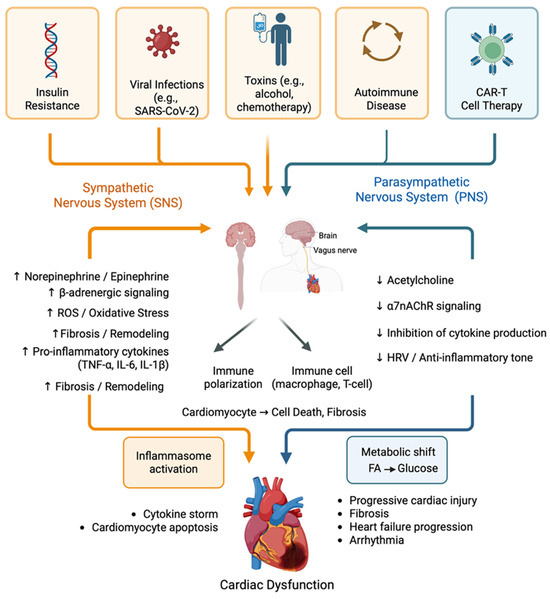
Figure 2.
Integrated schematic of the immunoneural axis in cardiomyopathy. The diagram illustrates the bidirectional interplay between the autonomic nervous system (sympathetic and parasympathetic branches) and the immune system (innate and adaptive) in cardiomyopathy pathogenesis. Systemic triggers, including insulin resistance, viral infections, toxins, autoimmune conditions, and CAR-T cell therapy, initiate dysregulation of this axis. Sympathetic overactivation promotes pro-inflammatory cytokine production (TNF-α, IL-1β, and IL-6), oxidative stress, metabolic dysfunction, and adverse cardiac remodelling. In parallel, parasympathetic withdrawal impairs the cholinergic anti-inflammatory pathway mediated by acetylcholine and α7-nicotinic receptors. These disruptions converge to drive immune polarization, inflammasome activation, cytokine storms, and progressive cardiac injury. The figure highlights the key feedback loops that reinforce neuroimmune dysregulation and create a self-sustaining cycle of inflammation and cardiac dysfunction. (Created with BioRender.com, Toronto, ON, Canada).
3.2. Parasympathetic Withdrawal and Loss of Anti-Inflammatory Control
The loss of parasympathetic restraint is not merely a consequence but also a driver of chronic inflammation in cardiomyopathy [23]. While sympathetic overactivation has received considerable attention, parasympathetic dysfunction is equally critical in the pathogenesis of cardiomyopathy.
3.2.1. Vagal Dysfunction and Cholinergic Anti-Inflammatory Control in Cardiomyopathy Pathophysiology
The vagus nerve mediates systemic anti-inflammatory control by releasing acetylcholine, which activates α7-nicotinic acetylcholine receptors (α7nAChRs) on innate immune cells to suppress pro-inflammatory cytokine production [24]. In cardiomyopathy, anti-inflammatory control is severely compromised. Reduced vagal signaling lowers acetylcholine availability, lifting the suppression of inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, within cardiac macrophages [25]. Clinically, this manifests as reduced heart rate variability (HRV), a surrogate marker of impaired baroreflex sensitivity, which correlates with disease severity and adverse outcomes.
Persistent systemic inflammation damages vagal afferent and efferent fibers, reinforcing dysfunction through a maladaptive feedback loop [26]. This becomes neurologically encoded through maladaptive neuroplasticity, remodelling of cardiac autonomic circuits, and sustaining inflammation, even after the resolution of the initial injury.
3.2.2. Baroreflex Dysfunction and Heart Rate Variability
Heart rate variability (HRV) analysis provides a non-invasive assessment of vagal function through beat-to-beat cardiac rhythm variations. Patients with cardiomyopathy demonstrate significantly reduced HRV, indicating compromised baroreflex sensitivity and impaired vagal anti-inflammatory capacity [27]. This creates another pathological feedback loop, in which diminished vagal tone permits inflammatory activation, whereas inflammatory mediators damage vagal fibers.
3.3. Central Nervous System Integration and Neuro-Immune Signaling
The integration of autonomic and immune responses occurs not only in the heart but also in sophisticated brain circuits that coordinate the body’s response to cardiac stress and inflammation. The central nervous system orchestrates neuroimmune integration through coordinated activity in the brainstem, hypothalamus, and cortical regions, which receive converging inputs from cardiac afferent nerves and circulating cytokines [28]. These central hubs decode peripheral inflammatory signals and modulate downstream autonomic outputs, exerting control over the cardiac electrophysiology. This neuroimmune crosstalk influences ion channel expression, repolarization dynamics, and conduction velocity within the myocardium, establishing a direct mechanistic link between central inflammatory processing and arrhythmogenic susceptibility in cardiomyopathy [29].
3.4. Inflammasome Activation and Pyroptosis
The NLRP3 inflammasome serves as a critical molecular sensor that integrates multiple danger signals related to cardiac stress and autonomic dysfunction, activating specific enzymes that produce mature inflammatory cytokines while simultaneously triggering a form of inflammatory cell death, called pyroptosis [30]. Sympathetic overactivation directly primes the inflammasome machinery, creating a molecular link between autonomic dysfunction and inflammatory activation. Excessive sympathetic β-adrenergic stimulation primes NLRP3 assembly, linking catecholaminergic stress to the amplification of inflammation. Simultaneously, the loss of parasympathetic restraint removes normal cholinergic inhibition of immune cells, further amplifying inflammatory responses.
Metabolic dysfunction, particularly insulin resistance, serves as an additional upstream driver that amplifies this inflammatory axis through multiple mechanisms. Defective insulin signaling in heart muscle cells enhances oxidative stress and mitochondrial dysfunction, both of which promote inflammasome activation. It also elevates free fatty acids in the bloodstream, which act as danger signals that activate immune pattern recognition receptors. Circulating free fatty acids act as DAMPs, activate TLR4, and initiate NF-κB–mediated cytokine expression [31]. Concurrently, advanced glycation end products (AGEs) engage RAGE receptors on cardiac macrophages, sustaining pro-inflammatory signaling and driving maladaptive fibrotic remodelling in diabetic cardiomyopathy.
4. Clinical Phenotypes and Neuro-Immune Signatures
The clinical manifestations of cardiomyopathy are shaped by specific neuroimmune signatures that can guide personalized management strategies.
4.1. Dilated Cardiomyopathy and Chronic Heart Failure
Dilated cardiomyopathy (DCM) is a prototypical neuroimmune disorder characterized by marked autonomic imbalance and systemic inflammation. DCM affects approximately 1:250 individuals globally and is the leading indication for cardiac transplantation [32]. The hallmark pathophysiology involves progressive ventricular dilatation and systolic dysfunction (ejection fraction <40%) driven by myocyte loss, interstitial fibrosis, and compensatory neurohormonal activation [33]. Genetic mutations account for 30–50% of cases, with titin (TTN) truncating variants being the most common cause (15–25%), followed by lamin A/C (LMNA) and desmin mutations [12]. Acquired causes include viral myocarditis (particularly coxsackievirus B3 and parvovirus B19), cardiotoxins (anthracyclines and alcohol), and peripartum cardiomyopathy [34]. Despite advances in neurohormonal blockade, which have achieved a 20–35% reduction in mortality, traditional hemodynamic-focused therapies fail to address the underlying neuroimmune dysfunction [35,36]. The hallmark of this condition is severe sympathetic nervous system overactivation, with norepinephrine levels directly correlating with the degree of heart muscle dysfunction and the patient outcomes [37]. Concurrently, patients display persistently elevated levels of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6, which serve as prognostic biomarkers of disease progression and mortality. Vagal withdrawal further contributes to autonomic dysregulation, as evidenced by significantly reduced heart rate variability (HRV) and impaired baroreflex function, both of which are independent predictors of sudden cardiac death and heart failure-related morbidity. This neuroimmune signature explains why conventional RAAS inhibition and beta-blockade, while providing symptomatic relief, often fail to halt disease progression in 60% of patients with treatment resistance [38].
4.2. Hypertrophic Cardiomyopathy and Genetic Neuro-Immune Interactions
Hypertrophic cardiomyopathy (HCM) affects approximately 1:500 individuals and is characterized by asymmetric left ventricular hypertrophy (wall thickness ≥15 mm) without a hemodynamic cause [39]. Over 1400 mutations in 11 sarcomeric genes have been identified, with MYH7 (β-myosin heavy chain) and MYBPC3 (myosin-binding protein C) accounting for approximately 70% of cases [40]. The pathophysiology centers on sarcomeric protein mutations, causing myofibrillar disarray, diastolic dysfunction, and dynamic outflow obstruction in 70% of cases [41]. Traditional management focuses on symptom control through negative inotropic agents (beta-blockers and calcium channel blockers) and septal reduction therapy; however, these approaches do not prevent fibrotic progression or address arrhythmia risk [42].
HCM presents a unique model of how genetic mutations in heart muscle proteins can trigger neuro-immune dysfunction through biomechanical stress, where inherited mutations in contractile proteins cause disorganized muscle architecture and mechanical stress, which activates local inflammatory responses. This mechanical stress promotes the recruitment of inflammatory cells and the activation of fibroblast cells that produce scar tissue, creating an electrical substrate that predisposes to dangerous arrhythmia [40]. LMNA mutations increase susceptibility to inflammation, elevating interleukin-6, tumor necrosis factor alpha, and macrophage infiltration, even in the early stages of the disease [43]. Autonomic imbalance further compounds this risk, with exaggerated sympathetic activity and impaired vagal modulation fostering an arrhythmogenic neuroimmune milieu that increases susceptibility to sudden cardiac death and adverse electrical remodelling. This mechanistic understanding explains why traditional negative inotropic therapy, while controlling symptoms, fails to modify the underlying inflammatory substrate driving disease progression [44].
4.3. Takotsubo Cardiomyopathy- an Acute Neuro-Immune Crisis
Takotsubo cardiomyopathy (TTC) presents as acute reversible cardiac dysfunction triggered by emotional or physical stress, predominantly affecting postmenopausal women (90% of cases), with plasma norepinephrine levels 2–3-fold higher than those in acute myocardial infarction [45]. TTC accounts for 1–2% of acute coronary syndrome presentations, with in-hospital mortality of 9% and a rate of recurrence reaching 2.9% per year [45]. The characteristic apical ballooning pattern reflects catecholamine-mediated myocardial stunning, with the traditional pathophysiology attributing the syndrome to coronary microvascular dysfunction and direct catecholamine toxicity [46]. Diagnosis follows the modified Mayo Clinic criteria of transient wall motion abnormalities extending beyond a single coronary territory, absence of obstructive coronary disease, and emotional/physical triggers [47]. TTC exemplifies acute neuroimmune dysregulation triggered by extreme emotional or physical stress, leading to a sudden surge in catecholamines. This surge directly damages the myocardial tissue and activates systemic inflammatory responses [48]. Acute-phase features include elevated inflammatory markers, including C-reactive protein, IL-6, and TNF-α, contributing to myocardial stunning and ballooning. This condition demonstrates how acute stress-induced sympathetic overactivation can overwhelm normal cardiac adaptive mechanisms, creating a cascade of neuroimmune activation that manifests as reversible cardiac dysfunction. The predominance in postmenopausal women reflects the loss of estrogen’s cardioprotective effects on autonomic regulation and inflammatory modulation, highlighting the intersection between hormonal status, neuroimmune function, and cardiac vulnerability [49].
4.4. Arrhythmogenic Cardiomyopathy
Arrhythmogenic cardiomyopathy (ACM) affects 1:2,000–5000 individuals, characterized by progressive fibrofatty replacement of the myocardium and a high risk of arrhythmia [50]. Mutations in five desmosomal genes (PKP2, DSG2, DSC2, DSP, JUP) account for 50–60% of cases, with plakophilin-2 (PKP2) most frequently involved [51]. Traditional pathophysiology focused on desmosomal protein dysfunction causing cell–cell adhesion defects and mechanical stress-induced myocyte death [52]. The right ventricle is predominantly affected (classical ARVC), though biventricular and left-dominant variants are increasingly recognized [53]. Exercise restriction is paramount given the role of physical activity in disease progression, though current therapies remain largely supportive. ACM is distinguished by complex neuroimmune pathophysiology driven by mutations in desmosomal proteins, which disrupt structural integrity and initiate inflammatory and autonomic dysregulation [54]. Histologically, ACM is marked by T-lymphocyte and macrophage infiltration, contributing to progressive cardiomyocyte loss and the hallmark fibrofatty myocardial replacement that predisposes patients to malignant arrhythmias [55].
This structural degeneration is accompanied by autonomic remodelling, characterized by abnormal sympathetic innervation and cycles of denervation and re-innervation, creating spatial heterogeneity in neural control. The neuroimmune axis plays a pivotal role in this process, where cytokine signaling influences neural sprouting and survival [56]. Notably, physical exertion amplifies disease progression by intensifying sympathetic output and mechanical stress, which in turn escalates local inflammation via neuro-immune crosstalk, accelerating fibrofatty transformation and arrhythmogenesis [57]. This mechanistic framework explains why exercise restriction remains a cornerstone of management and highlights the need for therapies targeting both the inflammatory pathways and autonomic dysfunction [58].
4.5. Pediatric and Developmental Cardiomyopathy
The incidence of pediatric cardiomyopathy is 0.57–1.13 per 100,000 children, with distinct etiological and prognostic profiles compared to adult disease [59]. Genetic causes predominate (60–70%), including metabolic disorders (glycogen storage diseases, mitochondrial cardiomyopathies) and sarcomeric mutations [60]. Infants present primarily with dilated cardiomyopathy (50% of cases), often secondary to metabolic or genetic disorders, while hypertrophic cardiomyopathy in children is frequently associated with inborn errors of metabolism or malformation syndromes [61]. Traditional management emphasizes heart failure optimization and consideration for mechanical support or transplantation, with 5-year survival rates of 65–75%, though diagnostic workup requires comprehensive metabolic screening and genetic testing [62].
It represents unique neuro-immune dynamics due to age-related differences in immune maturation, autonomic regulation, and genetic predisposition. Children are more susceptible to developing cardiomyopathy secondary to metabolic disorders, genetic syndromes, or post-infectious immune problems, with parvovirus B19 and human herpesvirus 6 being the main viral causes [63]. Multisystem Inflammatory Syndrome in Children (MIS-C) associated with SARS-CoV-2 represents a perfect example of neuro-immune cardiomyopathy, characterized by acute myocarditis and dilated cardiomyopathy through hyperinflammatory cytokine storm involving IL-6, IL-1β, and TNF-α [64,65]. Patients with MIS-C show profound autonomic dysfunction with reduced heart rate variability and elevated sympathetic tone, mirroring adult neuroimmune cardiomyopathy patterns [66]. Recent multicenter studies have revealed that 72% of patients with MIS-C develop low blood pressure and cardiac dysfunction, with 96% showing elevated brain natriuretic peptide levels and 64% demonstrating increased cardiac troponin levels [67]. Heightened neuroplasticity in pediatric autonomic circuits suggests unique opportunities for early intervention through vagal stimulation or targeted immunomodulatory strategies [68].
4.6. COVID-19 and Post-Viral Neuro-Immune Cardiomyopathy
The COVID-19 pandemic has provided unprecedented insight into the mechanisms through which viral infections can trigger persistent neuroimmune cardiac dysfunction. SARS-CoV-2–induced cardiomyopathy arises from a triad of direct viral cardio-tropism, systemic hyperinflammation, and severe autonomic imbalance. Viral entry via ACE2 receptors facilitate myocardial infiltration, and the ensuing cytokine storm exacerbates tissue injury and immune-mediated damage. COVID-19–associated cardiomyopathy affects 0.146% of hospitalized patients, with mortality rates reaching 13.3% [69,70]. The emerging evidence linking human papillomavirus infection with coronary artery disease through shared inflammatory and metabolic pathways, viral triggers such as SARS-CoV-2 may induce chronic cardiovascular vulnerability via immune-metabolic dysregulation [71,72]. The syndrome ranges from acute myocarditis to chronic dilated cardiomyopathy, with the traditional pathophysiology encompassing direct viral cytotoxicity via ACE2 receptors, cytokine storm-mediated injury, and microvascular dysfunction [73]. Acute presentations include fulminant myocarditis requiring mechanical support, whereas chronic manifestations resemble dilated cardiomyopathy. Beyond the acute phase, post-COVID cardiac symptoms affect 10–30% of survivors and are characterized by persistent dyspnea, reduced exercise tolerance, and autonomic dysfunction, including POTS-like syndromes. Cardiac MRI demonstrates persistent inflammatory changes in 60% of recovered patients [74]. This prolonged neuroimmune dysregulation demonstrates how viral triggers can establish persistent pathological circuits that maintain cardiac dysfunction long after viral clearance [75]. The mechanistic parallels between COVID-19 cardiomyopathy and other viral myocarditis suggest common neuroimmune pathways that could be therapeutically targeted across multiple etiologies. Table 1 summarizes the major immuno-neural signaling components implicated in cardiomyopathy pathophysiology, their key mediators, and mechanistic pathways. Shared neuroimmune mechanisms underlie disease progression across all major cardiomyopathy phenotypes, whereas the clinical outcomes remain highly variable. A summary of the etiology, prognosis, and current treatment strategies for each subtype is shown in Figure 3.

Table 1.
Key Neuro-immune Pathways in Cardiomyopathy.
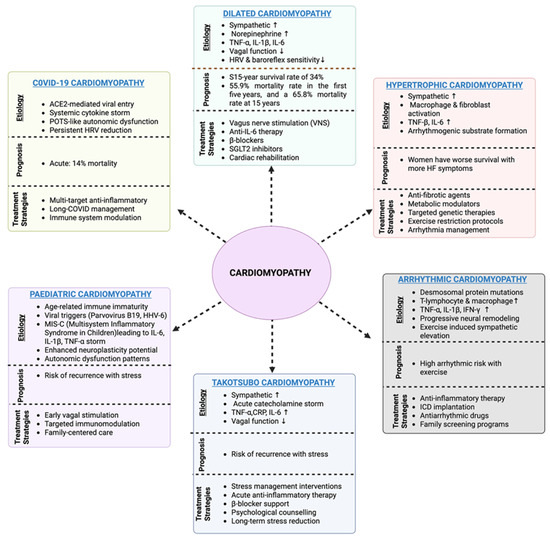
Figure 3.
Overview of cardiomyopathy subtypes, including their characteristic etiologies, prognostic implications, and current treatment strategies. Dilated cardiomyopathy is primarily driven by sympathetic overactivation, inflammatory cytokines, and vagal dysfunction, with poor long-term survival despite neurohormonal blockade therapy. Hypertrophic cardiomyopathy involves macrophage/fibroblast activation and arrhythmogenic remodelling, with a persistent risk of sudden death despite the use of β-blockers and myosin inhibitors. Arrhythmogenic cardiomyopathy is associated with desmosomal mutations, immune cell infiltration, and neural remodelling, with treatment centered on arrhythmia suppression and ICD implantation. Takotsubo cardiomyopathy reflects acute catecholamine surge and autonomic dysfunction, with recurrence risk linked to stress, whereas pediatric cardiomyopathies reflect immune immaturity and viral triggers, requiring age-specific immune modulation. COVID-19 cardiomyopathy combines viral entry via ACE2, cytokine storm, and persistent autonomic dysfunction, with long COVID sequelae complicating management. (Created with BioRender.com, Toronto, ON, Canada).
5. Therapeutic Interventions Targeting the Neuro-Immune Axis
These insights into the neuroimmune architecture set the stage for a deeper understanding of how targeted therapies, both pharmacologic and device-based, can modulate these interconnected systems to reverse or halt disease progression. The complex metabolic-immune-neural interactions described above have profound implications for therapeutic intervention. Rather than targeting individual pathways in isolation, optimal therapeutic strategies must address the interconnected nature of these systems, recognizing that successful intervention requires the simultaneous modulation of multiple nodes within this pathophysiological network.
5.1. Conventional Treatments of Cardiomyopathies and Their Limitations
Conventional management of cardiomyopathies remains phenotype-specific yet largely symptom-directed, with therapies that reduce mortality but fail to address the persistent neuroimmune dysfunction. In dilated cardiomyopathy (DCM), standard neurohormonal blockade with ACE inhibitors/ARBs, β-blockers, and mineralocorticoid receptor antagonists lowers mortality by 20–35% [91], with newer agents such as sacubitril–valsartan (ARNI therapy) and SGLT2 inhibitors offering incremental benefits [92]. Device therapies, including implantable cardioverter-defibrillators (ICDs) and cardiac resynchronization therapy (CRT), improve survival in selected patients [93]. However, up to 60% of patients remain refractory, 25% are readmitted within 30 days, and 5-year mortality exceeds 50% [94], reflecting unresolved inflammatory and autonomic dysregulation [95]. In hypertrophic cardiomyopathy (HCM), β-blockers and calcium channel blockers relieve obstruction, and myectomy or alcohol ablation is used in refractory cases [96]. The advent of mavacamten has provided significant symptomatic improvement [97]; however, fibrosis, inflammatory remodelling, and sudden death risk (1–2% annually) remain unresolved [98,99]. Similarly, arrhythmogenic cardiomyopathy (ACM) is managed with exercise restriction, antiarrhythmics, and ICDs [53,100]; however, disease progression continues with fibrofatty replacement and declining antiarrhythmic efficacy [101].
Other subtypes have similar therapeutic limitations. Takotsubo cardiomyopathy is treated with supportive care, including ACE inhibitors and β-blockers [102]; however, recurrence affects 10% of patients, and cardiogenic shock arises in 10–15% of cases [103], as stress-induced neuroimmune activation remains unaddressed [104]. In pediatric cardiomyopathies, extrapolated adult therapies such as ACE inhibitors, β-blockers, and diuretics [105] are often ineffective, with mechanical support or transplantation required in advanced disease [106]. The 5-year survival rate is only 59% [107], underscoring the unique neuroimmune biology of developing hearts. Finally, COVID-19–related cardiomyopathy is managed with conventional heart failure therapies and, in severe inflammatory cases, with corticosteroids or immunosuppression [108]. However, the combined effects of viral cardio-tropism, cytokine storm, and autonomic dysfunction generate sequelae that cannot be resolved by conventional treatments, including long-term autonomic dysregulation and exercise intolerance [109].
Together, these limitations reveal a shared gap across different cardiomyopathy phenotypes. Although conventional therapies improve hemodynamics and reduce mortality, they fail to disrupt the persistent inflammatory and autonomic circuits that drive disease progression. This therapeutic inadequacy highlights the urgent need for neuroimmune-targeted intervention.
5.2. Beyond Hemodynamics and Neuro-Immune Actions of Traditional Therapies
Traditional cardiovascular therapies exhibit pleiotropic effects that extend beyond their primary pharmacological targets. Beta-adrenergic blockers not only suppress sympathetic overactivation but also exert direct immunomodulatory effects by downregulating pro-inflammatory cytokine production, independent of their hemodynamic benefits. [110]. Similarly, renin–angiotensin–aldosterone system (RAAS) inhibitors confer neuro-immune advantages by enhancing vagal tone and interrupting angiotensin II-mediated inflammatory pathways, thereby contributing to autonomic rebalancing and myocardial immune quiescence in cardiomyopathy. These pleiotropic effects highlight the importance of reevaluating standard heart failure therapies through a neuroimmune lens.
5.3. Emerging Metabolic-Immune Modulators
Interestingly, a new class of medications originally developed for diabetes has emerged as powerful neuro-immune modulators in heart failure, exemplifying how targeting metabolic dysfunction can restore autonomic and immune balance. Metabolic modulators have emerged as potent neuro-immune therapies, with SGLT2 inhibitors exemplifying this therapeutic approach. Beyond glycemic control, they reduce oxidative stress, restore mitochondrial function, and suppress inflammation, targeting metabolic-immune-neural dysfunction [111]. Clinical evidence shows that SGLT2 inhibitors enhance vagal tone and decrease systemic inflammation in heart failure, independent of glucose lowering, indicating direct neuroimmune modulation [112,113]. This metabolic approach represents a significant advancement as it demonstrates how restoring cellular energy production can simultaneously improve autonomic balance, reduce inflammation, and promote beneficial cardiac remodelling.
5.4. Targeted Anti-Inflammatory Interventions
Building on our understanding of inflammatory pathways in cardiomyopathy, a new generation of therapies specifically targets immune dysfunction while simultaneously restoring autonomic function. The inflammatory arm of the neuroimmune axis has emerged as a compelling therapeutic target, and interventions that modulate both immune activation and autonomic function are gaining traction in clinical settings. Interleukin-1β antagonists are used because IL-1β is a key cytokine released during neuroimmune activation in cardiomyopathy. By blocking IL-1β, these drugs aim to disrupt the inflammatory loop, which is fueled by sympathetic overdrive. Interleukin-1 receptor antagonists reduce systemic inflammation while concurrently enhancing autonomic tone, as reflected by improved heart rate variability and reduced cardiovascular events [114]. These dual benefits demonstrate that immunomodulation can serve not only as an anti-inflammatory therapy but also as a method to rebalance neural circuits that control cardiac function.
Colchicine, a medication with ancient origins in the treatment of gout, has gained renewed attention in cardiology because of its ability to target specific inflammatory pathways that intersect with autonomic control mechanisms. Recent studies have highlighted the efficacy of the ketogenic diet in lowering inflammatory biomarkers and improving heart rate variability, underscoring its relevance when applied with precision to neuroimmune dysregulation [18]. These findings support a new therapeutic paradigm in which anti-inflammatory treatments serve not only as symptom reducers but also as comprehensive modulators of the neuroimmune interface.
5.5. Clinical Implications and Future Directions
Rather than targeting individual pathways in isolation, optimal therapeutic strategies must address the interconnected nature of these systems, recognizing that successful intervention requires simultaneous modulation of multiple nodes within this pathophysiological network. As illustrated in Figure 4, these therapeutic targets span the sympathetic and parasympathetic nervous systems, the immune interface, and central autonomic regulation, reflecting the need for integrated neuroimmune modulation in cardiomyopathy. These therapeutic innovations represent a shift from symptom management to targeting the underlying neuroimmune mechanisms driving cardiomyopathy. Integrating metabolic, autonomic, and immune-based strategies reflects a systems biology approach that enables more precise mechanism-guided interventions [115]. This understanding is relevant not only for cardiovascular conditions but also for systemic immune disorders, underscoring the need for therapies that address the neuroimmune axis.
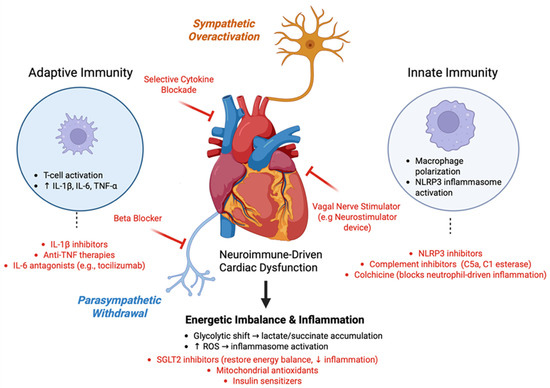
Figure 4.
Therapeutic Targets within the Neuroimmune Network. The figure illustrates the multidimensional therapeutic landscape of targeting the neuroimmune axis in cardiomyopathy. Sympathetic overactivation promotes inflammation via β-adrenergic signaling and cytokine release, which can be addressed by β-blockers and selective cytokine blockade. Adaptive immune activation, characterized by T-cell-mediated elevation of IL-1β, IL-6, and TNF-α, is targeted through monoclonal therapies, including IL-1β inhibitors, anti-TNF agents, and IL-6 antagonists such as tocilizumab. Concurrent parasympathetic withdrawal reduces vagal tone and cholinergic anti-inflammatory signaling, which is countered by vagal nerve stimulation. Innate immune dysregulation, including macrophage polarization and NLRP3 inflammasome activation, can be mitigated by NLRP3 inhibitors, complement inhibitors (e.g., C5a and C1 esterase), and colchicine. At the metabolic level, glycolytic shift and reactive oxygen species (ROS) drive further inflammation, which can be addressed by SGLT2 inhibitors, mitochondrial antioxidants, insulin sensitizers, and anti-inflammatory metabolic interventions. Together, these targeted therapies form a systems-based approach to restore the neuroimmune balance and prevent the progression of cardiac dysfunction. (Created with BioRender.com, Toronto, Ontario, Canada).
5.6. Targeted Immunotherapy
The therapeutic landscape of cardiomyopathy is shifting toward precision immunotherapy, moving beyond broad-spectrum suppression to targeted modulation of disease-driving inflammatory pathways. Interleukin-1β inhibition with canakinumab, a monoclonal antibody, has demonstrated significant cardiovascular benefits by attenuating systemic and myocardial inflammation [116]. Although its initial use was outside cardiology, its relevance in cardiomyopathy is increasingly recognized, warranting dedicated trials to define its utility in cardiac-specific inflammation [31,117].
Targeting tumor necrosis factor alpha (TNF-α) has yielded mixed results; early nonspecific blockade was largely ineffective [118], but emerging evidence suggests that subtype-specific or temporally precise TNF-α inhibition may benefit selected patients [119]. Interleukin-6 (IL-6) blockade, particularly via tocilizumab, has shown promise in inflammatory cardiomyopathies, such as myocarditis, in which IL-6 plays a dominant role [120]. In such settings, selective interruption of IL-6 signaling may be both protective and disease-modifying [121].
Inhibition of the complement system has emerged as a significant area of investigation in addressing myocardial inflammation across various cardiomyopathy phenotypes. The complement system, a key element of innate immunity, plays a pivotal role in mediating myocardial inflammation, as supported by evidence indicating its involvement in ischemia/reperfusion (I/R) injury and myocardial infarction [122,123]. Targeted complement blockade has been shown to disrupt self-perpetuating inflammatory loops that exacerbate cardiac dysfunction [124]. Specifically, inhibitors of complement components, such as C1 esterase inhibitors and C5a receptor antagonists, are currently being investigated for their potential to mitigate complement-mediated myocardial injury [125].
The complement cascade is central to the initial immune response, driving inflammatory processes within the myocardium, which affect various cardiomyopathy phenotypes. Research has demonstrated that Complement activation correlates with increased myocardial tissue damage during I/R, reinforcing the rationale for targeted complement inhibition as a therapeutic strategy [123,126]. Notably, specific agents targeting the complement system have demonstrated efficacy in preclinical models, supporting the potential clinical applications of immunotherapeutic interventions focused on tailored complement inhibition [127].
Moreover, the evolving landscape of treatment strategies underscores the urgent need for biomarker-driven patient stratification. As immunotherapy is integrated into cardiology, the success of such interventions will depend on accurately identifying the target populations, as highlighted in recent studies [128,129]. Effective selection based on immunological profiling and mechanistic understanding will be critical for optimizing patient outcomes in cardiomyopathy, suggesting that precise immunological metrics will serve as the cornerstone for advancing personalized therapeutic approaches.
5.7. Bioelectronic Medicine: The Future of Heart Healing
Bioelectronic medicine offers a novel approach to restoring the neuroimmune balance through autonomic modulation. Vagus nerve stimulation (VNS), which enhances parasympathetic tone and suppresses inflammatory cytokines, has improved the quality of life and exercise capacity in patients with HF, although mortality benefits remain uncertain. Clinical trials, including INOVATE HF, have demonstrated these benefits despite variability in long-term outcomes [80,130].
Non-invasive transcutaneous VNS provides autonomic and immunomodulatory effects while minimizing procedural risks. Baroreflex activation therapy (BAT) and thoracic spinal cord stimulation further expand therapeutic options by targeting sympathetic overdrive and restoring autonomic equilibrium [131].
5.8. Integrated Therapeutic Approaches–Healing Beyond Medication
Contemporary cardiomyopathy treatment paradigms have evolved to incorporate evidence-based lifestyle and behavioral interventions as adjunctive therapies to conventional pharmacological approaches. This integrated strategy recognizes the multifactorial nature of cardiomyopathy pathophysiology and targets both the direct cardiac effects and systemic modulatory pathways of cardiomyopathy.
5.8.1. Exercise-Based Cardiac Rehabilitation
Structured cardiac rehabilitation with supervised exercise training is a foundational component of cardiomyopathy management, supported by Level I evidence for key clinical outcomes. Meta analyses of randomized controlled trials consistently show that exercise interventions significantly improve peak oxygen consumption (VO2max), six-minute walk distance, and quality of life, with effect sizes ranging from 0.4 to 0.8 standard deviations compared to standard care [132]. These benefits are driven by multiple physiological mechanisms, including improved cardiac output, enhanced peripheral oxygen extraction, and favorable neurohormonal adaptation. Exercise increases heart rate variability, reduces sympathetic hyperactivation, and lowers inflammatory biomarkers such as C-reactive protein, interleukin 6, and TNF α [133]. These changes reflect integrated improvements in endothelial function, parasympathetic tone and modulation of the renin–angiotensin–aldosterone system.
5.8.2. Psychosocial Interventions and Stress Management
The bidirectional relationship between psychological stress and cardiomyopathy progression has prompted investigations into targeted psychosocial interventions in patients with cardiomyopathy. Systematic reviews indicate that cognitive-behavioral therapy, mindfulness-based stress reduction, and structured relaxation techniques may provide modest but clinically relevant benefits in selected cardiomyopathy populations. These interventions appear to operate through stress-mediated pathways involving hypothalamic–pituitary–adrenal axis modulation and a reduction in chronic inflammatory states [134]. However, the evidence base remains heterogeneous, with significant variability in the intervention protocols, outcome measures, and patient populations across studies. Further research is needed to establish the optimal intervention characteristics, identify patients who are most likely to benefit, and determine long-term clinical outcomes.
Lifestyle and behavioral strategies are increasingly recognized as vital, cost-effective approaches to modulate the neuro-immune axis in cardiomyopathy, enhancing disease management and patient outcomes [135]. Structured cardiac rehabilitation, with supervised exercise training, improves autonomic balance and reduces inflammation, as evidenced by enhanced heart rate variability, diminished sympathetic activity, and lower pro-inflammatory cytokines, highlighting physical activity as a key neuro-immune modulator [136]. Psychological stress reduction techniques, including cognitive behavioral therapy, mindfulness, and relaxation, show potential in attenuating neuroendocrine stress responses and their inflammatory consequences; however, further research is warranted to clarify their clinical impact on cardiomyopathy [137].
Emerging evidence on the gut–brain-heart axis suggests that targeted microbiome modulation through probiotics and dietary interventions may restore autonomic function and suppress systemic inflammation, offering innovative avenues to address the complex crosstalk between gut microbiota, neural pathways, and cardiac health [138]. Table 2 provides a comparative overview of the clinical therapies targeting the immuno-neural axis.
Table 2.
Clinical Interventions Targeting the Neuro-Immune Axis.
6. Precision Medicine and Future Directions
6.1. Biomarker Development
Comprehensive neuroimmune profiling in cardiomyopathy requires tools that assess both autonomic integrity and inflammatory status. Heart rate variability (HRV) and baroreflex sensitivity offer non-invasive insights into vagal tone and physiological adaptability, correlating strongly with disease outcomes [142]. Although microneurography remains the most precise method for assessing sympathetic activity, its invasiveness limits its clinical use [143]. Emerging tools now focus on combining wearable biosensors with cytokine profiling, enabling dynamic, real-time assessment of neuroimmune dysfunction for personalized therapeutic adaptation.
6.2. Genetic and Molecular Insights into Neuro-Immune Susceptibility
Genetic variations play a substantial role in determining individual susceptibility to neuro-immune dysfunction and therapeutic responsiveness in cardiomyopathy patients. LMNA mutations increase susceptibility to inflammation, elevating interleukin 6, tumor necrosis factor alpha, and macrophage infiltration, even in early disease stages [144]. These mutations disrupt nuclear envelope integrity, activating the cGAS-STING pathway and type I interferon responses, which promote cardiac inflammation [145]. Truncating variants in TTN impair calcium handling and mitochondrial function, thereby activating the NLRP3 inflammasome and driving fibrosis [146]. Polymorphisms in adrenergic receptors modulate sympathetic responsiveness, influencing arrhythmia risk and clinical outcomes [147]. Epigenetic regulators, such as microRNAs 155, 21, and 223, influence macrophage polarization and neurogenic inflammation, with microRNA 155 regulating sympathetic remodelling [148]. Single-cell RNA sequencing has revealed distinct inflammatory macrophage populations in genetic cardiomyopathy [149]. The integration of polygenic risk scores with autonomic function and inflammatory biomarkers enables personalized risk stratification and targeted neuroimmune therapies. Harnessing genetic and molecular insights can help delineate distinct neuroimmune phenotypes and identify actionable therapeutic targets.
6.3. Sex Based Considerations
Sex differences profoundly affect the risk, progression, and therapeutic response of cardiomyopathy through distinct immune and autonomic profiles in patients with cardiomyopathy. Females typically exhibit stronger humoral and T helper 1 immune responses, predisposing them to autoimmune myocarditis, whereas males display higher sympathetic tone and increased susceptibility to arrhythmias [150]. Female patients with hypertrophic cardiomyopathy demonstrate better survival but increased heart failure symptoms, reflecting divergent neuroimmune adaptations [151]. The predominance of females with Takotsubo cardiomyopathy is mechanistically linked to estrogen withdrawal and altered limbic autonomic regulation [152]. Estrogen receptor beta signaling enhances parasympathetic tone and suppresses nuclear factor kappa B-mediated inflammation, providing cardio protection [153]. In contrast, testosterone promotes pro-inflammatory responses and oxidative stress, accelerating cardiac remodelling in men [154].
6.4. Inflammatory Cardiomyopathy as a Spectrum of Immune and Neural Dysfunction
Inflammatory cardiomyopathies, including viral myocarditis, cardiac sarcoidosis, eosinophilic myocarditis, and peripartum cardiomyopathy, share common features of immune infiltration, dysregulated cytokine networks, and autonomic impairment. Viral myocarditis is characterized by T helper 1 and T helper 17 immune dominance with elevated interferon gamma, interleukin 17, and interleukin 1 beta levels that damage parasympathetic nerves [155]. Cardiac sarcoidosis involves granulomatous infiltration preferentially affecting conduction tissues, resulting in arrhythmias driven by macrophage-derived interleukin 12, interleukin 18, and tumor necrosis factor alpha-mediated sympathetic activation [156]. Peripartum cardiomyopathy, affecting one in one thousand to four thousand pregnancies, involves oxidative stress-induced cytokine activation, notably cleaved prolactin fragments that promote endothelial dysfunction and cardiomyocyte apoptosis [157]. These patients show elevated sympathetic drive and reduced parasympathetic function with autonomic status correlating with recovery potential.
Therapeutic strategies increasingly target both immune dysregulation and autonomic imbalance. For instance, bromocriptine modulates prolactin in peripartum cardiomyopathy alongside immunosuppressive therapy, while cardiac sarcoidosis treatment combines immunosuppression with autonomic support. Addressing both inflammation and neural remodelling is essential to optimize clinical outcomes. Table 3 summarizes the neuroimmune signatures across cardiomyopathy types, highlighting the differences in sympathetic tone, cytokine expression, and autonomic dysfunction.
Table 3.
Comparative Immuno-Neural Features Across Cardiomyopathy Types.
7. Conclusions
The neuroimmune axis is no longer a peripheral player in cardiomyopathy but a central determinant of disease progression, therapeutic resistance, and clinical outcomes. Autonomic dysfunction and immune dysregulation are not isolated phenomena but constitute an interwoven pathological network spanning all cardiomyopathy phenotypes. Sympathetic hyperactivation perpetuates chronic inflammation, whereas parasympathetic withdrawal dismantles endogenous anti-inflammatory control, fostering a self-sustaining cycle of cardiac injury. Conventional therapies targeting these systems in isolation are increasingly ineffective. Novel approaches, such as selective cytokine blockade and bioelectronic neuromodulation, are redefining the therapeutic landscape by modulating immune activity via the nervous system. In parallel, integrative omics platforms and real-time neuroimmune monitoring enhance patient stratification and enable dynamic treatment adaptations.
To fundamentally shift the trajectory of cardiomyopathy, future strategies must address inflammasome activation, autonomic remodelling, and metabolic immune crosstalk in a synchronized and personalized manner. The convergence of bioelectronic medicine, immunotherapy, and systems-level diagnostics holds the potential to not only delay disease progression but also restore physiological homeostasis. Emerging technologies, such as adaptive neuro-modulatory devices guided by immune biomarkers and single-cell immune profiling for individualized circuit mapping, may finally bridge the gap between mechanistic insight and clinical translation, transforming heart failure care from reactive to precisely anticipatory. This new framework positions the neuroimmune axis not as an accessory, but as a central node of pathogenesis and an essential therapeutic target in the future of cardiomyopathy care.
Author Contributions
This manuscript was conceptualized by D.S., P.D. and A.C., and planning and discussions were conducted collaboratively by all authors. All authors participated in writing the initial draft of this manuscript. A.C. supervised the project, provided resources, and led the review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research did not receive any external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The dataset supporting the findings of this study is included in this manuscript and its referenced sources, ensuring comprehensive access to the relevant data for further examination and analyses.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Savarese, G.; Becher, P.M.; Lund, L.H.; Seferovic, P.; Rosano, G.M.C.; Coats, A.J.S. Global burden of heart failure: A comprehensive and updated review of epidemiology. Cardiovasc Res. 2023, 118, 3272–3287. [Google Scholar] [CrossRef] [PubMed]
- Sapna, F.; Raveena, F.; Chandio, M.; Bai, K.; Sayyar, M.; Varrassi, G.; Khatri, M.; Kumar, S.; Mohamad, T. Advancements in Heart Failure Management: A Comprehensive Narrative Review of Emerging Therapies. Cureus. 2023, 15, e46486. [Google Scholar] [CrossRef] [PubMed]
- Riehle, C.; Abel, E.D. Insulin Signaling and Heart Failure. Circ. Res. 2016, 118, 1151–1169. [Google Scholar] [CrossRef] [PubMed]
- Khosravirad, Z.; Rostamzadeh, M.; Azizi, S.; Khodashenas, M.; Khodadoustan Shahraki, B.; Ghasemi, F.; Ghorbanzadeh, M. The Efficacy of Self-care Behaviors, Educational Interventions, and Follow-up Strategies on Hospital Readmission and Mortality Rates in Patients with Heart Failure. Galen Med. J. 2023, 12, e3116. [Google Scholar] [CrossRef]
- Carnevale, D. Neuroimmune axis of cardiovascular control: Mechanisms and therapeutic implications. Nat. Rev. Cardiol. 2022, 19, 379–394. [Google Scholar] [CrossRef]
- Koc, A.; Cagavi, E. Cardiac Immunology: A New Era for Immune Cells in the Heart. Adv. Exp. Med. Biol. 2021, 1312, 75–95. [Google Scholar] [CrossRef]
- Bahrar, H.; Bekkering, S.; Stienstra, R.; Netea, M.G.; Riksen, N.P. Innate immune memory in cardiometabolic disease. Cardiovasc. Res. 2024, 119, 2774–2786. [Google Scholar] [CrossRef]
- Asatryan, B.; Asimaki, A.; Landstrom, A.P.; Khanji, M.Y.; Odening, K.E.; Cooper, L.T.; Marchlinski, F.E.; Gelzer, A.R.; Semsarian, C.; Reichlin, T.; et al. Inflammation and Immune Response in Arrhythmogenic Cardiomyopathy: State-of-the-Art Review. Circulation 2021, 144, 1646–1655. [Google Scholar] [CrossRef]
- Cohen, C.D.; Rousseau, S.T.; Bermea, K.C.; Bhalodia, A.; Lovell, J.P.; Zita, M.D.; Cihakova, D.; Adamo, L. Myocardial Immune Cells: The Basis of Cardiac Immunology. J. Immunol. 2023, 210, 1198–1207. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic nervous system in heart failure: Pathophysiology and therapy. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef]
- Ziegler, K.A.; Engelhardt, S.; Carnevale, D.; McAlpine, C.S.; Guzik, T.J.; Dimmeler, S.; Swirski, F.K. Neural Mechanisms in Cardiovascular Health and Disease. Circ. Res. 2025, 136, 1233–1261. [Google Scholar] [CrossRef]
- Gigli, M.; Stolfo, D.; Merlo, M.; Sinagra, G.; Taylor, M.R.G.; Mestroni, L. Pathophysiology of dilated cardiomyopathy: From mechanisms to precision medicine. Nat. Rev. Cardiol. 2025, 22, 183–198. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Anderson, A.S. The sympathetic nervous system and heart failure. Cardiol. Clin. 2014, 32, 33–45. [Google Scholar] [CrossRef]
- Corbi, G.; Conti, V.; Russomanno, G.; Longobardi, G.; Furgi, G.; Filippelli, A.; Ferrara, N. Adrenergic signaling and oxidative stress: A role for sirtuins? Front. Physiol. 2013, 4, 324. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Bazoukis, G.; Stavrakis, S.; Armoundas, A.A. Vagus Nerve Stimulation and Inflammation in Cardiovascular Disease: A State-of-the-Art Review. J. Am. Heart Assoc. 2023, 12, e030539. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Wang, H.; Czura, C.J.; Friedman, S.G.; Tracey, K.J. The cholinergic anti-inflammatory pathway: A missing link in neuroimmunomodulation. Mol. Med. 2003, 9, 125–134. [Google Scholar] [CrossRef]
- Olivieri, F.; Biscetti, L.; Pimpini, L.; Pelliccioni, G.; Sabbatinelli, J.; Giunta, S. Heart rate variability and autonomic nervous system imbalance: Potential biomarkers and detectable hallmarks of aging and inflammaging. Ageing Res. Rev. 2024, 101, 102521. [Google Scholar] [CrossRef]
- Bellocchi, C.; Carandina, A.; Montinaro, B.; Targetti, E.; Furlan, L.; Rodrigues, G.D.; Tobaldini, E.; Montano, N. The Interplay between Autonomic Nervous System and Inflammation across Systemic Autoimmune Diseases. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef]
- Paulus, W.J.; Zile, M.R. From Systemic Inflammation to Myocardial Fibrosis: The Heart Failure With Preserved Ejection Fraction Paradigm Revisited. Circ. Res. 2021, 128, 1451–1467. [Google Scholar] [CrossRef]
- Pongratz, G.; Straub, R.H. The sympathetic nervous response in inflammation. Arthritis Res. Ther. 2014, 16, 504. [Google Scholar] [CrossRef] [PubMed]
- Wróbel-Nowicka, K.; Wojciechowska, C.; Jacheć, W.; Zalewska, M.; Romuk, E. The Role of Oxidative Stress and Inflammatory Parameters in Heart Failure. Medicina 2024, 60, 760. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, E.M. Neural regulation of innate immunity: A coordinated nonspecific host response to pathogens. Nat. Rev. Immunol. 2006, 6, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Huston, J.M.; Tracey, K.J. The pulse of inflammation: Heart rate variability, the cholinergic anti-inflammatory pathway and implications for therapy. J. Intern. Med. 2011, 269, 45–53. [Google Scholar] [CrossRef]
- Wu, Z.; Liao, J.; Liu, Q.; Zhou, S.; Chen, M. Chronic vagus nerve stimulation in patients with heart failure: Challenge or failed translation? Front. Cardiovasc. Med. 2023, 10, 1052471. [Google Scholar] [CrossRef]
- Komegae, E.N.; Farmer, D.G.S.; Brooks, V.L.; McKinley, M.J.; McAllen, R.M.; Martelli, D. Vagal afferent activation suppresses systemic inflammation via the splanchnic anti-inflammatory pathway. Brain Behav. Immun. 2018, 73, 441–449. [Google Scholar] [CrossRef]
- Siepmann, M.; Weidner, K.; Petrowski, K.; Siepmann, T. Heart Rate Variability: A Measure of Cardiovascular Health and Possible Therapeutic Target in Dysautonomic Mental and Neurological Disorders. Appl. Psychophysiol Biofeedback 2022, 47, 273–287. [Google Scholar] [CrossRef]
- Mohanta, S.K.; Yin, C.; Weber, C.; Godinho-Silva, C.; Veiga-Fernandes, H.; Xu, Q.J.; Chang, R.B.; Habenicht, A.J.R. Cardiovascular Brain Circuits. Circ. Res. 2023, 132, 1546–1565. [Google Scholar] [CrossRef]
- Yang, J.; Chen, S.; Duan, F.; Wang, X.; Zhang, X.; Lian, B.; Kou, M.; Chiang, Z.; Li, Z.; Lian, Q. Mitochondrial Cardiomyopathy: Molecular Epidemiology, Diagnosis, Models, and Therapeutic Management. Cells 2022, 11, 3511. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef]
- Lu, L.; Ahmed, F.S.; Akin, O.; Luk, L.; Guo, X.; Yang, H.; Yoon, J.; Hakimi, A.A.; Schwartz, L.H.; Zhao, B. Uncontrolled Confounders May Lead to False or Overvalued Radiomics Signature: A Proof of Concept Using Survival Analysis in a Multicenter Cohort of Kidney Cancer. Front. Oncol. 2021, 11, 638185. [Google Scholar] [CrossRef]
- Ware, J.S.; Cook, S.A. Role of titin in cardiomyopathy: From DNA variants to patient stratification. Nat. Rev. Cardiol. 2018, 15, 241–252. [Google Scholar] [CrossRef]
- Mahmaljy, H.; Yelamanchili, V.S.; Singhal, M. Dilated Cardiomyopathy; StatPearls Publishing: Orlando, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK441911/ (accessed on 18 July 2025).
- Yoshikawa, T. Contribution of acquired factors to the pathogenesis of dilated cardiomyopathy—The cause of dilated cardiomyopathy: Genetic or acquired? (Acquired-Side). Circ. J. 2011, 75, 1766–1773. [Google Scholar] [CrossRef]
- Khattab, E.; Myrianthefs, M.M.; Sakellaropoulos, S.; Alexandrou, K.; Mitsis, A. Precision medicine applications in dilated cardiomyopathy: Advancing personalized care. Curr. Probl. Cardiol. 2025, 50, 103076. [Google Scholar] [CrossRef] [PubMed]
- Bakalakos, A.; Ritsatos, K.; Anastasakis, A. Current perspectives on the diagnosis and management of dilated cardiomyopathy Beyond heart failure: A Cardiomyopathy Clinic Doctor’s point of view. Hellenic J. Cardiol. 2018, 59, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, H.P.; Fairweather, D.; Caforio, A.L.P.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated cardiomyopathy. Nat. Rev. Dis. Primers 2019, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Chen, C.; Wu, S.; Chen, J.; Han, Y.; Zhu, Q.; Xu, M.; Nie, Q.; Wang, L. Mechanism of angiotensin-converting enzyme inhibitors in the treatment of dilated cardiomyopathy based on a protein interaction network and molecular docking. Cardiovasc. Diagn. Ther. 2023, 13, 534–549. [Google Scholar] [CrossRef]
- Antunes, M.O.; Scudeler, T.L. Hypertrophic cardiomyopathy. Int. J. Cardiol. Heart Vasc. 2020, 27, 100503. [Google Scholar] [CrossRef]
- Glavaski, M.; Velicki, L.; Vucinic, N. Hypertrophic Cardiomyopathy: Genetic Foundations, Outcomes, Interconnections, and Their Modifiers. Medicina 2023, 59. [Google Scholar] [CrossRef]
- Nishimura, R.A.; Seggewiss, H.; Schaff, H.V. Hypertrophic Obstructive Cardiomyopathy: Surgical Myectomy and Septal Ablation. Circ. Res. 2017, 121, 771–783. [Google Scholar] [CrossRef]
- Rajan, D.; Zorner, C.R.; Hansen, M.L.; Tfelt-Hansen, J. Arrhythmias and Sudden Death: What is New in Hypertrophic Cardiomyopathy? Card. Fail. Rev. 2025, 11, e08. [Google Scholar] [CrossRef] [PubMed]
- Lillo, R.; Graziani, F.; Franceschi, F.; Iannaccone, G.; Massetti, M.; Olivotto, I.; Crea, F.; Liuzzo, G. Inflammation across the spectrum of hypertrophic cardiac phenotypes. Heart Fail. Rev. 2023, 28, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Maron, M.S.; Sherrid, M.V.; Rowin, E.J. Future Role of New Negative Inotropic Agents in the Era of Established Surgical Myectomy for Symptomatic Obstructive Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2022, 11, e024566. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, E.; Bairey Merz, C.N.; Mehta, P.K. Takotsubo Cardiomyopathy. Eur. Cardiol. 2015, 10, 25–30. [Google Scholar] [CrossRef]
- Komamura, K.; Fukui, M.; Iwasaku, T.; Hirotani, S.; Masuyama, T. Takotsubo cardiomyopathy: Pathophysiology, diagnosis and treatment. World J. Cardiol. 2014, 6, 602–609. [Google Scholar] [CrossRef]
- Buchmann, S.J.; Lehmann, D.; Stevens, C.E. Takotsubo Cardiomyopathy-Acute Cardiac Dysfunction Associated with Neurological and Psychiatric Disorders. Front. Neurol. 2019, 10, 917. [Google Scholar] [CrossRef]
- Barmore, W.; Patel, H.; Harrell, S.; Garcia, D.; Calkins, J.B., Jr. Takotsubo cardiomyopathy: A comprehensive review. World J. Cardiol. 2022, 14, 355–362. [Google Scholar] [CrossRef]
- Kuo, B.T.; Choubey, R.; Novaro, G.M. Reduced estrogen in menopause may predispose women to takotsubo cardiomyopathy. Gend. Med. 2010, 7, 71–77. [Google Scholar] [CrossRef]
- Richter, T.; Nestler-Parr, S.; Babela, R.; Khan, Z.M.; Tesoro, T.; Molsen, E.; Hughes, D.A. Rare Disease Terminology and Definitions—A Systematic Global Review: Report of the ISPOR Rare Disease Special Interest Group. Value. Health 2015, 18, 906–914. [Google Scholar] [CrossRef]
- Kyriakopoulou, E.; Versteeg, D.; de Ruiter, H.; Perini, I.; Seibertz, F.; Doring, Y.; Zentilin, L.; Tsui, H.; van Kampen, S.J.; Tiburcy, M.; et al. Therapeutic efficacy of AAV-mediated restoration of PKP2 in arrhythmogenic cardiomyopathy. Nat. Cardiovasc. Res. 2023, 2, 1262–1276. [Google Scholar] [CrossRef]
- Basso, C.; Bauce, B.; Corrado, D.; Thiene, G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2011, 9, 223–233. [Google Scholar] [CrossRef]
- Migliore, F.; Mattesi, G.; Zorzi, A.; Bauce, B.; Rigato, I.; Corrado, D.; Cipriani, A. Arrhythmogenic Cardiomyopathy-Current Treatment and Future Options. J. Clin. Med. 2021, 10. [Google Scholar] [CrossRef]
- Fan, X.; Yang, G.; Duru, F.; Grilli, M.; Akin, I.; Zhou, X.; Saguner, A.M.; Ei-Battrawy, I. Arrhythmogenic Cardiomyopathy: From Preclinical Models to Genotype-phenotype Correlation and Pathophysiology. Stem. Cell Rev. Rep. 2023, 19, 2683–2708. [Google Scholar] [CrossRef]
- Maione, A.S.; Pilato, C.A.; Casella, M.; Gasperetti, A.; Stadiotti, I.; Pompilio, G.; Sommariva, E. Fibrosis in Arrhythmogenic Cardiomyopathy: The Phantom Thread in the Fibro-Adipose Tissue. Front. Physiol. 2020, 11, 279. [Google Scholar] [CrossRef] [PubMed]
- Chavan, S.S.; Pavlov, V.A.; Tracey, K.J. Mechanisms and Therapeutic Relevance of Neuro-immune Communication. Immunity 2017, 46, 927–942. [Google Scholar] [CrossRef] [PubMed]
- Kohela, A.; van Rooij, E. Fibro-fatty remodelling in arrhythmogenic cardiomyopathy. Basic Res. Cardiol. 2022, 117, 22. [Google Scholar] [CrossRef] [PubMed]
- Meraviglia, V.; Alcalde, M.; Campuzano, O.; Bellin, M. Inflammation in the Pathogenesis of Arrhythmogenic Cardiomyopathy: Secondary Event or Active Driver? Front. Cardiovasc. Med. 2021, 8, 784715. [Google Scholar] [CrossRef]
- Malinow, I.; Fong, D.C.; Miyamoto, M.; Badran, S.; Hong, C.C. Pediatric dilated cardiomyopathy: A review of current clinical approaches and pathogenesis. Front. Pediatr. 2024, 12, 1404942. [Google Scholar] [CrossRef]
- Gerull, B.; Brodehl, A. Insights Into Genetics and Pathophysiology of Arrhythmogenic Cardiomyopathy. Curr. Heart Fail. Rep. 2021, 18, 378–390. [Google Scholar] [CrossRef]
- Lodato, V.; Parlapiano, G.; Cali, F.; Silvetti, M.S.; Adorisio, R.; Armando, M.; El Hachem, M.; Romanzo, A.; Dionisi-Vici, C.; Digilio, M.C.; et al. Cardiomyopathies in Children and Systemic Disorders When Is It Useful to Look beyond the Heart? J. Cardiovasc. Dev. Dis. 2022, 9. [Google Scholar] [CrossRef]
- Iezzi, L.; Sorella, A.; Galanti, K.; Gallina, S.; Chahal, A.A.; Bauce, B.; Cipriani, A.; Providencia, R.; Lopes, L.R.; Ricci, F.; et al. Arrhythmogenic cardiomyopathy diagnosis and management: A systematic review of clinical practice guidelines and recommendations with insights for future research. Eur. Heart J. Qual. Care. Clin. Outcomes 2025. [Google Scholar] [CrossRef]
- Krych, S.; Jeczmyk, A.; Jurkiewicz, M.; Zurek, M.; Jekielek, M.; Kowalczyk, P.; Kramkowski, K.; Hrapkowicz, T. Viral Myocarditis as a Factor Leading to the Development of Heart Failure Symptoms, Including the Role of Parvovirus B19 Infection-Systematic Review. Int. J. Mol. Sci. 2024, 25, 8127. [Google Scholar] [CrossRef]
- Cantarutti, N.; Battista, V.; Adorisio, R.; Cicenia, M.; Campanello, C.; Listo, E.; Campana, A.; Trocchio, G.; Drago, F. Cardiac Manifestations in Children with SARS-CoV-2 Infection: 1-Year Pediatric Multicenter Experience. Children 2021, 8, 717. [Google Scholar] [CrossRef]
- Akansel, S.; Kofler, M.; Van Praet, K.M.; Unbehaun, A.; Sundermann, S.H.; Jacobs, S.; Falk, V.; Kempfert, J. Minimally invasive mitral valve surgery after failed transcatheter mitral valve repair in an intermediate-risk cohort. Interact. Cardiovasc. Thorac. Surg. 2022, 35, ivac163. [Google Scholar] [CrossRef] [PubMed]
- Beaudry, J.T.; Dietrick, B.; Lammert, D.B.; Constas, A.; McCaw, J.; Hammond, J.; Buendia, M.; Stein, J.E.; Pekosz, A.; Schuette, J.; et al. Fatal SARS-CoV-2 Inflammatory Syndrome and Myocarditis in an Adolescent: A Case Report. Pediatr. Infect. Dis. J. 2021, 40, e72–e76. [Google Scholar] [CrossRef] [PubMed]
- Thom, K.; Kahl, B.; Wagner, T.; van Egmond-Frohlich, A.; Krainz, M.; Frischer, T.; Leeb, I.; Schuster, C.; Ehringer-Schetitska, D.; Minkov, M.; et al. SARS-CoV-2 Associated Pediatric Inflammatory Multisystem Syndrome With a High Prevalence of Myocarditis–A Multicenter Evaluation of Clinical and Laboratory Characteristics, Treatment and Outcome. Front. Pediatr. 2022, 10, 896252. [Google Scholar] [CrossRef] [PubMed]
- Gargus, M.; Ben-Azu, B.; Landwehr, A.; Dunn, J.; Errico, J.P.; Tremblay, M.E. Mechanisms of vagus nerve stimulation for the treatment of neurodevelopmental disorders: A focus on microglia and neuroinflammation. Front. Neurosci. 2024, 18, 1527842. [Google Scholar] [CrossRef]
- Sattar, Y.; Sandhyavenu, H.; Patel, N.; Victor, V.; Patel, D.; Hussain, B.; Titus, A.; Thyagaturu, H.; Alraiyes, M.; Atti, L.; et al. In-Hospital Outcomes of COVID-19 Associated Myocarditis (from a Nationwide Inpatient Sample Database Study). Am. J. Cardiol. 2023, 192, 39–44. [Google Scholar] [CrossRef]
- Boehmer, T.K.; Kompaniyets, L.; Lavery, A.M.; Hsu, J.; Ko, J.Y.; Yusuf, H.; Romano, S.D.; Gundlapalli, A.V.; Oster, M.E.; Harris, A.M. Association Between COVID-19 and Myocarditis Using Hospital-Based Administrative Data–United States, March 2020-January 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 1228–1232. [Google Scholar] [CrossRef]
- Saha, D.; Dutta, P.; Rebello, K.R.; Shankar, A.; Chakraborty, A. Exploring the potential link between human papillomavirus infection and coronary artery disease: A review of shared pathways and mechanisms. Mol. Cell. Biochem. 2025, 480, 3971–3994. [Google Scholar] [CrossRef]
- Dutta, P.; Saha, D.; Earle, M.; Prasad, C.P.; Singh, M.; Darswal, M.; Aggarwal, V.; Naik, N.; Yadav, R.; Shankar, A.; et al. Unveiling HPV’s hidden link: Cardiovascular diseases and the viral intrigue. Indian Heart J. 2024, 76, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Pannucci, P.; Jefferson, S.R.; Hampshire, J.; Cooper, S.L.; Hill, S.J.; Woolard, J. COVID-19-Induced Myocarditis: Pathophysiological Roles of ACE2 and Toll-like Receptors. Int. J. Mol. Sci. 2023, 24, 5374. [Google Scholar] [CrossRef] [PubMed]
- Kusumawardhani, N.Y.; Putra, I.C.S.; Kamarullah, W.; Afrianti, R.; Pramudyo, M.; Iqbal, M.; Prameswari, H.S.; Achmad, C.; Tiksnadi, B.B.; Akbar, M.R. Cardiovascular Disease in Post-Acute COVID-19 Syndrome: A Comprehensive Review of Pathophysiology and Diagnosis Approach. Rev. Cardiovasc. Med. 2023, 24, 28. [Google Scholar] [CrossRef] [PubMed]
- Castiello, T.; Georgiopoulos, G.; Finocchiaro, G.; Claudia, M.; Gianatti, A.; Delialis, D.; Aimo, A.; Prasad, S. COVID-19 and myocarditis: A systematic review and overview of current challenges. Heart Fail. Rev. 2022, 27, 251–261. [Google Scholar] [CrossRef]
- Turpeinen, A.K.; Vanninen, E.; Magga, J.; Tuomainen, P.; Kuusisto, J.; Sipola, P.; Punnonen, K.; Vuolteenaho, O.; Peuhkurinen, K. Cardiac sympathetic activity is associated with inflammation and neurohumoral activation in patients with idiopathic dilated cardiomyopathy. Clin. Physiol. Funct. Imaging 2009, 29, 414–419. [Google Scholar] [CrossRef]
- Kawai, C.; Yui, Y.; Hoshino, T.; Sasayama, S.; Matsumori, A. Myocardial catecholamines in hypertrophic and dilated (congestive) cardiomyopathy: A biopsy study. J. Am. Coll. Cardiol. 1983, 2, 834–840. [Google Scholar] [CrossRef]
- Paur, H.; Wright, P.T.; Sikkel, M.B.; Tranter, M.H.; Mansfield, C.; O’Gara, P.; Stuckey, D.J.; Nikolaev, V.O.; Diakonov, I.; Pannell, L.; et al. High levels of circulating epinephrine trigger apical cardiodepression in a beta2-adrenergic receptor/Gi-dependent manner: A new model of Takotsubo cardiomyopathy. Circulation 2012, 126, 697–706. [Google Scholar] [CrossRef]
- Shah, R.M.; Shah, M.; Shah, S.; Li, A.; Jauhar, S. Takotsubo Syndrome and COVID-19: Associations and Implications. Curr. Probl. Cardiol. 2021, 46, 100763. [Google Scholar] [CrossRef]
- Gold, M.R.; Van Veldhuisen, D.J.; Hauptman, P.J.; Borggrefe, M.; Kubo, S.H.; Lieberman, R.A.; Milasinovic, G.; Berman, B.J.; Djordjevic, S.; Neelagaru, S.; et al. Vagus Nerve Stimulation for the Treatment of Heart Failure: The INOVATE-HF Trial. J. Am. Coll. Cardiol. 2016, 68, 149–158. [Google Scholar] [CrossRef]
- Wu, S.J.; Li, Y.C.; Shi, Z.W.; Lin, Z.H.; Rao, Z.H.; Tai, S.C.; Chu, M.P.; Li, L.; Lin, J.F. Alteration of Cholinergic Anti-Inflammatory Pathway in Rat with Ischemic Cardiomyopathy-Modified Electrophysiological Function of Heart. J. Am. Heart Assoc. 2017, 6, e006510. [Google Scholar] [CrossRef]
- Ruiz, P.; Gabarre, P.; Chenevier-Gobeaux, C.; Francois, H.; Kerneis, M.; Cidlowski, J.A.; Oakley, R.H.; Lefevre, G.; Boissan, M. Case report: Changes in the levels of stress hormones during Takotsubo syndrome. Front. Cardiovasc. Med. 2022, 9, 931054. [Google Scholar] [CrossRef]
- Wang, E.; Zhou, R.; Li, T.; Hua, Y.; Zhou, K.; Li, Y.; Luo, S.; An, Q. The Molecular Role of Immune Cells in Dilated Cardiomyopathy. Medicina 2023, 59, 1246. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qin, L.; Xu, X.; Chen, R.; Zhang, G.; Wang, B.; Li, B.; Chu, X.M. Immune modulation: The key to combat SARS-CoV-2 induced myocardial injury. Front. Immunol. 2025, 16, 1561946. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Duan, F.; Hu, J.; Luo, B.; Huang, B.; Lou, X.; Sun, X.; Li, H.; Zhang, X.; Yin, S.; et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox. Biol. 2020, 34, 101523. [Google Scholar] [CrossRef]
- Higashikuni, Y.; Liu, W.; Numata, G.; Tanaka, K.; Fukuda, D.; Tanaka, Y.; Hirata, Y.; Imamura, T.; Takimoto, E.; Komuro, I.; et al. NLRP3 Inflammasome Activation Through Heart-Brain Interaction Initiates Cardiac Inflammation and Hypertrophy During Pressure Overload. Circulation 2023, 147, 338–355. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef]
- Zwaka, T.P.; Manolov, D.; Ozdemir, C.; Marx, N.; Kaya, Z.; Kochs, M.; Hoher, M.; Hombach, V.; Torzewski, J. Complement and dilated cardiomyopathy: A role of sublytic terminal complement complex-induced tumor necrosis factor-alpha synthesis in cardiac myocytes. Am. J. Pathol. 2002, 161, 449–457. [Google Scholar] [CrossRef]
- Mavroidis, M.; Davos, C.H.; Psarras, S.; Varela, A.; Athanasiadis, N.C.; Katsimpoulas, M.; Kostavasili, I.; Maasch, C.; Vater, A.; van Tintelen, J.P.; et al. Complement system modulation as a target for treatment of arrhythmogenic cardiomyopathy. Basic Res. Cardiol. 2015, 110, 27. [Google Scholar] [CrossRef]
- Anderson, F.L.; Port, J.D.; Reid, B.B.; Hanson, G.; Kralios, A.C.; Hershberger, R.E.; Bristow, M.R. Effect of therapeutic dopamine administration on myocardial catecholamine and neuropeptide Y concentrations in the failing ventricles of patients with idiopathic dilated cardiomyopathy. J. Cardiovasc. Pharmacol. 1992, 20, 800–806. [Google Scholar]
- Enzan, N.; Matsushima, S.; Ide, T.; Tohyama, T.; Funakoshi, K.; Higo, T.; Tsutsui, H. The use of angiotensin II receptor blocker is associated with greater recovery of cardiac function than angiotensin-converting enzyme inhibitor in dilated cardiomyopathy. ESC Heart Fail. 2022, 9, 1175–1185. [Google Scholar] [CrossRef]
- Mo, X.; Lu, P.; Yang, X. Efficacy of sacubitril-valsartan and SGLT2 inhibitors in heart failure with reduced ejection fraction: A systematic review and meta-analysis. Clin. Cardiol. 2023, 46, 1137–1145. [Google Scholar] [CrossRef]
- Kumar, P.; Schwartz, J.D. Device therapies: New indications and future directions. Curr. Cardiol. Rev. 2015, 11, 33–41. [Google Scholar] [CrossRef]
- Baris, R.O.; Tabit, C.E. Heart Failure Readmission Prevention Strategies-A Comparative Review of Medications, Devices, and Other Interventions. J. Clin. Med. 2025, 14, 5894. [Google Scholar] [CrossRef] [PubMed]
- Kehat, I. Novel strategies for the treatment of heart failure. Rambam Maimonides Med. J. 2012, 3, e0011. [Google Scholar] [CrossRef] [PubMed]
- Dicorato, M.M.; Citarelli, G.; Mangini, F.; Alemanni, R.; Albanese, M.; Cicco, S.; Greco, C.A.; Forleo, C.; Basile, P.; Carella, M.C.; et al. Integrative Approaches in the Management of Hypertrophic Cardiomyopathy: A Comprehensive Review of Current Therapeutic Modalities. Biomedicines 2025, 13, 1256. [Google Scholar] [CrossRef]
- Savsin, H.; Tokarek, T. Comprehensive Review: Mavacamten and Aficamten in Hypertrophic Cardiomyopathy. Biomedicines 2025, 13, 1619. [Google Scholar] [CrossRef]
- Alnour, F.; Beuthner, B.E.; Hakroush, S.; Topci, R.; Vogelgesang, A.; Lange, T.; Seidler, T.; Kutschka, I.; Toischer, K.; Schuster, A.; et al. Cardiac fibrosis as a predictor for sudden cardiac death after transcatheter aortic valve implantation. EuroIntervention 2024, 20, e760–e769. [Google Scholar] [CrossRef]
- Adabag, A.S.; Luepker, R.V.; Roger, V.L.; Gersh, B.J. Sudden cardiac death: Epidemiology and risk factors. Nat. Rev. Cardiol. 2010, 7, 216–225. [Google Scholar] [CrossRef]
- Wolowiec, L.; Grzesk, G.; Osiak, J.; Wijata, A.; Medlewska, M.; Gaborek, P.; Banach, J.; Wolowiec, A.; Glowacka, M. Beta-blockers in cardiac arrhythmias-Clinical pharmacologist’s point of view. Front. Pharmacol. 2022, 13, 1043714. [Google Scholar] [CrossRef]
- Coscarella, I.L.; Landim-Vieira, M.; Pinto, J.R.; Chelko, S.P. Arrhythmogenic Cardiomyopathy: Exercise Pitfalls, Role of Connexin-43, and Moving beyond Antiarrhythmics. Int. J. Mol. Sci. 2022, 23, 8753. [Google Scholar] [CrossRef]
- Rodriguez Mejia, R.A.; Singh, A.; Bahekar, A.; Gupta, A. Efficacy of beta-blocker therapy in Takotsubo cardiomyopathy: A systematic review and meta-analysis. Int. J. Cardiol. 2025, 437, 133483. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Khan, H.; Gamble, D.T.; Scally, C.; Newby, D.E.; Dawson, D. Takotsubo Syndrome: Pathophysiology, Emerging Concepts, and Clinical Implications. Circulation 2022, 145, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- Zarate, S.M.; Kirabo, A.; Hinton, A.O., Jr.; Santisteban, M.M. Neuroimmunology of Cardiovascular Disease. Curr. Hypertens Rep. 2024, 26, 339–347. [Google Scholar] [CrossRef]
- Bogle, C.; Colan, S.D.; Miyamoto, S.D.; Choudhry, S.; Baez-Hernandez, N.; Brickler, M.M.; Feingold, B.; Lal, A.K.; Lee, T.M.; Canter, C.E.; et al. Treatment Strategies for Cardiomyopathy in Children: A Scientific Statement From the American Heart Association. Circulation 2023, 148, 174–195. [Google Scholar] [CrossRef]
- Velleca, A.; Shullo, M.A.; Dhital, K.; Azeka, E.; Colvin, M.; DePasquale, E.; Farrero, M.; Garcia-Guereta, L.; Jamero, G.; Khush, K.; et al. The International Society for Heart and Lung Transplantation (ISHLT) guidelines for the care of heart transplant recipients. J. Heart Lung Transplant 2023, 42, e1–e141. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Cochran, T.R.; Briston, D.A.; Brown, S.R.; Sambatakos, P.J.; Miller, T.L.; Carrillo, A.A.; Corcia, L.; Sanchez, J.E.; Diamond, M.B.; et al. Pediatric cardiomyopathies: Causes, epidemiology, clinical course, preventive strategies and therapies. Future Cardiol. 2013, 9, 817–848. [Google Scholar] [CrossRef]
- Kamarullah, W.; Nurcahyani; Mary Josephine, C.; Bill Multazam, R.; Ghaezany Nawing, A.; Dharma, S. Corticosteroid Therapy in Management of Myocarditis Associated with COVID-19; a Systematic Review of Current Evidence. Arch. Acad. Emerg. Med. 2021, 9, e32. [Google Scholar] [CrossRef]
- Mukkawar, R.V.; Reddy, H.; Rathod, N.; Kumar, S.; Acharya, S. The Long-Term Cardiovascular Impact of COVID-19: Pathophysiology, Clinical Manifestations, and Management. Cureus. 2024, 16, e66554. [Google Scholar] [CrossRef]
- Ticinovic Kurir, T.; Milicevic, T.; Novak, A.; Vilovic, M.; Bozic, J. Adropin–Potential Link in Cardiovascular Protection for Obese Male Type 2 Diabetes Mellitus Patients Treated with Liraglutide. Acta. Clin. Croat. 2020, 59, 344–350. [Google Scholar] [CrossRef]
- Li, J.; Zhou, L.; Gong, H. New insights and advances of sodium-glucose cotransporter 2 inhibitors in heart failure. Front. Cardiovasc. Med. 2022, 9, 903902. [Google Scholar] [CrossRef]
- Huang, S.; Zhao, Q. The Trend of Immunotherapy Combined with Nanomedicine. Curr. Med. Chem. 2022, 29, 3817–3818. [Google Scholar] [CrossRef]
- Berezutsky, V. Clinical reasoning: Sherlock Holmes or Dr. Joseph Bell. Med. Teach. 2023, 45, 114. [Google Scholar] [CrossRef]
- Papaioannou, V.; Pneumatikos, I.; Maglaveras, N. Association of heart rate variability and inflammatory response in patients with cardiovascular diseases: Current strengths and limitations. Front. Physiol. 2013, 4, 174. [Google Scholar] [CrossRef]
- Chakraborty, A.; Dutta, P.; Saha, D.; Singh, M.; Prasad, C.P.; Pushpam, D.; Shankar, A.; Saini, D. Chimeric antigen receptor CAR-T therapy on the move: Current applications and future possibilities. Curr. Tissue Microenviron. Rep. 2023, 4, 29–40. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Glynn, R.J.; Koenig, W.; Libby, P.; Everett, B.M.; Lefkowitz, M.; Thuren, T.; Cornel, J.H. Inhibition of Interleukin-1beta by Canakinumab and Cardiovascular Outcomes in Patients With Chronic Kidney Disease. J. Am. Coll. Cardiol. 2018, 71, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Carter, K.T.; Palei, A.C.; Spradley, F.T.; Witcher, B.M.; Martin, L.; Hester, R.L.; Kutcher, M.E. A rat model of orthopedic injury-induced hypercoagulability and fibrinolytic shutdown. J. Trauma Acute Care Surg. 2020, 89, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.M.; Cooper, L.T.; Kem, D.C.; Stavrakis, S.; Kosanke, S.D.; Shevach, E.M.; Fairweather, D.; Stoner, J.A.; Cox, C.J.; Cunningham, M.W. Cardiac myosin-Th17 responses promote heart failure in human myocarditis. JCI insight 2016, 1, e85851. [Google Scholar] [CrossRef]
- Jang, D.I.; Lee, A.H.; Shin, H.Y.; Song, H.R.; Park, J.H.; Kang, T.B.; Lee, S.R.; Yang, S.H. The Role of Tumor Necrosis Factor Alpha (TNF-alpha) in Autoimmune Disease and Current TNF-alpha Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Del Cornò, M.; Donninelli, G.; Varano, B.; Da Sacco, L.; Masotti, A.; Gessani, S. HIV-1 gp120 activates the STAT3/interleukin-6 axis in primary human monocyte-derived dendritic cells. J. Virol. 2014, 88, 11045–11055. [Google Scholar] [CrossRef]
- Panagiotou, A.; Trendelenburg, M.; Osthoff, M. The lectin pathway of complement in myocardial ischemia/reperfusion injury—Review of its significance and the potential impact of therapeutic interference by C1 esterase inhibitor. Front. Immunol. 2018, 9, 1151. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.-W. The role of complement in myocardial infarction reperfusion injury: An underappreciated therapeutic target. Front. Cell Dev. Biol. 2020, 8, 606407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, L.; Wang, S.; Cheng, H.; Xu, L.; Pei, G.; Wang, Y.; Fu, C.; Jiang, Y.; He, C.; et al. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct. Target. Ther. 2022, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Fernandes, S.M.; Davis, A.E., 3rd. The effect of C1 inhibitor on myocardial ischemia and reperfusion injury. Cardiovasc. Pathol. 2013, 22, 75–80. [Google Scholar] [CrossRef]
- Torp, M.-K.; Ranheim, T.; Schjalm, C.; Hjorth, M.; Heiestad, C.M.; Dalen, K.T.; Nilsson, P.H.; Mollnes, T.E.; Pischke, S.; Lien, E. Intracellular complement component 3 attenuated ischemia-reperfusion injury in the isolated buffer-perfused mouse heart and is associated with improved metabolic homeostasis. Front. Immunol. 2022, 13, 870811. [Google Scholar] [CrossRef]
- Cheng, X.-F.; He, S.-T.; Zhong, G.-Q.; Meng, J.-J.; Wang, M.; Bi, Q.; Tu, R.-H. Exosomal HSP90 induced by remote ischemic preconditioning alleviates myocardial ischemia/reperfusion injury by inhibiting complement activation and inflammation. BMC Cardiovasc. Disord. 2023, 23, 58. [Google Scholar] [CrossRef]
- Ivanova, M.M.; Dao, J.; Friedman, A.; Kasaci, N.; Goker-Alpan, O. Sex Differences in Circulating Inflammatory, Immune, and Tissue Growth Markers Associated with Fabry Disease-Related Cardiomyopathy. Cells 2025, 14, 322. [Google Scholar] [CrossRef]
- Velikkakam, T.; Gollob, K.J.; Dutra, W.O. Double-negative T cells: Setting the stage for disease control or progression. Immunology 2022, 165, 371–385. [Google Scholar] [CrossRef]
- Kolman, B.S.; Verrier, R.L.; Lown, B. The effect of vagus nerve stimulation upon vulnerability of the canine ventricle: Role of sympathetic-parasympathetic interactions. Circulation 1975, 52, 578–585. [Google Scholar] [CrossRef]
- Gronda, E.; Seravalle, G.; Trevano, F.Q.; Costantino, G.; Casini, A.; Alsheraei, A.; Lovett, E.G.; Vanoli, E.; Mancia, G.; Grassi, G. Long-term chronic baroreflex activation: Persistent efficacy in patients with heart failure and reduced ejection fraction. J. Hypertens. 2015, 33, 1704–1708. [Google Scholar] [CrossRef]
- Rees, K.; Taylor, R.S.; Singh, S.; Coats, A.J.; Ebrahim, S. Exercise based rehabilitation for heart failure. Cochrane Database Syst. Rev. 2004, 3, CD003331. [Google Scholar] [CrossRef]
- Daniela, M.; Catalina, L.; Ilie, O.; Paula, M.; Daniel-Andrei, I.; Ioana, B. Effects of Exercise Training on the Autonomic Nervous System with a Focus on Anti-Inflammatory and Antioxidants Effects. Antioxidants 2022, 11, 350. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Dou, X.; Chen, L.; Hu, F.; Li, Z.; Gao, H.; Li, X.; Zhang, M.; Hu, Z. The Utilization and Potential of Mindfulness-Based Stress Reduction Therapy in Individuals Diagnosed with Acute Coronary Syndrome. Rev. Cardiovasc. Med. 2024, 25, 277. [Google Scholar] [CrossRef] [PubMed]
- Catalina, G.R.; Gheorman, V.; Gheorman, V.; Fortofoiu, M.C. The Role of Neuroinflammation in the Comorbidity of Psychiatric Disorders and Internal Diseases. Healthcare 2025, 13, 837. [Google Scholar] [CrossRef]
- Nascimentoa, P.M.; Vieiraa, M.C.; Sperandeib, S.; Serraa, S.M. Supervised exercise improves autonomic modulation in participants in cardiac rehabilitation programs. Rev. Port. Cardiol. 2016, 35, 19–24. [Google Scholar] [CrossRef]
- Lee, S.C.; Tsai, P.H.; Yu, K.H.; Chan, T.M. Effects of Mind–Body Interventions on Immune and Neuroendocrine Functions: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Healthcare 2025, 13, 952. [Google Scholar] [CrossRef]
- Merino Del Portillo, M.; Clemente-Suarez, V.J.; Ruisoto, P.; Jimenez, M.; Ramos-Campo, D.J.; Beltran-Velasco, A.I.; Martinez-Guardado, I.; Rubio-Zarapuz, A.; Navarro-Jimenez, E.; Tornero-Aguilera, J.F. Nutritional Modulation of the Gut–Brain Axis: A Comprehensive Review of Dietary Interventions in Depression and Anxiety Management. Metabolites 2024, 14, 549. [Google Scholar] [CrossRef]
- Jordan, J.; Tank, J.; Heusser, K.; Reuter, H. Baroreflex activation therapy through electrical carotid sinus stimulation. Auton. Neurosci. 2024, 256, 103219. [Google Scholar] [CrossRef]
- Liao, S.Y.; Liu, Y.; Zuo, M.; Zhang, Y.; Yue, W.; Au, K.W.; Lai, W.H.; Wu, Y.; Shuto, C.; Chen, P.; et al. Remodelling of cardiac sympathetic re-innervation with thoracic spinal cord stimulation improves left ventricular function in a porcine model of heart failure. Europace 2015, 17, 1875–1883. [Google Scholar] [CrossRef]
- Vargas-Uricoechea, H.; Castellanos-Pinedo, A.; Urrego-Noguera, K.; Vargas-Sierra, H.D.; Pinzon-Fernandez, M.V.; Barcelo-Martinez, E.; Ramirez-Giraldo, A.F. Mindfulness-Based Interventions and the Hypothalamic-Pituitary-Adrenal Axis: A Systematic Review. Neurol. Int. 2024, 16, 1552–1584. [Google Scholar] [CrossRef]
- Gitler, A.; Bar Yosef, Y.; Kotzer, U.; Levine, A.D. Harnessing non-invasive vagal neuromodulation: HRV biofeedback and SSP for cardiovascular and autonomic regulation (Review). Med. Int. 2025, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Knudsen, B.; Gholston, A.; Skubal, A.; Blanz, S.; Settell, M.; Frank, J.; Trevathan, J.; Ludwig, K. Microneurography as a minimally invasive method to assess target engagement during neuromodulation. J. Neural. Eng. 2023, 20. [Google Scholar] [CrossRef] [PubMed]
- Gerbino, A.; Forleo, C.; Milano, S.; Piccapane, F.; Procino, G.; Pepe, M.; Piccolo, M.; Guida, P.; Resta, N.; Favale, S.; et al. Pro-inflammatory cytokines as emerging molecular determinants in cardiolaminopathies. J. Cell Mol. Med. 2021, 25, 10902–10915. [Google Scholar] [CrossRef]
- Oduro, P.K.; Zheng, X.; Wei, J.; Yang, Y.; Wang, Y.; Zhang, H.; Liu, E.; Gao, X.; Du, M.; Wang, Q. The cGAS-STING signaling in cardiovascular and metabolic diseases: Future novel target option for pharmacotherapy. Acta Pharm. Sin. B 2022, 12, 50–75. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Matuskova, L.; Czippelova, B.; Turianikova, Z.; Svec, D.; Kolkova, Z.; Lasabova, Z.; Javorka, M. Beta-adrenergic receptors gene polymorphisms are associated with cardiac contractility and blood pressure variability. Physiol. Res. 2021, 70, S327–S337. [Google Scholar] [CrossRef]
- Sprenkle, N.T.; Serezani, C.H.; Pua, H.H. MicroRNAs in Macrophages: Regulators of Activation and Function. J. Immunol. 2023, 210, 359–368. [Google Scholar] [CrossRef]
- Yang, Y.L.; Li, X.W.; Chen, H.B.; Tang, Q.D.; Li, Y.H.; Xu, J.Y.; Xie, J.J. Single-cell transcriptomics reveals writers of RNA modification-mediated immune microenvironment and cardiac resident Macro-MYL2 macrophages in heart failure. BMC Cardiovasc. Disord. 2024, 24, 432. [Google Scholar] [CrossRef]
- Fairweather, D.; Frisancho-Kiss, S.; Rose, N.R. Sex differences in autoimmune disease from a pathological perspective. Am. J. Pathol. 2008, 173, 600–609. [Google Scholar] [CrossRef]
- Geske, J.B.; Ong, K.C.; Siontis, K.C.; Hebl, V.B.; Ackerman, M.J.; Hodge, D.O.; Miller, V.M.; Nishimura, R.A.; Oh, J.K.; Schaff, H.V.; et al. Women with hypertrophic cardiomyopathy have worse survival. Eur. Heart. J. 2017, 38, 3434–3440. [Google Scholar] [CrossRef]
- Sethi, Y.; Murli, H.; Kaiwan, O.; Vora, V.; Agarwal, P.; Chopra, H.; Padda, I.; Kanithi, M.; Popoviciu, M.S.; Cavalu, S. Broken Heart Syndrome: Evolving Molecular Mechanisms and Principles of Management. J. Clin. Med. 2022, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Luo, Y.; Tai, R.; Zhang, N. Estrogen receptor beta suppresses inflammation and the progression of prostate cancer. Mol. Med. Rep. 2019, 19, 3555–3563. [Google Scholar] [CrossRef]
- Tostes, R.C.; Carneiro, F.S.; Carvalho, M.H.; Reckelhoff, J.F. Reactive oxygen species: Players in the cardiovascular effects of testosterone. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R1–R14. [Google Scholar] [CrossRef]
- Fairweather, D.; Frisancho-Kiss, S.; Yusung, S.A.; Barrett, M.A.; Davis, S.E.; Gatewood, S.J.; Njoku, D.B.; Rose, N.R. Interferon-gamma protects against chronic viral myocarditis by reducing mast cell degranulation, fibrosis, and the profibrotic cytokines transforming growth factor-beta 1, interleukin-1 beta, and interleukin-4 in the heart. Am. J. Pathol. 2004, 165, 1883–1894. [Google Scholar] [CrossRef]
- Vereckei, A.; Besenyi, Z.; Nagy, V.; Radics, B.; Vago, H.; Jenei, Z.; Katona, G.; Sepp, R. Cardiac Sarcoidosis: A Comprehensive Clinical Review. Rev. Cardiovasc. Med. 2024, 25, 37. [Google Scholar] [CrossRef]
- Bollen, I.A.; Van Deel, E.D.; Kuster, D.W.; Van Der Velden, J. Peripartum cardiomyopathy and dilated cardiomyopathy: Different at heart. Front. Physiol. 2014, 5, 531. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).