
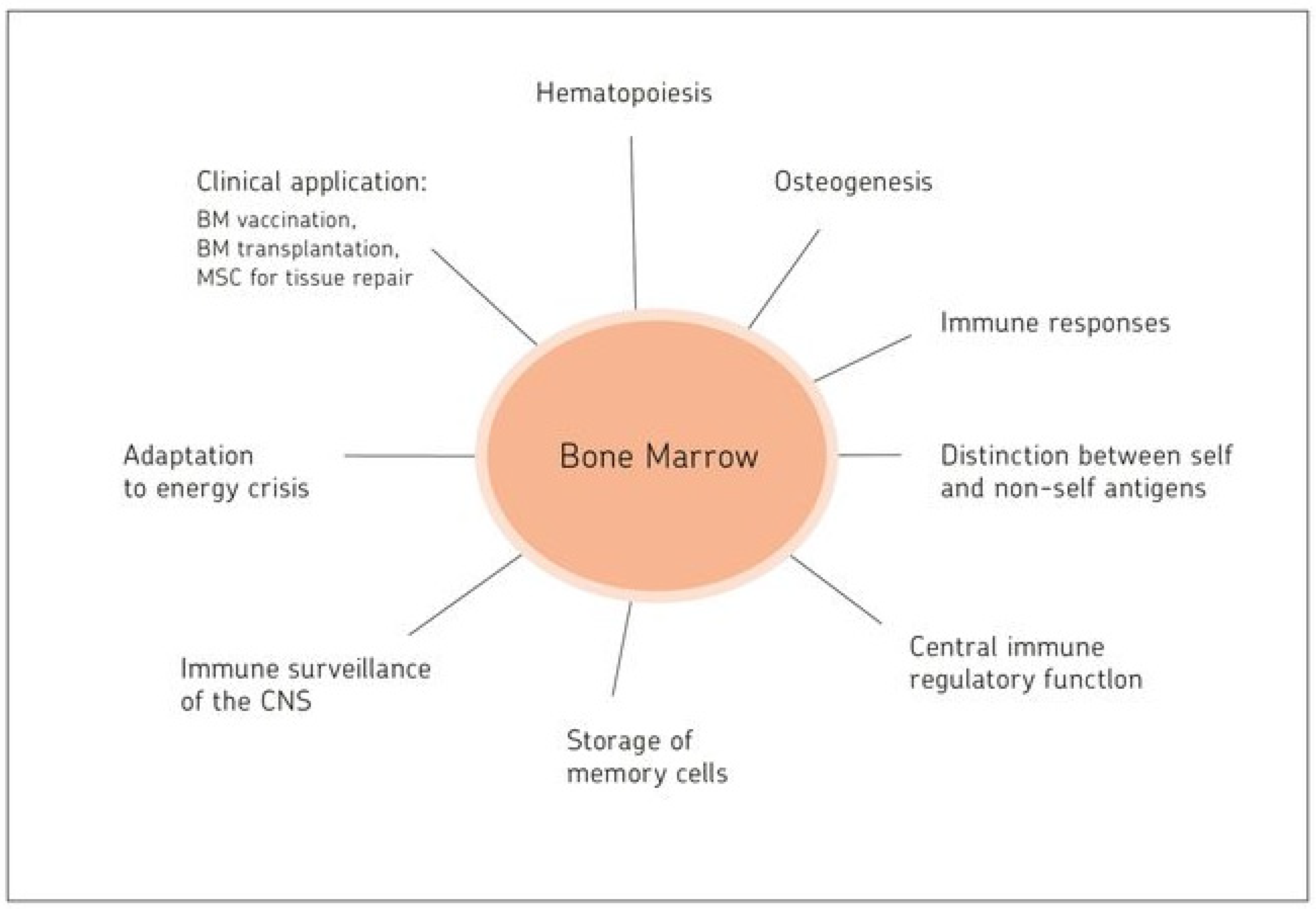
Bone Marrow: The Central Immune System


Abstract
1. Introduction
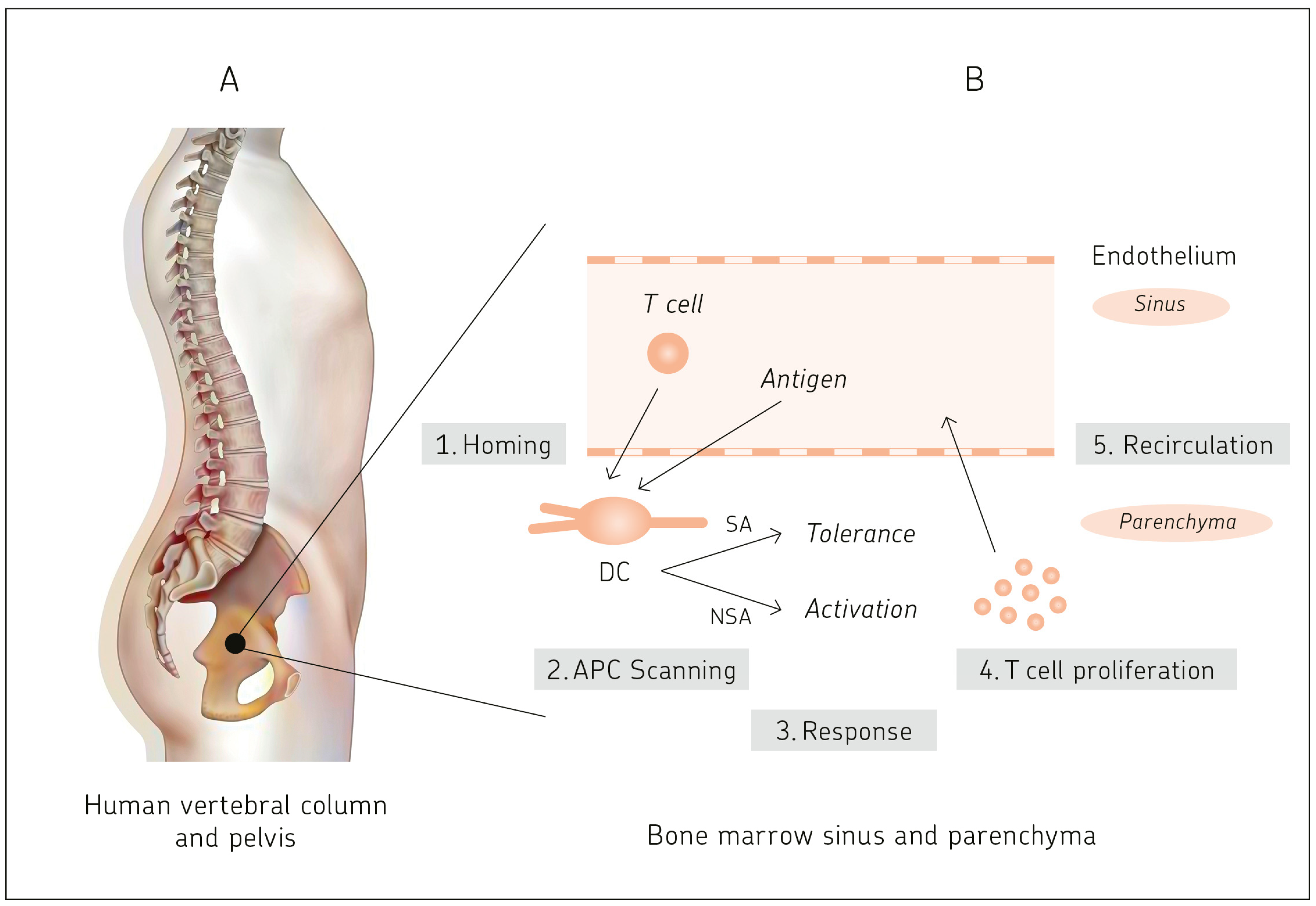
2. Bone Marrow: A Hematopoietic and Antigen-Responsive Lymphatic Organ
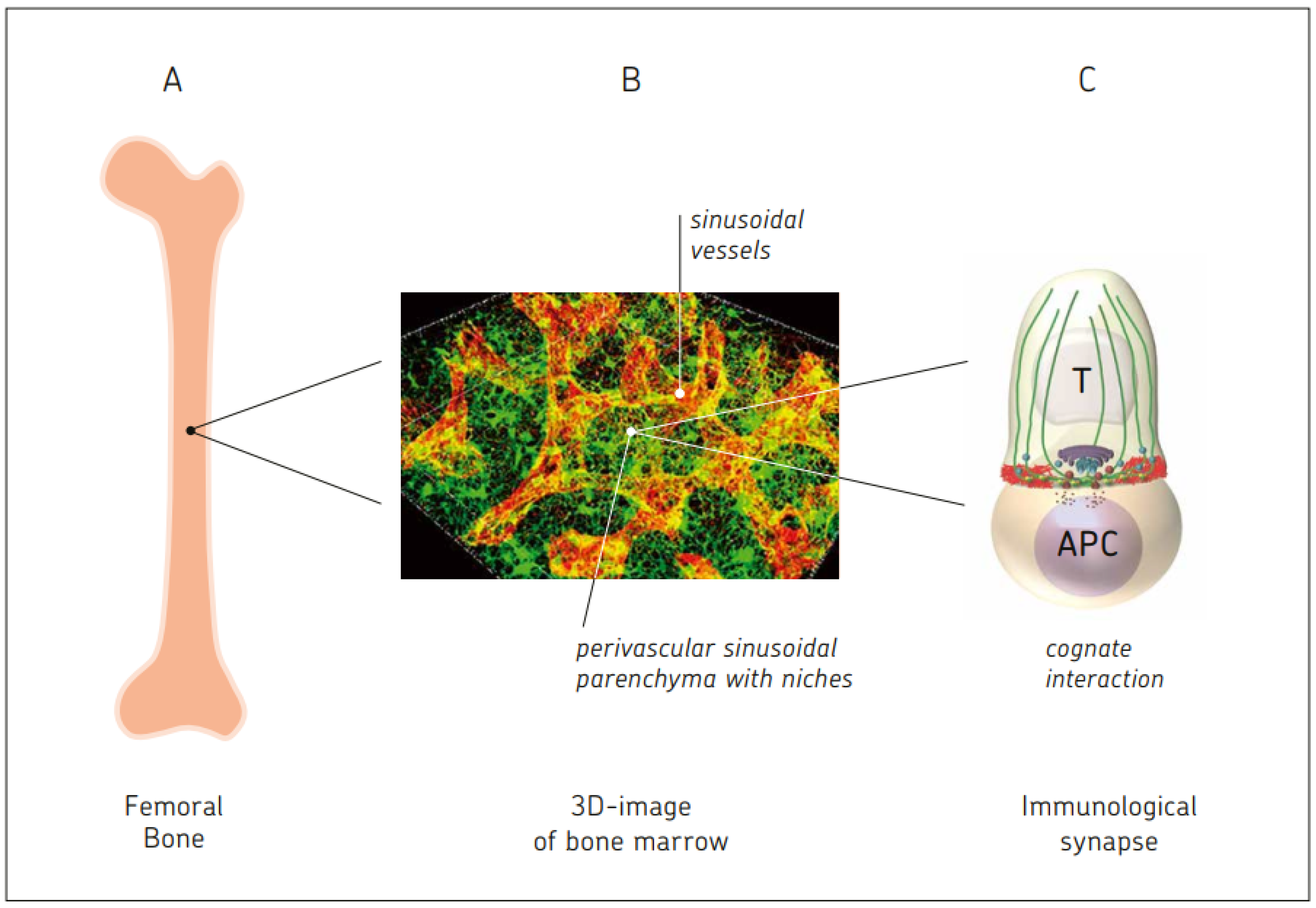
2.1. BM: A Central Organ Protected by Bone
2.2. BM: A Central Hematopoietic Organ
2.3. BM: A Central Antigen Responsive Lymphatic Organ
2.4. Active Control of Proliferating Tumor Cells by CD8+ Memory T Cells Leading to Tumor Dormancy in BM
2.4.1. Antigen-Presentation Capacity of BM DCs to Naïve T Cells
2.4.2. Primary T Cell Responses in Bone Marrow
2.4.3. Two-Photon Dynamic Imaging Revealing Cross-Presentation of Blood-Borne Antigens to Naïve T Cells in BM
2.4.4. Targeting Glycan Modified OVA to DC-SIGN
2.4.5. Cluster Formation in BM Parenchyma of Antigen-Presenting DCs and Antigen-Specific T Cells
3. Comparison between BM and Blood
3.1. Comparison of DC Generation from Mononuclear Cells and Their Function
3.2. Enrichment of Memory T Cells in BM of Breast Cancer Patients
3.3. Generation of Tumor-Specific CTL from BM, but Not PBL, of Breast Cancer Patients
3.4. Superior Therapeutic Efficiency In Vivo of Reactivated MTCs from BM in Comparison to Blood of Breast Cancer Patients
4. BM Storage Capacity for Memory B and Memory T Cells
4.1. Survival Niches for Memory B and Memory Plasma Cells in BM Parenchyma
4.2. Memory T Cells
4.3. Survival Niches for Memory T Cells in BM Parenchyma
4.4. Tissue-Resident Memory T Cells in BM Parenchyma
4.5. Stem-like Memory T Cells in BM Parenchyma
4.6. Enrichment of Virus-Specific MTCs in Human BM Parenchyma
4.7. Cognate Re-Activation of TA-Specific BM MTCs Ex Vivo and In Situ
4.8. Hypotheses for the Maintenance of Long-Term Memory in the BM
- Quiescence: Following the successful resolution of an immune reaction, antibody-secreting memory plasma cells and memory B and T cells persist as quiescent cells (non-proliferative, non-migratory) in dedicated survival niches organized by BM stromal cells. The immune memory cells dock individually onto dedicated stromal cells, which control their maintenance. The number of available dedicated stromal cells defines the size of the memory compartment [61].
- Cognate re-activation of BM memory cells. Upon re-encounter with the antigen, which enters directly via the blood into the vascularized BM or is transported there by APCs, antigen-specific memory B and MTCs are re-activated. MTCs proliferate locally, form immune clusters, and provide local protection. Others exit the BM and contribute to secondary immune reactions in the periphery. BM clusters in the parenchyma can develop into large follicles. These include memory B and memory plasma cells in addition to CD4+ MTCs, suggesting T–B cell interaction [62]. Once a BCR binds its T cell-dependent antigen, the antigen is taken up into the B cell through receptor-mediated endocytosis. This is then degraded and presented to T cells as a p-MHC II complex at the cell membrane. Memory B cells in immune follicles might receive stimulatory signals from antigen-specific helper T cells upon T–B synapse formation. More than one antigenic determinant of a protein is required for such antigen-specific T-B cell interactions [63], one interacting with the BCR, the other with the TCR. Thus, activated memory B cells may directly differentiate into antibody-secreting cells in the BM, providing rapid enhancement of humoral immunity [61,62].
5. Bone Marrow Vaccination or Allogeneic BM Cell Injection: Novel Approaches to Enhance or Reduce Antigen-Specific Immunity
6. Interactions in BM between Three Types of Stem Cells and Immune Cells
6.1. Hematopoietic Stem Cells (HSCs) in Cross-Talk with T Cells and DCs
6.2. Extramedullary HSCs in Meninges of Adult Mice Providing Immune Surveillance of the CNS
6.3. BM Neural Crest-Derived Stem Cells Affecting B Cell Lymphopoiesis
6.4. Mesenchymal Stem Cells in Cross-Talk with T Cells
7. Effect of Dietary Restriction (DR) on the BM
7.1. Effect of DR on Monocytes from the BM
7.2. Effect of DR on Mucosal Immune Responses: Migration of Naïve B Cells from PPs to BM
7.3. Effects of DR on Memory T Cells: BM as a Refuge for Immune Memory
8. Blood-Borne Antigens, Circulating Cells, and Subcellular Particles
8.1. Self and Non-Self Antigens
8.2. Circulating Tumor Cells, Tumor-Associated Antigens, and Immunogenic Cell Death
8.3. Circulatory Antigen-Presenting DCs and Their Homing to BM
8.4. Circulatory Naïve T and Memory T Lymphocyte Subsets
8.5. CNS-Derived Antigens, CNS Immunosurveillance, and Cells Traveling through Cerebrospinal Fluid into Venous Blood
9. Neuro-Immune and Neuro-Osteogenic Links, Pathologies, and Interventions
9.1. Neuro-Immune Links
9.2. Pathologies and Interventions
- (i)
- CNS lymphoma. In primary CNS lymphoma, attention has turned to the long-term outcomes of consolidation therapies, and recent studies have highlighted the excellent disease control afforded by high-dose chemotherapy and stem cell transplantation [105]. Also, in patients with primary CNS lymphoma, chemoimmunotherapy with methotrexate, cytarabine, thiotepa, and rituximab (MATRix regimen) achieved impressive increases in complete remission rates [106].
- (ii)
- Malignant glioma (GBM). The glioma immune landscape has been described as a double-edged sword for treatment [107]. There are the effects of tumor cells on the tumor microenvironment, the immunosuppressive effects of myeloid immune cells, and the lymphocyte responses against the glioma cells [107]. Clinical and translational advances in malignant glioma immunotherapy have been summarized recently [108]. The review includes vaccine-based therapies, adoptive cell therapies, technical innovation, and outlook [108]. Synergy between temozolomide chemotherapy and individualized multimodal immunotherapy has been reported to improve the overall survival of IDH1 wild-type MGMT promoter-unmethylated GBM [109]. The concept of randomized controlled immunotherapy clinical trials for GBM has been challenged [110].
- (iii)
- Neuro-degenerative and neuro-autoimmune diseases. The role of T cells in brain inflammation has been reviewed [111]. The immune system is deeply involved in autoimmune diseases of the CNS, such as multiple sclerosis (MS), n-methyl-d-aspartate (NMDA) receptor encephalitis, and narcolepsy [111]. Additionally, the immune system is involved in neuro-degenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) [111]. The review focuses on the role of T cells, including CD4+ T cells, CD8+ T cells, and regulatory T cells (Tregs) in cerebral infarction and neuro-degenerative diseases [111]. Myelin oligodendrocyte glycoprotein (MOG) is an important auto (self)-antigen (SA) in inflammatory demyelinating diseases of the CNS [112].
- (iv)
- Chimeric antigen-receptor (CAR) T cells. Chimeric antigen receptor (CAR) T-cell therapy is a new and emerging cell therapy which has achieved remarkable success in the treatment of hematological malignancies [113]. The side effects include prolonged cytopenia (PC). Cytokine analysis after CAR T-cell infusion showed that CXCL12 and stem cell factor were significantly decreased in the BM of patients with PC, suggesting reduced niche cell function [114]. Another study revealed apoptosis of HSCs contributing to BM suppression following CAR T-cell therapy [115]. New technologies involving CAR-cytokine-induced killer cells (CAR-CIK) [116] and synapse-tuned CARs [117] demonstrated selective homing to BM niches [86i] and enhanced anti-tumor immune cell activity [116,117].
9.3. Neuro-Osteogenic Network
10. Bone Marrow–Blood Interaction
10.1. BM Capacity for Cognate T Cell–APC Interactions
10.2. Effects of MSC-Derived Stromal Cells in the BM
- (i)
- Stromal cell–immune cell contact-dependent PI3K and APRIL induces NF-kB signaling and prevents mitochondrial and ER stress of memory plasma cells [140];
- (ii)
- Stromal cell CD80/CD86 expression provides CD28 stimulation in BM-resident plasma cells, leading to sustained antibody responses [141];
- (iii)
- Stromal cells providing superior bio-availability of IL-15 cause upregulation of glucocortioid-induced TNF receptor (GITR) on CD8 MTCs [142];
- (iv)
- Stromal-cell-derived Il-7 mediates homeostasis of naïve and memory CD8 T cells in vivo [143];
- (v)
- Stromal-cell-expressed VCAM-1 holds immune cells in the niche and maintains plasma cell longevity [144].
10.3. Autonomous BM-Derived Adaptive Immune Response
- (i)
- Animals devoid of secondary lymphatic organs are viable, while animals devoid of BM cannot survive and, therefore, do not exist;
- (ii)
- BM is self-sufficient with regard to T cell-mediated immune responses against blood-borne antigens.
11. Bone Marrow: T Regulatory Cells and Dendritic Cells Reacting to Cancer and Microbial Infection
11.1. Epigenetic Regulation
11.2. BM and Treg Cells
11.3. GvL without GvH
11.4. Treg Cells in BM of Ewing Sarcoma Patients
11.5. Tumor-Specific BM Treg Cells in Breast Cancer Patients
11.6. Effect of Microbial Infection and Inflammation on BM
11.6.1. Sepsis
11.6.2. Severe Malaria
11.6.3. Colitis
12. Tumor Dormancy in BM and Maintenance of Tumor-Specific MTCs
13. Clinical Studies Revealing the Importance of Stem-like MTCs
14. Bone Marrow Mesenchymal Stem Cells and Stromal Cells
14.1. Effects on Tumors
14.2. Effects in the Liver: Hepatic Stellate Cells, MTCs, and Liver Fibrosis
14.3. Effects in Regenerative Medicine
- (i)
- Heart failure: Human MSCs are being used to treat patients for heart failure such as cardiomyopathy. A systematic review and meta-analysis revealed that this is an effective treatment modality [185].
- (ii)
- Neurological diseases: Intracerebral hemorrhage (ICH) is a common acute nervous system disease with high mortality and causing severe disability. BM MSC transplantation alleviates brain injury after ICH in mice through the Hippo signaling pathway [186]. Polarized anti-inflammatory MSCs increase hippocampal neurogenesis and improve cognitive function in aged mice [187]. The transplantation of nasal olfactory mucosa MSCs showed benefits in the AD mouse model of Alzheimer’s disease (AD) [188]. An exosomal microRNA (miR-146a) secreted from BM-MSCs is taken up into astrocytes [189]. Intracerebra-ventricularly injected BM-MSCs improve cognitive impairment by increasing the expression of miR-146a in the hippocampus [190]. This is due to an effect of astrocytes, which are key cells for the formation of synapses. Thus, restoration of astrocyte function may lead to synaptogenesis and correction of cognitive impairment [191]. MSCs are likely able to cross the blood–brain barrier. Therefore, MSC-based therapy is regarded as an important means of ameliorating neurological impairment [187,188,189,190,191].
- (iii)
- Lung injury and fibrosis: MSC-derived EVs attenuate radiation-induced lung injury via miRNA-214-3p [192]. MSCs with downregulated Hippo signaling are better than normal MSCs in terms of attenuating lung injury in mice with acute respiratory distress syndrome (ARDS) [193]. The therapeutic efficacy of MSCs has been reported for post-COVID pulmonary fibrosis [194].
- (iv)
- Hepatobiliary diseases and sepsis: Adipose-derived MSCs (AD-MSCs) have the potential to modulate inflammation, ameliorate ischemia–reperfusion injury, and support liver and biliary tract regeneration [195]. The anti-inflammatory potential of these cells also has paramount importance in the treatment of sepsis [196].
- (v)
- Kidney injury and diseases: Murine MSCs protect against sepsis-associated acute kidney injury [196]. Rat MSCs ameliorate chronic kidney disease injury via regulating the Nrf2-keap1/p53 pathway [197]. A multi-therapeutic role of MSCs has been reported in diabetic nephropathy [198], and engineered BM stem cell-sheets were shown to alleviate renal damage in a rat model of chronic glomerulonephritis [199].
- (vi)
- Osteoarthritis: Rheumatic diseases such as osteoarthritis (OA) are a major social and economic burden. MSCs provide a cell source for cartilage regeneration due to numerous advantages, comprising relative ease to isolate and culture, chondrogenic capacity, and anti-inflammatory effects. Preclinical studies for articular cartilage repair have been reviewed [200]. Clinical-grade embryonic stem cell-derived mesenchymal stromal cells were shown to ameliorate the progression of OA in a rat model [201].
- (vii)
- Vascular regeneration: A mechanical conditioning technique applied to human MSCs enhances vascular regeneration [202].
- (viii)
- (ix)
- Autoimmunity: Transplantation of human umbilical cord MSCs ameliorated systemic lupus erythematosus (SLE) in MRL/lpr mice [205]. MSC-derived exosomes ameliorated lupus in the same mouse model by inducing M2 macrophage polarization and Treg cell expansion [206]. Melatonin treatment improved human MSC therapy in a mouse model of type II diabetes mellitus (T2DM) via the PI3K/AKT signaling pathway [207]. Transplanted MSCs ameliorated the clinical course of experimental autoimmune encephalomyelitis (EAE) in a mouse model of multiple sclerosis (MS) [191]. Human umbilical cord-derived MSCs ameliorated psoriasis-like dermatitis by suppressing IL-17 producing γ, δ T cells [208].
15. Highlights and Perspectives
- (i)
- Antigen presentation capacity;
- (ii)
- T-APC interaction capacity;
- (iii)
- Memory cell storage capacity;
- (iv)
- Capacity of maintaining self-tolerance and immune homeostasis;
- (v)
- Capacity for tissue repair;
- (vi)
- Capacity to adapt to periods of energy crisis;
- (vii)
- Ability to sense signals via adrenergic nerve fibers from the autonomous peripheral nervous system;
- (viii)
- Capacity of immunosurveillance of the central nervous system.
| 1. Central: |
|
| 2. Multifunctional: |
|
| 3. Protective: |
|
| Feature | Description |
| Hematopoiesis | HSCs program the major lineages of blood cells Proliferation and maturation of committed precursor cells via colony-stimulating factors, growth factors, cytokines, and chemokines BM stromal cells providing signals for the development of lymphocyte progenitors from HSCs and for subsequent differentiation of pre-B/B cells and pre-T cells Egress to the thymus of BM-derived pre-T cells for further T cell differentiation with positive and negative selection Homeostatic hematopoiesis provides the organism with oxygen, energy, and immune protective capacity |
| Osteogenesis | MSCs program the major lineages of osteogenesis Osteogenesis, like hematopoiesis, is constantly maintained at a steady state HSC-derived osteoclasts interact in balance with MSC-derived osteoblasts and osteocytes |
| Antigen-specific T cell response to blood-borne antigens | Microenvironment facilitates T-APC interactions between BM and blood Priming of naïve T cells Responses resulting in the generation of CTL activity, protective anti-tumor immunity, and immunological memory Cognate antigen-specific re-activation of memory T cells |
| Storage and protection of immunological memory | Multiple niches created by stromal cells providing quiescence and survival signals Stem-like MTCs with special properties |
| Adaptation to energy crisis (transient dietary restriction) | Mobilization of monocytes from the BM Migration of B cells from PPs to BM Recruitment of MTCs from the periphery Increase in T cell homing factors, erythropoiesis, and adipogenesis Reorganization of the BM compartment |
| Central regulatory function within the immune system | Maintenance of homeostasis in concerted interaction of regulatory immune cells with the neuronal network Feedback from the periphery (lymph nodes) leads to central regulatory means in the BM |
| Autonomous life-protecting function | Secondary immune functions of BM independent from the lymph nodes and spleen No animal model without BM exists No bone cortex without BM MSCs Most prominent source of de novo cell generation in the body In communication with the autonomous peripheral nervous system, BM is autonomous in the maintenance of homeostasis |
| BM mesenchymal stem cells and stromal cells for tissue repair and niche formation | MSCs: great potential in regenerative medicine, e.g., heart failure, neurological diseases, lung injury and fibrosis, liver regeneration, kidney injury, vascular regeneration, and autoimmune diseases Stromal cells: Important role in niche formation, e.g., niches for HSCs, MSCs, memory B cells, memory plasma cells, memory CD4+ T cells, memory CD8+ T cells, stem-like memory T cells, and neuronal stem cells |
- As a major immune organ, the BM is a treasure and should receive more attention. Many of its secrets have not yet been resolved and require intensive research efforts. We know more about the immune functions in the lymph nodes and spleen than about those in the BM. It would be fascinating, for instance, to find out how many T-APC immune synapses occur in the BM at any time point and to compare this with other immune organs. Also, the question of adjuvant requirement for vaccination responses in lymph nodes and BM requires further investigations. If the requirements are less for the BM, intra-BM vaccination might reduce the risk of adjuvant-associated side effects.
- Most of the cytostatic anti-cancer drugs that have been approved in the past have been proven to have detrimental effects on the BM (e.g., chlorambucil, melphalan, busulfan, thioguanin, cyclophosphamide, ifosfamide, imatinib, vinblastin, cisplatin, etoposid, chloramphenicol). New drug approval should minimize detrimental drug effects on the BM.
- Among the available immunotherapeutic anti-cancer drugs, the most important are immune checkpoint inhibitors, anti-cancer vaccines, oncolytic viruses, and adoptive T cell therapies. All of these have to be assessed with regard to their side effects on the BM. Cancer vaccines and oncolytic viruses have been found to exert profoundly lower side effects in cancer patients than other systemic therapies.
- Adoptive T cell therapies against cancer should make use of BM-enriched cancer-reactive MTCs, including stem-like MTCs, and their synergistic effects with dendritic cells as APCs and hematopoietic stem cells.
- To achieve improvements to the graft-versus-leukemia reactivity of allogeneic donor cells, vaccination of donors against host tumor cells is recommended. Donor-derived immune BM mononuclear cells from vaccinated donors are likely to contain cancer-reactive MTCs and primed T regulatory cells, preventing graft-versus-host disease.
- Given the fact that hematopoietic and mesenchymal stem cells from the BM interact with T cells and DCs, it is conceivable that in BM, stem cell-derived committed precursors and late precursors also interact with mature immune cells and build informative feedback-regulatory networks.
- Viral and microbial infections, inflammatory reactivities by the immune system, and auto-immune reactivities are of major medical concern. Experimental models of sepsis, malaria, and colitis have revealed effects on BM-derived DCs. New possibilities for central intervention should be explored.
- BM mesenchymal stem cells have great potential for regenerative medicine. This review presents many examples from research conducted within the last few years.
- The BM and the brain are the two centers (i) of learning by experience, (ii) of memory establishment, and (iii) of memory storage.
- The fascinating area of communication between networks of the immune system and those of the neuronal system is a promising future research area in which artificial intelligence (AI) could be helpful. Deep learning in AI attempts to simulate nature’s neuronal network learning process. Positive impacts can be expected, in particular in the fields of neuro-degenerative and neuro-autoimmune diseases.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgements
Conflicts of Interest
Abbreviations
References
- Flajnik, M.F.; Kasahara, M. Origin and evolution of the adaptive immunity system: Genetic events and selective pressures. Nat. Rev. Genet. 2010, 11, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Pancer, Z.; Amemiya, C.T.; Ehrhardt, G.R.A.; Ceitlin, J.; Gartland, G.L.; Cooper, M.D. Somatic diversification of variable lymphocyte receptors in the agnathan sea lamprey. Nature 2004, 430, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Estefa, J.; Tafforeau, P.; Clement, A.M.; Klembara, J.; Niedzwiedzki, G.; Berruyer, C.; Sanchez, S. New light shed on the early evolution of limb-bone growth plate and bone marrow. eLife 2021, 10, e51581. [Google Scholar] [CrossRef] [PubMed]
- Belyavsky, A.; Petinati, N.; Drize, N. Hematopoiesis during ontogenesis, adult life, and aging. Int. J. Mol. Med. 2021, 22, 9231. [Google Scholar] [CrossRef]
- Osmond, D.G. Production and selection of B lymphocytes in bone marrow: Lymphostromal interactions and apoptosis in normal, mutant and transgenic mice. Adv. Exp. Med. Biol. 1994, 355, 15–20. [Google Scholar]
- Koni, P.A.; Joshi, S.K.; Temann, U.A.; Olson, D.; Burkly, I.; Flavell, R.A. Conditional vascular cell adhesion molecule 1 depletion in mice impaired lymphocyte migration to bone marrow. J. Exp. Med. 1994, 355, 15–20. [Google Scholar]
- Abbas, A.; Lichtman, A.; Pillai, S. (Eds.) Cellular and Molecular Immunology, 10th ed.; Elsevier: Philadelphia, PA, USA, 2022. [Google Scholar]
- Shahrabi, S.; Rezaeeyan, H.; Ahmadzadeh, A.; Shahjahani, M.; Saki, N. Bone marrow blood vessels: Normal and neoplastic niche. Oncol. Rev. 2016, 10, 72–77. [Google Scholar] [CrossRef][Green Version]
- Marenzana, M.; Arnett, T.B. The key role of the blood supply to bone. Bone Res. 2013, 1, 203–215. [Google Scholar] [CrossRef]
- Nombela-Arrieta, C.; Manz, M.G. Quantification and three-dimensional microanatomical organization of the bone marrow. Blood Adv. 2017, 1, 407–416. [Google Scholar] [CrossRef]
- Wu, K.; Li, R.; Zhang, Y.; Liu, Y.; Wang, M.; Huang, J.; Zhu, C.; Zhang, J.; Yuan, X.; Liu, Q. The discovery of a new type of innervation in lymphoid organs. Physiol. Rep. 2023, 11, e15604. [Google Scholar] [CrossRef]
- Pabst, R. The bone marrow is not only a primary lymphoid organ: The critical role for T lymphocyte migration and housing of long-term plasma cells. Eur. J. Immunol. 2018, 48, 1096–1100. [Google Scholar] [CrossRef]
- Feuerer, M.; Rocha, M.; Bai, L.; Umansky, V.; Solomayer, E.F.; Bastert, G.; Diel, I.J.; Schirrmacher, V. Einrichment of memory T cells and other profound immunological changes in the bone marrow from untreated breast cancer Patients. Int. J. Cancer 2001, 92, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Waxenbaum, J.A.; Reddy, V.; Futterman, B. Anatomy, back, thoracic vertebrae. In StatPearls (Internet); StatPearls Publishing: Treasure Islands, FL, USA, 2023; Bookshelf ID: NBK459153. [Google Scholar] [PubMed]
- Ordovas-Montanes, J.; Rakoff-Nahoum, S.; Huang, S.; Riol-Blanco, L.; Barreiro, O.; von Andrian, U.H. The regulation of immunological processes by peripheral neurons in homeostasis and disease. Trends Immunol. 2015, 36, 578–604. [Google Scholar] [CrossRef] [PubMed]
- Comazzetto, S.; Murphy, M.M.; Berto, S.; Jeffery, E.; Zhao, Z.; Morrison, S.J. Restricted hematopoietic progenitors and erythropoiesis require SCF from Leptin receptor+ niche cells in the bone marrow. Cell Stem Cell 2019, 24, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Beckhove, P.; Garbi, N.; Mahnke, Y.; Limmer, A.; Hommel, M.; Hämmerling, G.J.; Kyewski, B.; Hamann, A.; Umansky, V.; et al. Bone marrow as a priming site for T-cell responses to blood-borne antigen. Nat. Med. 2003, 9, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Mazo, I.B.; von Andrian, U.H. Adhesion and homing of blood-borne cells in bone marrow microvessels. J. Leukoc. Biol. 1999, 66, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Beckhove, P.; Bai, L.; Solomayer, E.F.; Bastert, G.; Diel, I.J.; Pedain, C.; Oberniedermayr, M.; Schirrmacher, V.; Umansky, V. Therapy of human tumors in NOD/SCID mice with patient-derived reactivated memory T cells from bone marrow. Nat. Med. 2001, 7, 452–458. [Google Scholar] [CrossRef]
- Feuerer, M.; Beckhove, P.; Mahnke, Y.; Hommel, M.; Kyewsky, B.; Hamann, A.; Umansky, V.; Schirrmacher, V. Bone marrow microenvironment facilitating dendritic cell: CD4 T cell interactions and maintenance of CD4 memory. Int. J. Oncol. 2004, 25, 867–876. [Google Scholar]
- Khazaie, K.; Prifti, S.; Beckhove, P.; Griesbach, A.; Russel, S.; Collins, M.; Schirrmacher, V. Persistence of dormant tumor cells in the bone marrow of tumor cell-vaccinated mice correlates with long-term immunological protection. Proc. Natl. Acad. Sci. USA 1994, 91, 7430–7434. [Google Scholar] [CrossRef]
- Müller, M.; Gounari, F.; Prifti, S.; Hacker, H.J.; Schirrmacher, V.; Khazaie, K. EblacZ tumor dormancy in bone marrow and lymph nodes: Active control of proliferating tumor cells by CD8+ immune T cells. Cancer Res. 1998, 58, 5439–5446. [Google Scholar]
- Milo, I.; Sapoznikov, A.; Kalchenko, V.; Tal, O.; Krauthammer, R.; van Rooijen, N.; Dudziak, D.; Jung, S.; Shakkar, G. Dynamic imaging reveals promiscuous crosspresentation of blood-borne antigens to naïve CD8+ T cells in the bone marrow. Blood 2013, 122, 193–208. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Singh, S.K.; Stephani, J.; Schaefer, M.; Kalay, H.; Garcia-Vallejo, J.J.; den Haan, J.; Saeland, E.; Sparwasser, T.; van Kooyk, Y. Targeting glycan modified OVA to murine DC-SIGN transgenic dendritic cells enhances MHC class I and II presentation. Mol. Immunol. 2009, 47, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Feuerer, M.; Fournier, P.; Ahlert, T.; Umansky, V.; Beckhove, P. T-cell priming in bone marrow: The potential for long-lasting protective anti-tumor immunity. Trends Mol. Med. 2003, 9, 526–534. [Google Scholar] [CrossRef]
- Schirrmacher, V. Cancer-reactive memory T cells from bone marrow: Spontaneous induction and therapeutic potential (Review). Int. J. Oncol. 2015, 47, 2005–2016. [Google Scholar] [CrossRef]
- Bai, L.; Feuerer, M.; Beckhove, P.; Umansky, V.; Schirrmacher, V. Generation of dendritic cells from human bone marrow mononuclear cells: Advantages for clinical application in comparison to peripheral blood monocyte derived cells. Int. J. Oncol. 2002, 20, 247–253. [Google Scholar] [CrossRef]
- Sommerfeldt, N.; Schütz, F.; Sohn, C.; Förster, J.; Schirrmacher, V.; Beckhove, P. The shaping of a polyvalent and highly individual T-cell repertoire in the bone marrow of breast cancer patients. Cancer Res. 2006, 66, 8258–8265. [Google Scholar] [CrossRef]
- Sommerfeldt, N.; Beckhove, P.; Ge, Y.; Schütz, F.; Choi, C.; Bucur, M.; Domschke, C.; Sohn, C.; Schneeweis, A.; Rom, J.; et al. Heparanase: A new metastasis-associated antigen recognized in breast cancer patients by spontaneously induced memory T lymphocytes. Cancer Res. 2006, 66, 7716–7723. [Google Scholar] [CrossRef]
- Yoshida, T.; Mei, H.; Dörner, T.; Hiepe, F.; Radbruch, A.; Fillatreau, S.; Hoyer, B.F. Memory B and memory plasma cells. Immunol. Rev. 2010, 237, 117–139. [Google Scholar] [CrossRef] [PubMed]
- Slamanig, S.A.; Nolte, M.A. The bone marrow as sanctuary for plasma cells and memory T-cells: Implications for adaptive immunity and vaccinology. Cells 2021, 10, 1508. [Google Scholar] [CrossRef]
- Schirrmacher, V. New insights into mechanisms of long-term protective anti-tumor immunity induced by cancer vaccines modified by virus infection. Biomedicines 2020, 8, 55. [Google Scholar] [CrossRef]
- Di Rosa, F.; Pabst, R. The bone marrow: A nest for migratory memory T cells. Trends Immunol. 2005, 26, 360–366. [Google Scholar] [PubMed]
- Mazo, I.B.; Honczarenko, M.; Leung, H.; Cavanagh, L.L.; Bonasio, R.; Weninger, W.; Engelke, K.; Xia, L.; McEver, R.P.; Koni, P.A.; et al. Bone marrow as a major reservoir and site of recruitment for central memory CD8+ T cells. Immunity 2005, 22, 259–270. [Google Scholar] [CrossRef]
- Becker, T.C.; Coley, S.M.; Wherry, E.J.; Ahmed, R. Bone marrow is a preferred site for homeostatic proliferation of memory CD8 T cells. J. Immunol. 2005, 174, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, H.; Lin, W.; Voss, S.; Hinkley, L.; Westergren, M.; Tian, G.; Berry, D.; Lewellen, D.; Vile, R.G.; et al. Human bone marrow: A reservoir for “enhanced effector memory” CD8+ T cells with potent recall function. J. Immunol. 2006, 177, 6730–6737. [Google Scholar] [CrossRef] [PubMed]
- Murao, A.; Oka, Y.; Tsuboi, A.; Elisseeva, O.A.; Tanaka-Harada, Y.; Fujiki, F.; Nakajima, H.; Nishida, S.; Hosen, N.; Shirakata, T.; et al. High frequencies of less differentiated and more proliferative WT1-specific CD8+ T cells in bone marrow in tumor-bearing patients: An important role of bone marrow as a secondary lymphoid organ. Cancer Sci. 2010, 101, 848–854. [Google Scholar] [CrossRef]
- Schmitz-Winnenthal, F.H.; Volk, C.; Z’graggen, K.; Galindo, L.; Nummer, D.; Ziouta, Y.; Bucur, M.; Weitz, J.; Schirrmacher, V.; Büchler, M.W.; et al. High frequencies of functional tumor-reactive T cells in bone marrow and blood of pancreatic cancer patients. Cancer Res. 2005, 65, 10079–10087. [Google Scholar] [CrossRef]
- Choi, C.; Witzens, M.; Bucur, M.; Feuerer, M.; Sommerfeld, N.; Trojan, A.; Ho, A.; Schirrmacher, V.; Goldschmidt, H.; Beckhove, P. Enrichment of functional CD8 memory T cells specific for MUC1 in bone marrow of patients with multiple myeloma. Blood 2005, 105, 2132–2134. [Google Scholar] [CrossRef]
- Sung, J.H.; Zhang, H.; Moseman, E.A.; Alvarez, D.; Iannacone, M.; Henrickson, S.E.; de la Torre, J.C.; Groom, J.R.; Luster, A.D.; von Andrian, U.H. Chemokine guidance of central memory T cells is critical for antiviral recall responses in lymph nodes. Cell 2012, 150, 1249–1263. [Google Scholar] [CrossRef]
- Hanazawa, A.; Hayashizaki, K.; Shinoda, K.; Yagita, H.; Okumura, K.; Lohning, M.; Hara, T.; Tani-ichi, S.; Ikuta, K.; Eckes, B.; et al. CD49b-dependent establishment of T helper cell memory. Immunol. Cell Biol. 2013, 91, 524–531. [Google Scholar] [CrossRef]
- Alp, S.Ö.; Durlanik, S.; Schulz, D.; McGrath, M.; Grün, J.R.; Bardua, M.; Ikuta, K.; Sgouroudis, E.; Riedel, R.; Zehentmeier, S.; et al. Memory CD8+ T cells colocalize with IL-7+ stromal cells in bone marrow and rest in terms of proliferation and transcription. Europ. J. Immunol. 2015, 45, 975–987. [Google Scholar]
- Herndler-Brandstetter, D.; Landgraf, K.; Jenewein, B.; Tzankow, A.; Brunauer, R.; Brunner, S.; Parson, W.; Kloss, F.; Gassner, R.; Lepperdinger, G.; et al. Human bone marrow hosts polyfunctional memory CD4+ and CD8+ T cells with close contact to IL-15 producing cells. J. Immunol. 2011, 186, 6965–6971. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A.; Waldmann, R.; Lin, J.-X.; Leonard, W.J. The implications of IL-15 trans-presentation on the immune response. Adv. Immunol. 2022, 156, 103–132. [Google Scholar] [CrossRef] [PubMed]
- Pascutti, M.F.; Geerman, S.; Collins, N.; Brasser, G.; Nota, B.; Stark, R.; Behr, F.; Oja, A.; Slot, E.; Panagioti, E.; et al. Peripheral and systemic antigens elicit an expandable pool of resident memory CD8+ T cells in the bone marrow. Eur. J. Immunol. 2019, 49, 853–872. [Google Scholar] [CrossRef]
- Wang, S.; Wang, L.; Liu, Y.; Zhu, Y.; Liu, Y. Characteristics of T-cell receptor repertoire of stem cell-like memory CD4+ T cells. PeerJ. 2021, 9, e11987. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Reinke, S.; Shen, Y.; Schollmeyer, L.; Liu, Y.-C.; Wang, Z.; Hardt, S.; Hipfl, C.; Hoffmann, U.; Frischbutter, S.; et al. A ubiquitous bone marrow reservoir of preexisting SARS-CoV-2-reactive memory CD4+ T lymphocytes in unexposed individuals. Front. Immunol. 2022, 13, 1004656. [Google Scholar] [CrossRef]
- Kudernatsch, R.F.; Letsch, A.; Guerreiro, M.; Löbel, M.; Bauer, S.; Volk, H.D.; Scheibenbogen, C. Human bone marrow contains a subset of quiescent early memory CD8+ T cells characterized by high CD127 expression and efflux capacity. Eur. J. Immunol. 2014, 44, 3532–3542. [Google Scholar] [CrossRef]
- Wu, K.; Li, Y.; Zhang, S.; Zhou, N.; Liu, B.; Pan, T.; Zhang, X.; Luo, H.; Huang, Z.; Li, X.; et al. Preferential homing of tumor-specific and functional CD8+ stem cell-like memory T cells to the bone marrow. J. Immunother. 2019, 42, 197–207. [Google Scholar] [CrossRef]
- Wu, K.; Wang, F.; Guo, G.; Li, Y.; Qiu, L.-J.; Li, X. CD4+ TSCMs in the bone marrow assist in maturation of antibodies against Influenza in mice. Mediat. Inflamm. 2019, 2019, 3231696. [Google Scholar] [CrossRef]
- Chi, X.; Luo, S.; Ye, P.; Hwang, W.-L.; Cha, J.-H.; Yan, X.; Yang, W.-H. T-cell exhaustion and stemness in anti-tumor immunity: Characteristics, mechanisms, and implications. Front. Immunol. 2023, 20, 4771. [Google Scholar] [CrossRef]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef]
- Cieri, N.; Camissa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Prosvasi, F.; Bondanza, A.; Bordignon, C.; Peccatori, J.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem cells from naïve precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Imura, Y.; Ando, M.; Chikuma, S.; Yoshimura, A. In Vitro conversion of activated T cells into stem cell memory-like T cells. Methods Mol. Biol. 2019, 2048, 41–51. [Google Scholar]
- Palendira, U.; Chinn, R.; Raza, W.; Piper, K.; Pratt, G.; Machado, L.; Bell, A.; Khan, N.; Hislop, A.D.; Steyn, R.; et al. Selective accumulation of virus-specific CD8+ T cells with unique homing phenotype within human bone marrow. Blood 2008, 112, 3293–3302. [Google Scholar] [CrossRef]
- Pangrazzi, L.; Naismith, E.; Meryk, A.; Keller, M.; Jenewein, B.; Trieb, K.; Grubeck-Loebenstein, B. Increased IL-15 production and accumulation of highly differentiated CD8+ effector/memory T cells in the bone marrow of persons with cytomegalovirus. Front. Immunol. 2017, 8, 715. [Google Scholar] [CrossRef]
- Racanelli, V.; Frassanito, M.A.; Leone, P.; Brunetti, C.; Ruggien, S.; Dammacco, F. Bone marrow of persistently hepatitis C virus-infected individuals accumulates memory CD8+ T cells specific for current and historical viral antigens: A study in patients with benign hematological disorders. J. Immunol. 2007, 179, 5387–5398. [Google Scholar] [CrossRef]
- Na, I.K.; Letsch, A.; Guerreio, M.; Bauer, S.; Noack, I.; Geginat, J.; Reinke, P.; Loesch, M.; Kienapfel, H.; Thiel, E.; et al. Human bone marrow as a source to generate CMV-specific CD4+ T cells with multifunctional capacity. J. Immunother. 2009, 32, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Lineburg, K.E.; Martins, J.P.; Ambalathingal, G.R.; Neller, M.A.; Morrison, B.; Matthews, K.K.; Rehan, S.; Crooks, P.; Panikkar, A.; et al. Autologous CMV-specific T cells are a safe adjuvant immunotherapy for primary glioblastoma multiforme. J. Clin. Investig. 2020, 130, 6041–6053. [Google Scholar] [CrossRef] [PubMed]
- DiRosa, F. Two niches in the bone marrow: A hypothesis on life-long T cell memory. Trends Immunol. 2016, 17, 503–512. [Google Scholar] [CrossRef]
- Chang, H.-D.; Radbruch, A. Maintenance of quiescent immune memory in the bone marrow. Eur. J. Immunol. 2021, 51, 1592–1601. [Google Scholar] [CrossRef]
- Chang, H.-D.; Tokoyoda, K.; Radbruch, A. Immunological memories of the bone marrow. Immunol. Rev. 2018, 283, 86–98. [Google Scholar] [CrossRef]
- Rajewsky, K.; Schirrmacher, V.; Nase, S.; Jerne, N. The requirement for more than one antigenic determinant for immunogenicity. J. Exp. Med. 1969, 129, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Fresnay, S.; Zhang, X.; Strome, S.; Sewell, D.A. Bone marrow vaccination: A novel approach to enhance antigen specific antitumor immunity. Vaccine 2011, 29, 8599–8605. [Google Scholar] [CrossRef]
- Kushida, T.; Inaba, M.; Ichioka, N.; Esumi, T.; Ogawa, R.; Iida, H.; Ikehara, S. Intra-bone marrow injection of allogeneic bone marrow cells: A powerful new strategy for treatment of intractable autoimmune diseases in MRL/lpr mice. Blood 2001, 97, 3292–3299. [Google Scholar] [CrossRef]
- Kushida, T.; Inaba, M.; Ichioka, N.; Esumi, T.; Ogawa, R.; Iida, H.; Ikehara, S. Crucial role of donor-derived stromal cells in successful treatment for intractable autoimmune diseases in mrl/lpr mice by bmt via portal vein. Stem Cells 2001, 19, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Wildes, T.J.; Grippin, A.; Dyson, K.A.; Wummer, B.M.; Damiani, D.; Abraham, R.S.; Flores, C.T.; Mitchell, D.A. Cross-talk between T cells and hematopoietic stem cells during adopitive cellular therapy for malignant glioma. Clin. Cancer Res. 2019, 24, 3955–3966. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yao, J.-C.; Oetjen, K.A.; Krambs, J.R.; Xia, J.; Zhang, J.; Schmidt, A.P.; Helton, N.M.; Fulton, R.S.; Heath, S.E.; et al. Il-1β expression in bone marrow dendritic cells is induced by TLR2 agonists and regulates HSC function. Blood 2022, 140, 1607–1620. [Google Scholar] [CrossRef]
- Niu, C.; Yu, J.; Zou, T.; Lu, Y.; Deng, L.; Yun, H.; Si, C.-Y.; Wu, X.; Jiang, H.; Guo, T.; et al. Identification of hematopoietic stem cells residing in the meninges of adult mice at steady state. Cell Rep. 2022, 41, 111592. [Google Scholar] [CrossRef]
- Tsunokuma, N.; Yamane, T.; Matsumoto, C.; Tsuneto, M.; Isono, K.; Imanaka-Yoshida, K.; Yamazaki, H. Depletion of neural crest-derived cells leads to reduction in plasma noradrenaline and alters B lymphopoiesis. J. Immunol. 2017, 198, 156–169. [Google Scholar] [CrossRef]
- Ott, L.C.; Han, C.Y.; Mueller, J.L.; Rahman, A.A.; Hotta, R.; Goldstein, A.M.; Stavely, R. Bone marrow stem cells derived from nerves have neurogenic properties and potential uitility for regenerative therapy. Int. J. Mol. Sci. 2023, 24, 5211. [Google Scholar] [CrossRef]
- Coste, C.; Neirinckx, V.; Sharma, A.; Agirman, G.; Rogister, B.; Foguenne, J.; Lallemend, F.; Gothot, A.; Wislet, S. Human bone marrow harbors cells with neural crest-associated characteristics like human adipose and dermis tissues. PLoS ONE 2017, 12, e0177962. [Google Scholar] [CrossRef]
- Khan, A.A.; Huat, T.J.; Mutery, A.A.; El-Serafi, A.T.; Kacem, H.H.; Abdullah, S.H.; Reza, M.F.; Abdullah, J.M.; Jaafar, H. Significant transcriptomic changes are associated with differentiation of bone marrow-derived mesenchymal stem cells into neural progenitor-like cells in the presence of bFGF and EGF. Cell Biosci. 2020, 10, 126. [Google Scholar] [CrossRef]
- Akhter, W.; Nakhle, J.; Vaillant, L.; Garcin, G.; Le Saout, C.; Simon, M.; Crozet, C.; Djouad, F.; Jorgensen, C.; Vignais, M.-L.; et al. Transfer of mesenchymal stem cell mitochondria to CD4+ T cells contributes to repress Th1 differentiation by downregulating T-bet expression. Stem Cell Res. Ther. 2023, 14, 12. [Google Scholar] [CrossRef]
- Schirrmacher, V. Mitochondria at work: New insights into regulation and dysregulation of cellular energy supply and metabolism. Biomedicines 2020, 8, 526. [Google Scholar] [CrossRef]
- Jordan, S.; Tung, N.; Casanova-Acebes, M.; Chang, C.; Cantoni, C.; Zhang, D.; Wirtz, T.H.; Naik, S.; Rose, S.A.; Brocker, C.N.; et al. Dietary intake regulates the circulating inflammatory monocyte pool. Cell 2019, 178, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Noguchi, R.; Takahashi, D.; Morikawa, T.; Koshida, K.; Komiyama, S.; Ishihara, N.; Yamada, T.; Kawamura, Y.I.; Muroi, K.; et al. Fasting-refeeding impacts immune cell dynamics and mucosal immune responses. Cell 2019, 178, 1072–1087. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 178, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Collins, N.; Han, S.-J.; Enamorado, M.; Link, V.M.; Huang, B.; Moseman, A.; Kishton, R.J.; Shannon, J.P.; Dixit, D.; Schwab, S.R.; et al. The bone marrow protects and optimizes immunological memory during dietary restriction. Cell 2019, 178, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Vollmann, E.H.; Rattay, K.; Barreiro, O.; Thiriot, A.; Fuhlbrigge, R.A.; Vrbanac, V.; Kim, K.-W.; Jung, S.; Tager, A.M.; von Andrian, U.H. Specialized transendothelial dendritic cells mediate thymic T-cell selection agains blood-borne macromolecules. Nat. Commun. 2021, 12, 6230. [Google Scholar] [CrossRef]
- Matioubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef]
- Waller, K.M.; De La Mata, N.L.; Kelly, P.J.; Ramachandran, V.; Rawlinson, W.D.; Wyburn, K.R.; Webster, A.C. Residual risk of infection with blood-borne viruses in potential organ donors at increased risk of infection: Systematic review and meta-analysis. Med. J. Aust. 2019, 211, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Cariappa, A.; Mazo, I.B.; Chase, C.; Shi, H.N.; Liu, H.; Rose, H.; Leung, H.; Cherayil, B.J.; Russell, P.; von Andrian, U.; et al. Perisinusoidal B cells in the bone marrow participate in T-independent responses to blood-borne microbes. Immunity 2005, 23, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kometani, K.; Minato, N.; Hamazaki, Y. Bone marrow endothelial cells take up blood-borne immune complexes via Fcg receptor IIb2 in an erythropoitin-dependent manner. J. Immunol. 2020, 205, 2008–2015. [Google Scholar] [CrossRef]
- He, S.; Ding, L.; Yuan, H.; Zhan, G.; Yang, X.; Wu, Y. A review of sensors for classification and subtype discrimination of cancer: Insights into circulating tumor cells and tumor-derived extracellular vesicles. Anal. Chim. Acta 2023, 1244, 340703. [Google Scholar] [CrossRef] [PubMed]
- Rak, J.; Strzadala, L. Heterogeneity of extracellular vesicles and particles: Molecular voxels in the blood borne “hologram” of organ function, disfunction and cancer. Arch. Immunol. Ther. Exp. 2023, 71, 5. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, J.; Hao, H.; Lu, F.; Wang, J.; Ma, M.; Jia, B.; Zhou, M.; Wang, J.; Chi, Y.; et al. Secreted proteins MDK, WFDC2, and CXCL14 as candidate biomarkers for early diagnosis of lung adenocarcinoma. BMC Cancer 2023, 23, 110. [Google Scholar] [CrossRef]
- Tripathy, A.; John, V.; Wadden, J.; Kong, S.; Sharba, S.; Koschmann, C. Liquid biopsy in pedriatic brain tumors. Front. Genet. 2023, 13, 1114762. [Google Scholar] [CrossRef]
- Stergiopoulou, D.; Markou, A.; Strati, A.; Zavridou, M.; Tzanikou, E.; Mastoraki, S.; Kallergi, G.; Georgoulias, V.; Lianidou, E. Comprehensive liquid biopsy analysis as a tool for the early detection of minimal residual disease in breast cancer. Sci. Rep. 2023, 13, 1258. [Google Scholar] [CrossRef]
- Someya, M.; Hasegawa, T.; Tsuchiya, T.; Kitagawa, M.; Fukushima, Y.; Gocho, T.; Mafune, S.; Ikeuchi, Y.; Kozuka, Y.; Idogawa, M.; et al. Predictive value of an exosomal microRNA-based signature for tumor immunity in cervical cancer patients treated with chemoradiotherapy. Med. Mol. Morphol. 2023, 56, 38–45. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Tokita, S.; Hirama, T.; Kochin, V.; Nakatsugawa, M.; Shinkawa, T.; Hirohashi, Y.; Tsukahara, T.; Hata, F.; Takemasa, I.; et al. CD8+ T-cell immune surveillance against a tumor antigen encoded by the oncogenic long noncoding RNA PVT1. Cancer Immunol. Res. 2021, 9, 1342–1353. [Google Scholar] [CrossRef]
- Galluzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef]
- Garg, A.D.; Vanderberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool, S.W.; Agostinis, P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci. Transl. Med. 2016, 8, 328ra27. [Google Scholar] [CrossRef]
- Cavanagh, L.L.; Bonasio, R.; Mazo, I.B.; Halin, C.; Cheng, G.; van der Welden, A.W.M.; Cariappa, A.; Chase, C.; Russell, P.; Starnbach, M.N.; et al. Activation of bone marrow-resident memory T cells by circulating, antigen-bearing dendritic cells. Nat. Immunol. 2005, 6, 1029–1037. [Google Scholar] [CrossRef]
- Alvarez, D.; Vollmann, E.H.; von Andrian, U.H. Mechanisms and consequences of dendritic cell migration. Immunity 2008, 29, 325. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, A.; Li, W.; Liu, Y.; Zhang, G.; Ye, S.; Zhao, Z.; Shi, J.; Jia, Y.; Liu, X.; et al. Reference range of naïve T and T memory lymphocyte subsets in peripheral blood of healthy adult. Clin. Trial. 2022, 207, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Li, W.; Li, Y.; Yu, J. The clinical value of the changes of peripheral lymphocyte subsets absolute counts in patients with non-small cell lung cancer. Transl. Oncol. 2020, 13, 849. [Google Scholar] [CrossRef] [PubMed]
- Mollgard, K.; Beinlich, F.R.M.; Kusk, P.; Miyakoshi, L.M.; Delle, C.; Pla, V.; Hauglund, N.L.; Esmail, T.; Rasmussen, M.K.; Gomolka, R.S.; et al. A mesothelium divides the subarachnoid space into functional compartments. Science 2023, 378, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Mills, W.A., 3rd; Coburn, M.A.; Eyo, U.B. The emergence of the calvarial hematopoietic niche in health and disease. Immunol. Rev. 2022, 311, 26–38. [Google Scholar] [CrossRef]
- Petry, P.; Oschwald, A.; Kierdorf, K. Microglia tissue surveillance: The never-resting gardener in the developing and adult CNS. Eur. J. Immunol. 2023, 53, e2250232. [Google Scholar] [CrossRef]
- Arutyunov, A.; Klein, R.S. Microglia at the scene of the crime: What their transcriptomics reveal about brain health. Curr. Opin. Neurol. 2023, 36, 207–213. [Google Scholar] [CrossRef]
- Dzyubenko, E.; Hermann, D.M. Role of glia and extracellular matrix in controlling neuroplasticity in the central nervous system. Semin. Immunopathol. 2023, 45, 377–387. [Google Scholar] [CrossRef]
- Klimov, V.; Cherevko, N.; Klimov, A.; Novikov, P. Neuronal-immune cell units in allergic inflammation in the nose. Int. J. Mol. Sci. 2022, 23, 6938. [Google Scholar] [CrossRef]
- Ji, H.; Lai, D.; Tou, J. Neuroimmune regulation in Hirschsprung’s disease associated enterocolitis. Front. Immunol. 2023, 14, 1127375. [Google Scholar] [CrossRef] [PubMed]
- Barden, M.M.; Omuro, A.M. Top advances of the year: Neuro-oncology. Cancer 2023, 129, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Ferren, A.J.M.; Cwynarski, K.; Pulczynski, E.; Ponzoni, M.; Deckert, M.; Politi, L.S.; Torri, V.; Fox, C.P.; Rosée, P.L.; Schorb, E.; et al. Chemoimmunotherapy with methotrexate, cytarabine, thiotepa, and rituximab (MATRix regimen) in patients with primary CNS lymphoma: Results of the first randomisation of the International extranodal lymphoma study group-32 (IELSG32) phase 2 trial. Lancet Hematol. 2016, 3, e217–e227. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.; Schmidt, M.H.H.; Schuman, U. The glioma immune landscape: A double-edged sword for treatment regimens. Cancers 2023, 15, 2024. [Google Scholar] [CrossRef]
- Bunse, L.; Bunse, T.; Krämer, C.; Chih, Y.-C.; Platten, M. Clinical and translational advances in glioma immunotherapy. Neurotherapeutics 2022, 19, 1799–1817. [Google Scholar] [CrossRef]
- Van Gool, S.W.; Makalowski, J.; Bitar, M.; Van de Vliet, P.; Schirrmacher, V.; Stuecker, W. Synergy between TMZ and individualized multimodal immunotherapy to improve overall survival of IDH1 wild-type MGMT promoter-unmethylated GBM patients. Genes Immun. 2022, 23, 255–259. [Google Scholar] [CrossRef]
- Van Gool, S.W.; Makalowski, J.; Fiore, S.; Sprenger, T.; Prix, L.; Schirrmacher, V.; Stuecker, W. Randomized-controlled immunotherapy clinical trials for GBM challenged. Cancers 2020, 13, 32. [Google Scholar] [CrossRef]
- Yoshimura, A.; Ohyagi, M.; Ito, M. T cells in the brain inflammation. Adv. Immunol. 2023, 157, 29–58. [Google Scholar] [CrossRef]
- Eliseeva, D.D.; Zakharova, M.N. Myelin oligodendrocyte glycoprotein as an autoantigen in inflammatory demyelinating disease of the Central nervous system. Biochemistry 2023, 88, 551–563. [Google Scholar] [CrossRef]
- Hu, Y.; Feng, J.; Gu, T.; Wang, L.; Wang, Y.; Zhou, L.; Hong, R.; Yin, E.T.S.; Zhang, M.; Lu, P.; et al. CAR T-cell therapies in China: Rapid evolution and a bright future. Lancet Hematol. 2022, 9, e930–e941. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, W.; Asada, N.; Naoi, Y.; Abe, M.; Fujiwara, H.; Ennishi, D.; Nishimori, H.; Fujii, K.; Fujii, N.; Matsuoka, K.-I.; et al. Bone marrow microenvironment disruption and sustained inflammation with prolonged haematologic toxicity after CAR T-cell therapy. Br. J. Haematol. 2023, 202, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Read, J.A.; Rouce, R.H.; Mo, F.; Mamonkin, M.; King, K.Y. Apoptosis of hematopoietic stem cells contributes to bone marrow suppression following chimeric antigen receptor T cell therapy. Transplant. Cell Ther. 2023, 29, e1–e165. [Google Scholar] [CrossRef] [PubMed]
- Biondi, M.; Tettamanti, S.; Galimberti, S.; Cerina, B.; Tomasoni, C.; Piazza, R.; Donsante, S.; Bido, S.; Perriello, V.M.; Broccoli, V.; et al. Selective homing of CAR-CIK cells to the bone marrow niche enhances control of the Acute myeloid leukemia burden. Blood 2023, 141, 2587–2598. [Google Scholar] [CrossRef]
- Chockley, P.J.; Ibanez-Vega, J.; Krenciute, G.; Talbot, L.J.; Gottschalk, S. Synapse-tuned CARs enhance immune cell anti-tumor activity. Nat. Biotechnol. 2023, 2013, 1–12. [Google Scholar] [CrossRef]
- Tao, R.; Mi, B.; Hu, Y.; Lin, S.; Xiong, Y.; Lu, X.; Panayi, A.C.; Li, G.; Liu, G. Hallmarks of peripheral nerve function in bone regeneration. Bone Res. 2023, 11, 6. [Google Scholar] [CrossRef]
- Liu, S.; Liu, S.; Li, S.; Liang, B.; Han, X.; Liang, Y.; Wie, X. Nerves within bone and their application in tissue engineering of bone regeneration. Front. Neurol. 2023, 13, 1085560. [Google Scholar] [CrossRef]
- Manz, B.N.; Jackson, B.L.; Petit, R.S.; Dustion, M.L.; Groves, J. T-cell triggering thresholds are modulated by the number of antigen within individual T-cell clusters. Proc. Natl. Acad. Sci. USA 2011, 108, 9089–9094. [Google Scholar] [CrossRef]
- Henrickson, S.E.; Perro, M.; Loughhead, S.M.; Senman, B.; Stutte, S.; Quigley, M.; Alexe, G.; Iannacone, M.; Flynn, M.P.; Omid, S.; et al. Antigen availability determines CD8+ T cell-dendritic cell interaction kinetics and memory fate decisions. Immunity 2013, 39, 496–507. [Google Scholar] [CrossRef]
- Borger, J.; Zamoyska, R.; Gakamsky, D.M. Proximity of TCR and its CD8 coreceptor controls sensitivity of T cells. Immunol. Lett. 2014, 157, 16–22. [Google Scholar] [CrossRef]
- Sedwick, C.E.; Morgan, M.M.; Jusino, L.; Cannon, J.L.; Miller, J.; Burkhardt, J.K. TCR, LFA-1, and CD28 play unique and complementary roles in signaling T cell cytoskeletal reorganization. J. Immunol. 1999, 162, 1367–1375. [Google Scholar] [CrossRef]
- Gaud, G.; Lesourne, R.; Love, P.E. Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol. 2018, 18, 485–497. [Google Scholar] [CrossRef]
- Geltink, R.I.K.; O’Sullivan, D.; Corrado, M.; Bremser, A.; Buck, M.D.; Buescher, J.M.; Firat, E.; Zhu, X.; Niedermann, G.; Caputa, G.; et al. Mitochondrial priming by CD28. Cell 2017, 171, 385–397.e11. [Google Scholar] [CrossRef]
- Panda, A.K.; Kim, Y.; Shevach, E.M. Control of memory phenotype T lymphocyte homeostasis: The role of costimulation. J. Immunol. 2022, 208, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Martin-Cófreces, N.B.; Valpuesta, J.M.; Sánchez-Madrid, F. Folding for the immune synapse: CCT chaperonin and the cytoskeleton. Front. Cell Dev. Biol. 2021, 9, 658460. [Google Scholar] [CrossRef] [PubMed]
- Lanzavecchia, A.; Sallusto, F. From synapses to immunological memory: The role of sustained T cell stimulation. Curr. Opin. Immunol. 2000, 12, 92–98. [Google Scholar] [CrossRef]
- Waldman, M.M.; Rahkola, J.T.; Sigler, A.L.; Chung, J.W.; Willett, B.A.S.; Kedl, R.M.; Friedman, R.S.; Jacobelli, J. Ena/VASP protein-mediated actin polymerization contributes to naïve CD8+ T cell activation and expansion by promoting T cell-APC interactions in vivo. Front. Immunol. 2022, 13, 856977. [Google Scholar] [CrossRef]
- Kaitao, L.; William, R.; Zhou, Y.; Cheng, Z. Single-molecule investigations on T-cell activation. Curr. Opin. Biomed. Eng. 2019, 12, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Cassioli, C.; Baldari, C.T. Lymphocyte polarization during immune synapse assembly: Centrosomal actin joins the game. Front. Immunol. 2022, 13, 830835. [Google Scholar] [CrossRef] [PubMed]
- Mastrogiovanni, M.; Juzanz, M.; Alcover, A.; Di Bartolo, V. Coordinating cytoskeleton and molecular traffic in T cell migration, activation, and effector functions. Front. Cell Dev. Biol. 2020, 8, 591348. [Google Scholar] [CrossRef] [PubMed]
- Manes, T.D.; Pober, J.S. T cell receptor-driven transendothelial migration of human effector memory CD4 T cells involves Vav, Rac, and Myosin IIA. J. Immunol. 2013, 190, 3079–3088. [Google Scholar] [CrossRef] [PubMed]
- Manes, T.D.; Wang, V.; Pober, J.S. Costimulators expressed on human endothelial cells modulate antigen-dependent recruitment of circulating T lymphocytes. Front. Immunol. 2022, 13, 1016361. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Schlude, C.; Weitz, J.; Beckhove, P. Strong T-cell costimulation can reactivate tumor antigen-specific T cells in late-stage metastasized colorectal carcinoma patients: Results from a phase I clinical study. Int. J. Oncol. 2015, 46, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Göhring, J.; Schrangl, L.; Schütz, G.J.; Huppa, J.B. Mechanosurveillance: Tiptoeing T cells. Front. Immunol. 2022, 13, 886326. [Google Scholar] [CrossRef]
- Basu, R.; Whitlock, B.M.; Husson, J.; Le Floc’h, A.; Jin, W.; Oyler-Yaniv, A.; Dotiwala, F.; Giannone, G.; Hivroz, C.; Biais, N.; et al. Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell 2016, 165, 100–110. [Google Scholar] [CrossRef]
- Kim, H.-R.; Jun, C.-D. T cell microvilli: Sensors or senders? Front. Immunol. 2019, 10, 1753. [Google Scholar] [CrossRef]
- Bai, L.; Beckhove, P.; Feuerer, M.; Umansky, V.; Choi, C.; Solomayer, F.S.; Diel, I.J.; Schirrmacher, V. Cognate interactions between memory T cells and tumor antigen presenting dendritic cells from bone marrow of breast cancer patients: Bidirectional cell stimulation, survival and antitumor activity in vivo. Int. J. Cancer 2003, 103, 73–83. [Google Scholar] [CrossRef]
- Cornelis, R.; Hahne, S.; Taddeo, A.; Petkau, G.; Malko, D.; Durek, P.; Thiem, M.; Heiberger, L.; Peter, L.; Mohr, R.; et al. Stromal cell-contact dependent PI3K and APRIL induced NF-kB signaling prevent mitochondrial and ER stress induced death of memory plasma cells. Cell Rep. 2020, 32, 107982. [Google Scholar] [CrossRef]
- Rozanski, C.H.; Arens, R.; Carlson, L.M.; Nair, J.; Boise, L.H.; Chanan-Khan, A.A.; Schoenberger, S.P.; Lee, K.P. Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. J. Exp. Med. 2011, 208, 1435–1446. [Google Scholar] [CrossRef]
- Snell, L.M.; Lin, G.H.Y.; Watts, T.H. IL-15-dependent upregulation of GITR on CD8 memory phenotype T cells in the bone marrow relative to spleen and lymph node suggests the bone marrow as a site of superior bioavailability of IL-15. J. Immunol. 2012, 188, 5915–5923. [Google Scholar] [CrossRef]
- Schluns, K.S.; Kieper, W.C.; Jameson, S.C.; Lefrancois, L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat. Immunol. 2000, 1, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Wols, H.A.M.; Underhill, G.H.; Kansas, G.S.; Witte, P.L. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J. Immunol. 2002, 169, 4213–4221. [Google Scholar] [CrossRef] [PubMed]
- Delacher, M.; Imbusch, C.D.; Weichenhan, D.; Breiling, A.; Wagenblatt, A.H.; Träger, U.; Hofer, A.-C.; Kägebein, D.; Wang, Q.; Frauhammer, F.; et al. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat. Immunol. 2017, 18, 1160–1172. [Google Scholar] [CrossRef] [PubMed]
- Ashman, J.; Mutsonziwa, N.; Romano, M.M.; Kordasti, S.; Lombardi, G.; Shangaris, P. Regulatory T cell niche in the bone marrow, a new player in hematopoietic stem cell transplantation. Blood Rev. 2022, 31, 101030. [Google Scholar] [CrossRef]
- Tikka, C.; Beasley, L.; Xu, C.; Yang, J.; Cooper, S.; Lechner, J.; Gutch, S.; Kaplan, M.H.; Capitano, M.; Yang, K. BATF sustains homeostasis and functionality of bone marrow Treg cells to preserve homeostatic regulation of hematopoiesis and development of B cells. Front. Immunol. 2023, 14, 1026368. [Google Scholar] [CrossRef]
- Nicholls, J.; Cao, B.; Le Texier, L.; Xiong, L.Y.; Hunter, C.R.; Llanes, G.; Aguliar, E.G.; Schroder, W.A.; Phipps, S.; Lynch, J.P.; et al. Bone marrow regulatory T cells are a unique population, supported by niche-specific cytokines and plasmacytoid dendritic cells, and required for chronic graft-versus-host disease control. Front. Cell Dev. Biol. 2021, 9, 737880. [Google Scholar] [CrossRef]
- Wang, X.; Hanuffa, M.A.; Holtick, U.; Collin, M.P.; Jackson, G.; Hilkens, C.M.U.; Holler, E.; Edinger, M.; Hoffmann, P.; Dickinson, A.M. Regulatory T-cell suppression of CD8+ T-cell-mediated graft-versus-host reaction requires their presence during priming. Transplantation 2009, 88, 188–197. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Beckhove, P.; Choi, C.; Griesbach, A.; Mahnke, Y. Tumor-immune memory T cells from the bone marrow exert GvL without GvH reactivity in advanced metastasized cancer. Int. J. Oncol. 2005, 27, 1141–1149. [Google Scholar] [CrossRef]
- Zhang, Y.; Joe, G.; Hexner, E.; Zhu, J.; Emerson, S.G. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat. Med. 2005, 11, 1299–1305. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Beckhove, P.; Krüger, A.; Rocha, M.; Umansky, V.; Fichtner, K.-P.; Hull, W.E.; Zangemeister-Wittke, U.; Griesbach, A.; Jurianz, K.; et al. Effective immune rejection of advanced metastasized cancer. Int. J. Oncol. 1995, 6, 505–521. [Google Scholar] [CrossRef]
- Lee, K.; Hacker, H.; Umansky, V.; Schirrmacher, V.; Rocha, M. Changes in liver glycogen and lipid metabolism during transient graft-versus-host (GvH) and graft-versus-leukenia (GvL) reactivity. Int. J. Oncol. 1996, 9, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. T cell-mediated immunotherapy of metastases: State of the art in 2005. Expert Opin. Biol. Ther. 2005, 5, 1051–1068. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Meng, H.B.; Hua, J.; Song, Z.S.; He, Z.G.; Zhou, B.; Qian, M.P. The SDF-1/CXCR4 axis regulates migration of transplanted bone marrow mesenchymal stem cells towards the pancreas in rats with acute pancreatitis. Mol. Med. Rep. 2014, 9, 1575–1582. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Brinkrolf, P.; Landmeier, S.; Altvater, B.; Chen, C.; Pscherer, S.; Rosemann, A.; Ranft, A.; Dirksen, U.; Juergens, H.; Rossig, C. A high proportion of bone marrow T cells with regulatory phenotype (CD4+CD25hiFoxP3+) in Ewing sarcoma patients is associated with metastatic disease. Int. J. Cancer 2009, 125, 879–886. [Google Scholar] [CrossRef]
- Koch, J.; Schober, S.J.; Hindupur, S.V.; Schöning, C.; Klein, F.G.; Mantwill, K.; Ehrenfeld, M.; Schillinger, U.; Hohnecker, T.; Qi, P.; et al. Targeting the Retinoblastoma/E2F repressive complex by CDK4/6 inhibitors amplifies oncolytic potency of an oncolytic adenovirus. Nat. Commun. 2022, 13, 4689. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Böhm, H.-H.; Rathinasamy, A.; Xydia, M.; Hu, X.; Pincha, M.; Umansky, L.; Breyer, C.; Hillier, M.; Bonertz, A.; et al. Tumor-specific regulatory T cells from the bone marrow orchestrate antitumor immunity in breast cancer. Cancer Immunol. Res. 2019, 7, 1998–2012. [Google Scholar] [CrossRef]
- Cho, D.S.; Schmitt, R.E.; Dasgupta, A.; Ducharme, A.M.; Doles, J.D. Acute and sustained alterations to the bone marrow immune microenvironment following polymicrobial infection. Shock 2022, 58, 45–55. [Google Scholar] [CrossRef]
- Peng, Y.; Wu, Q.; Liu, H.; Zhang, J.; Han, Q.; Yin, F.; Wang, L.; Chen, Q.; Zhang, F.; Feng, C.; et al. An immune-related gene signature predicts the 28-day mortality in patients with sepsis. Front. Immunol. 2023, 14, 1152117. [Google Scholar] [CrossRef]
- Tu, Q.; Li, Y.; Zhu, J.; Guo, L.; Liu, C.; Liu, L.; Yuan, Y.; Zou, Y.; Chen, F.; Yao, L.; et al. Mitochondrial DNA mediates immunoparalysis of dendritic cells in sepsis via STING signalling. Cell Prolif. 2022, 55, e13328. [Google Scholar] [CrossRef]
- Moore-Fried, J.; Paul, M.; Jing, Z.; Fooksman, D.; Lauvau, G. CD169+ macrophages orchestrate plasmacytoid dendritic cell arrest and retention for optimal priming in the bone marrow of malaria-infected mice. eLife 2022, 11, e78873. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Xu, Z.; Zhang, Z.; Mo, Y.; Deng, Y.; Li, L.; Fei, S.; Wu, J.; Wang, K.; Zhang, Q.; et al. RNA-Seq approach to investigate the effects of melatonin on bone marrow-derived dendritic cells from dextran sodium-induced colitis mice. Toxicology 2022, 481, 153354. [Google Scholar] [CrossRef] [PubMed]
- Flores-Guzmaán, F.; Utikal, J.; Umansky, V. Dormant tumor cells interact with memory CD8+ T cells in RET transgenic mouse melanoma model. Cancer Lett. 2020, 474, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, Y.D.; Schirrmacher, V. A novel tumor model system for the study of long-term protective immunity and immune T cell memory. Cell. Immunol. 2005, 221, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, Y.D.; Schwendemann, J.; Beckhove, P.; Schirrmacher, V. Maintenance of lon-term tumour-specific T-cell memory by residual dormant tumour cells. Immunology 2005, 115, 325–336. [Google Scholar] [CrossRef]
- Anadon, C.M.; Yu, X.; Hänggi, K.; Biswas, S.; Chaurio, R.A.; Martin, A.; Payne, K.K.; Mandal, G.; Innamarato, P.; Harro, C.M.; et al. Ovarian cancer immunogenicity is governed by a narrow subset of progenitor tissue-resident memory T cells. Cancer Cell 2022, 40, 545–557.e13. [Google Scholar] [CrossRef]
- Mizukoshi, E.; Nakagawa, H.; Tamai, T.; Kitahara, M.; Fushimi, K.; Nio, K.; Terashima, T.; Lida, N.; Arai, K.; Vamashita, T.; et al. Peptide vaccine-treated, long-term surviving cancer patients harbor self-renewing tumor-specific CD8+ T cells. Nat. Commun. 2022, 13, 3123. [Google Scholar] [CrossRef]
- Schuetz, F.; Ehlert, K.; Ge, Y.; Schneeweiss, A.; Rom, J.; Inzkinweli, N.; Sohn, C.; Schirrmacher, V.; Beckhove, P. Treatment of advanced metastasized breast cancer with bone marrow-derived tumour-reactive memory T cells: A pilot clinical study. Cancer Immunol. Immunother. 2009, 58, 887–900. [Google Scholar] [CrossRef]
- Domschke, C.; Ge, Y.; Bernhardt, I.; Schott, S.; Keim, S.; Juenger, S.; Bucur, M.; Mayer, L.; Blumenstein, M.; Rom, J.; et al. Long-term survival after adoptive bone marrow T cell therapy of advanced metastasized breast cancer: Follow-up analysis of a clinical pilot trial. Cancer Immunol. Immunother. 2013, 62, 1053–1060. [Google Scholar] [CrossRef]
- Poliwoda, S.; Noor, N.; Downs, E.; Schaaf, A.; Cantwell, A.; Ganti, L.; Kaye, A.D.; Mosel, L.I.; Carroll, C.B.; Viswanath, O.; et al. Stem cells: A comprehensive review of origins and emerging clinical roles in medical practice. Orthop. Rev. 2022, 14, 37498. [Google Scholar] [CrossRef]
- Senkal, S.; Hayal, T.B.; Sagrac, D.; Sisli, H.B.; Asutay, A.B.; Kirath, B.; Sümer, E.; Rizvanov, A.A.; Sahin, F.; Dogan, A. Human ESC-derived neuromesodermal progenitors (NMPs) successfully differentiate into mesenchymal stem cells (MSCs). Stem Cell Rev. Rep. 2022, 18, 278–293. [Google Scholar] [CrossRef] [PubMed]
- Saidova, A.A.; Vorobjev, I.A. Lineage commitment, signaling pathways, and the cytoskeleton systems in mesenchymal stem cells. Tissue Eng. Part B Rev. 2020, 26, 13–25. [Google Scholar] [CrossRef]
- Qi, S.; Zhong, Z.; Zhu, Y.; Wang, Y.; Ma, M.; Wang, Y.; Liu, X.; Jin, R.; Jiao, Z.; Zhu, R.; et al. Two Hippo signaling modules orchestrate liver size and tumorigenesis. EMBO J. 2023, 42, e112126. [Google Scholar] [CrossRef]
- Yan, Y.-C.; Li, Y.-H.; Xiao, B.-G.; Wang, J.; Xi, J.-Y.; Yu, W.-B. Cellular and molecular mechanisms underly the combined treatment of fasudil and bone marrow derived-neuronal stem cells in a Parkinson’s disease mouse model. Mol. Neurobiol. 2023, 60, 1826–1835. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.-W.; Ding, D.-C. Role of cancer-associated mesenchymal stem cells in the tumor microenvironment: A review. Tzu Chi Med. J. 2022, 35, 24–30. [Google Scholar] [CrossRef]
- Keshavarz, M.; Ebrahimzadeh, M.S.; Mirt, S.M.; Dianat-Moghadam, H.; Ghorbanhosseini, S.S.; Mohebbi, S.R.; Keyvani, H.; Ghaemi, A. Oncolytic Newcastle disease virus delivered by mesenchymal stem cell-engineered system enhances the therapeutic effects altering tumor microenvironment. Virol. J. 2020, 17, 64. [Google Scholar] [CrossRef]
- Schirrmacher, V.; van Gool, S.; Stuecker, W. Counteracting immunosuppression in the tumor microenvironment by oncolytic Newcastle disease virus and cellular immunotherapy. Int. J. Mol. Sci. 2022, 23, 13050. [Google Scholar] [CrossRef]
- Schirrmacher, V. Cancer vaccines and oncolytic viruses exert profoundly lower side effects in cancer patients than other systemic therapies: A comparative analysis. Biomedicines 2020, 8, 61. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Su, Y.-C.; Or, Y.-E.; Cheng, C.-F.; Kung, T.K. CD8+ T cell memory is sustained in mice by hepatic stellate cells. Hepatology 2022, 77, 1486–1498. [Google Scholar] [CrossRef]
- Sun, M.; Kisseleva, T. Reversibility of liver fibrosis. Clin. Res. Hepatol. Gastroenterol. 2015, 39 (Suppl. S1), 60–63. [Google Scholar] [CrossRef]
- Li, F.; Ma, N.; Zhao, R.; Wu, G.; Zhang, Y.; Qiao, Y.; Han, D.; Xu, Y.; Xiang, Y.; Yan, B.; et al. Overexpression of mir-483-5p/3p cooperates to inhibit mouse liver fibrosis by suppressing the TGF-β stimulated HSCs in transgenic mice. J. Cell. Mol. Med. 2014, 18, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-L.; Wu, J.; Wei, D.; Zhang, D.-W.; Feng, H.; Chen, Z.-N.; Bian, H. Newcastle disease virus represses the activation of human hepatic stellate cells and reverses the development of hepatic fibrosis in mice. Liver Int. 2009, 29, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Kalou, Y.; Al-Khani, A.M.; Haider, K.H. Bone marrow mesenchymal stem cells for heart failure treatment: A systematic review and Meta-analysis. Heart Lung Circ. 2023, 32, 870–880. [Google Scholar] [CrossRef]
- Chen, X.; Xu, C.-X.; Liang, H.; Xi, Z.; Pan, J.; Yang, Y.; Sun, Q.; Yang, G.; Sun, Y.; Bian, L. Bone marrow mesenchymal stem cells transplantation alleviates brain injury after intracerebral hemorrhage in mice through the Hippo signaling pathway. Aging 2020, 12, 6306–6323. [Google Scholar] [CrossRef] [PubMed]
- Tfilin, M.; Gobshtis, N.; Fozailoff, D.; Fraifeld, V.E.; Turgeman, G. Polarized anti-inflammatory mesenchymal stem cells increase hippocampal neurogenesis and improve cognitive function in aged mice. Int. J. Mol. Sci. 2023, 24, 4490. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.-G.; Chen, M.-L.; Duan, R.; Wang, X.; Pang, Z.-L.; Ge, L.-T.; Lu, M.; Xie, H.; Liu, Z.-Z. Transplantation of nasal olfactory mucosa mesenchymal stem cells benefits Alzheimer’s disease. Mol. Neurobiol. 2022, 59, 7323–7336. [Google Scholar] [CrossRef]
- Qu, M.; Xing, F.; Xing, N. Mesenchymal stem cells for the treatment of cognitive impairment caused by neurological diseases. Biotechnol. Lett. 2022, 44, 903–916. [Google Scholar] [CrossRef]
- Nakano, M.; Kubota, K.; Kobayashi, E.; Chikenji, T.S.; Saito, Y.; Konan, N.; Fujimiya, M. Bone marrow-derived mesenchymal stem cells improve cognitive impairment in an Alzheimer’s disease model by increasing the expression of microRNA-146a in hippocampus. Sci. Rep. 2020, 10, 10772. [Google Scholar] [CrossRef]
- Vigo, T.; Voulgari-Kokota, A.; Errede, M.; Girolamo, F.; Ortolan, J.; Mariani, M.C.; Ferrara, G.; Virgintino, D.; Buffo, A.; de Rosbo, N.K.; et al. Mesenchymal stem cells instruct a beneficial phenotype in reactive astrocytes. Glia 2021, 69, 1204–1215. [Google Scholar] [CrossRef]
- Lei, X.; He, N.; Zhu, L.; Zhou, M.; Zhang, K.; Wang, C.; Huang, H.; Chen, S.; Li, Q.; Han, Z.; et al. Mesenchymal stem cell-derived extracellular vesicles attenuate radiation-induced lung injury via miRNA-214-3p. Antiox. Redox Signal. 2021, 35, 849–862. [Google Scholar] [CrossRef]
- Li, L.; Dong, L.; Zhang, J.; Gao, F.; Hui, J.; Yan, J. Mesenchymal stem cells with downregulated Hippo signalling attenuate lung injury in mice with lipopolysaccharide-induced acute respiratory distress syndrome. Int. J. Mol. Sci. 2019, 43, 1241–1252. [Google Scholar] [CrossRef]
- Vishnupriya, M.; Naveenkumar, M.; Manjima, K.; Sooryasree, N.V.; Saranya, T.; Ramya, S.; Winster, S.H.; Paulpandi, M.; Arul, B.N. Post-COVID pulmonary fibrosis: Therapeutic efficacy using with mesenchymal stem cells—How the lung heals. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 2748–2751. [Google Scholar] [CrossRef] [PubMed]
- Satilmis, B.; Cicek, G.S.; Cicek, E.; Akbulut, S.; Sahin, T.T.; Yilmaz, S. Adipose-derived stem cells in the treatment of hepatobiliary diseases and sepsis. World J. Clin. Cases 2022, 10, 4348–4356. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Luo, F.; Che, L.; Zhang, H.; Zhao, L.; Zhang, W.; Man, X.; Bu, Q.; Luan, H.; Zhou, B.; et al. Mesenchymal stem cells protect against sepsis-associated acute kidney injury by inducing Gal-9/Tim-3 to remodel immune homeostasis. Ren. Fail. 2023, 45, 2187229. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Fang, Q.; Shi, C.; Zhang, Y.; Xia, C.; Zhang, Y.; Wang, J.; Chen, F. Bone marrow mesenchymal stem cells inhibit ferroptosis via regulating the Nrf2-keap1/p53 pathway to ameliorate chronic kidney disease injury in the rats. J. Recept. Signal Transduct. Res. 2023, 43, 1–10. [Google Scholar] [CrossRef]
- Wang, Y.; Shan, S.-K.; Guo, B.; Li, F.; Zheng, M.-H.; Lei, L.-M.; Xu, Q.-S.; Ullah, M.H.E.; Xu, F.; Lin, X.; et al. The multi-therapeutic role of MSCs in diabetic nephropathy. Front. Endocrinol. 2021, 12, 671566. [Google Scholar] [CrossRef]
- Wang, B.; Kim, K.; Tian, M.; Kameishi, S.; Zhuang, L.; Okano, T.; Huang, Y. Engineered bone marrow stem cell-sheets alleviate renal damage in a rat chronic glomerulonephritis model. Int. J. Mol. Sci. 2023, 24, 3711. [Google Scholar] [CrossRef]
- Fernandez-Pernas, P.; Barrachina, L.; Marquina, M.; Rodellar, C.; Arufe, M.C.; Costa, C. Mesenchymal stromal cells for articular cartilage repair: Preclinical studies. Eur. Cell Mater. 2020, 40, 88–114. [Google Scholar] [CrossRef]
- Xing, D.; Wang, K.; Wu, J.; Zhao, Y.; Liu, W.; Li, J.J.; Gao, T.; Yan, D.; Wang, L.; Hao, J.; et al. Clinical-grade human embryonic stem-cell-derived mesenchymal stromal cells ameliorate the progression of osteoarthritis in a rat model. Molecules 2021, 26, 604. [Google Scholar] [CrossRef]
- Massidda, M.W.; Im, B.; Lee, J.; Baker, A.B. Mechanical conditioning of human mesenchymal stem cells for enhancing vascular regeneration. STAR Protoc. 2023, 4, 102103. [Google Scholar] [CrossRef]
- Hackel, A.; Vollmer, S.; Bruderek, K.; Lang, S.; Brandau, S. Immunological priming of mesenchymal stromal/stem cells and their extracellular vesicles augment their therapeutic benefits in experimental graft-versus-host disease via engagement of PD-1 ligands. Front. Immunol. 2023, 14, 1078551. [Google Scholar] [CrossRef] [PubMed]
- Sang, H.; Zhao, R.; Lai, G.; Deng, Z.; Zhuang, W.; Wu, M.; Wu, J. Bone marrow mesenchymal stem-cell-derived exosomes attenuate the maturation of dendritic cells and reduce the rejection of allogeneic transplantation. Adv. Clin. Exp. Med. 2023, 32, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Chun, W.; Tian, J.; Zhang, Y. Transplantation of mesenchymal stem cells ameliorates systemic lupus erythematosus and upregulates B10 cells through TGF-β1. Stem Cell Res. Ther. 2021, 12, 512. [Google Scholar] [CrossRef]
- Sun, W.; Yan, S.; Yang, C.; Yang, J.; Wang, H.; Li, C.; Zhang, L.; Zhao, L.; Zhang, J.; Cheng, M.; et al. Mesenchymal stem cells-derived exosomes ameliorate lupus by inducing M2 macrophage polarization and regulatory T cell expansion in MRL/lpr mice. Immunol. Investig. 2022, 51, 1785–1803. [Google Scholar] [CrossRef]
- Aierken, A.; Li, B.; Liu, P.; Cheng, X.; Kou, Z.; Tan, N.; Zhang, M.; Yu, S.; Shen, Q.; Du, X.; et al. Melatonin treatment improves human umbilical cord mesenchymal stem cell therapy in a mouse model of type II diabetes mellitus via the PI3K/AKT signaling pathway. Stem Cell Res. Ther. 2022, 13, 164. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, Y.; Zhou, X.; Zhao, Z.; Yu, Q.; Chen, Z.; Wang, Y.; Xu, P.; Yu, Z.; Guo, C.; et al. Human umbilical cord-derived mesenchymal stem cells ameliorate psoriasis-like dermatitis by suppressing IL-17-producing gd T cells. Cell Tissue Res. 2022, 388, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, Y.; Takagi, T.; Takeda, Y.; Rajbhandari, S.; Yoshida, Y.; Nakagomi, T.; Yoshimura, S. Identification of novel multipotent stem cells in mouse spinal cord following traumatic injury. Stem Cell Dev. 2022, 31, 555–568. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Feature | Description | Ref. | Year |
|---|---|---|---|
| Generative lymphatic organ | -Organ of hematopoiesis and osteogenesis: from stem cells via committed precursors to late precursors and mature forms | [7] [9] | 2022 2013 |
| -Maintaining a constantly steady state of cells from the bone cortex and marrow | [8] | 2016 | |
| -Most prominent source of de novo cellular generation in the body: 4–5 × 1011/day | [10] | 2017 | |
| Antigen responsive lymphatic organ | -Bone marrow as a priming site for T-cell responses to blood-borne antigens | [17] | 2003 |
| -Two photon dynamic imaging in situ revealing T cell priming | [23] | 2013 | |
| Functional comparison to blood | -12% of all human lymphocytes found in the BM at any time compared to 2% in the peripheral blood | [9] | 2013 |
| -BM: 4–5% TBW; lymph nodes and spleen: 1–1.5% TBW | [10] | 2017 | |
| -Tumor dormancy in bone marrow | [21] | 1994 | |
| -Active control in BM by CD8+ memory T cells | [22] | 1998 | |
| -Enrichment of MTCs in BM of breast cancer patients | [13] | 2001 | |
| -Generation of CTLs from BM, but not PBL, of breast cancer patients | [17] | 2003 | |
| [13] | 2001 | ||
| -Superior therapeutic efficiency of BM MTCs from breast cancer patients | [19] | 2001 | |
| Survival niches for memory lymphocytes organized by bone marrow stromal cells in the parenchyma | -IgG+ memory B cells, IgG+ memory plasma cells, | [61] | 2021 |
| -IgA+ memory plasma cells | [62] | 2018 | |
| -Memory T: unique homing phenotype: CCR5 + CXCR6 + CXCR3- | [55] | 2008 | |
| -Memory CD4+ T cells, CD69+ | [20] | 2004 | |
| -Memory CD8+ T cells, CD69+ | [17] | 2003 | |
| -Stem cell-like memory T cells, CD69+ CD127+ | [47] | 2022 | |
| -Cognate re-activation of TA-specific BM-MTCs: ex vivo | [19] | 2001 | |
| in situ by vaccination with ATV-NDV | [32] | 2020 | |
| -Enrichment in BM of virus-specific MTCs: EBV | [55] | 2008 | |
| CMV | [58] | 2009 | |
| HCV | [47] | 2022 | |
| SARS-Cov-2 | [46] | 2021 | |
| Hematopoietic stem cell cross-talk with T cells and DCs | -Cross-talk with T cells: adoptive T cell therapy and transfer of HSC leading to potent intratumoral DCs in malignant glioma | [67] | 2019 |
| -Cross-talk with BM DCs: Il-1β regulating HSC function | [68] | 2022 | |
| Effect of dietary restriction | -Monocyte mobilization from BM | [76] | 2019 |
| -Migration of B cells from PPs to BM | [77] | 2019 | |
| -Memory T cells collapse in secondary lymphatic organs; in contrast, memory T cell accumulates in BM | [79] | 2019 | |
| -Adipocytes and CXCR4-CXCL12 interactions contribute to enhanced T cell accumulation and remodeling of the BM compartment | [79] | 2019 |
| Feature | Description | Ref | Year |
|---|---|---|---|
| Transport of blood-borne antigens | -Peripheral and systemic antigens | [45] | 2019 |
| -Immune complexes | [84] | 2020 | |
| -Macromolecules (for central tolerance) | [80] | 2021 | |
| -Tumor-secreted proteins | [87] | 2023 | |
| Homing of blood circulating cells | -Blood-borne viruses, e.g., HCV, HBV, and HIV | [82] | 2019 |
| -Circulatory tumor cells and derived extracellular vesicles, apoptotic bodies, and tumor-associated proteins | [85] | 2023 | |
| -Circulating antigen-presenting cells | [94] | 2005 | |
| Egress of blood circulating cells | -Circulatory naïve and memory T cells | [96] | 2022 |
| Antigen presentation capacity | -BM DCs, prepulsed in vivo with a model tumor antigen, primed naïve T cells in vivo and induced long-lasting systemic tumor antigen-specific resistance. | [25] | 2003 |
| T–APC interaction capacity | -APC scanning | [120] | 2011 |
| -Immunological synapse formation | [128] | 2000 | |
| [127] | 2021 | ||
| -T cell costimulation and signaling | [126] | 2022 | |
| -Mitochondrial priming by CD28 | [125] | 2017 | |
| -Cytoskeletal reorganization | [123] | 1999 | |
| -Response polarization | [131] | 2022 | |
| -Bidirectional cell stimulation | [139] | 2003 | |
| -Follicular immune cluster formation in parenchyma | [17] | 2003 | |
| -T cell activation and proliferation | [20] | 2004 | |
| -Memory B cell activation and antibody production | [31] | 2021 | |
| Storage capacity of memory cells in BM survival niches | -Stromal-cell-expressed VCAM-1 holding immune cells in the niche | [144] | 2002 |
| -Stromal-cell-secreted IL-7 and IL-15 mediating survival of MTCs | [6] | 1994 | |
| -Stromal-cell-expressed CD80/CD86 binding CD28 on T cells and plasma cells | [143] | 2000 | |
| -Stromal cell contact and secreted APRIL/BAFF preventing mitochondrial and ER stress of plasma cells | [144] [141] | 2002 2011 | |
| Autonomic adaptive immune response | -Independence from secondary lymphoid organs | [17] | 2003 |
| -Map3k14aly/aly splenectomized mouse response similar to that of BM in normal mice | |||
| Neuro-immune links | -Allergic inflammation | [103] | 2022 |
| -Neuro-oncology | [105] | 2023 | |
| -Neuro-osteogenic network | [118] | 2023 |
| Feature | Description | Ref. | Year |
|---|---|---|---|
| Tissue-regulatory cells | -Maintenance of self-tolerance and support of organ homeostasis | [145] | 2017 |
| -Epigenetic regulation | |||
| BM niche for Treg cells | -Immunosuppression and maintenance of HSCs | [146] | 2022 |
| -Future role in HSC transplantation | |||
| BM Tregs being unique | -Supported by niche-specific cytokines and pDCs | [148] | 2021 |
| -Future role in control of GvH disease | |||
| -Role of transcription factor BATF | [147] | 2023 | |
| GvL without GvH | -Tumor-immune MTCs from allogeneic MHC-identical bone marrow exerting GvL without GvH | [150] | 2005 |
| -Suppression of GvH requiring Treg priming | [151] | 2005 | |
| Tumor-specific BM Treg cells | -Orchestration of antitumor immunity in breast cancer | [159] | 2019 |
| -Tregs, selectively activated in BM, egressing into the blood | [159] | 2019 | |
| Osseous Ewing sarcoma (ES) | -High proportion of BM Treg cells | [157] | 2009 |
| -Association with metastatic disease | |||
| Effect of microbial infection and inflammation | -Widespread transcriptional changes in BM immune cells in peritonitis sepsis model | [160] | 2022 |
| -Mitochondrial DNA mediating immunoparalysis in BM DCs via STING signaling in sepsis model | [162] | 2022 | |
| -Optimal priming of BM pDCs by CD169+ macrophages in mouse model of severe malaria | [163] | 2022 | |
| -Melatonin affecting BM DCs in DSS-induced colitis mice | [164] | 2022 | |
| Tumor dormancy | -Colocalization in BM of melanoma cells and CD8+ MTCs | [165] | 2020 |
| -Maintenance of CD8+ MTCs in BM niches by residual ESb-Gal lymphoma cells | [167] | 2005 |
| Feature | Description | Ref. | Year |
|---|---|---|---|
| Heart failure | -Meta-analysis revealing BM-MSC treatment as an effective intervention | [185] | 2023 |
| Neurological diseases | -MSC treatment for cognitive impairment | [189] | 2022 |
| -Instruction of a beneficial phenotype in reactive astrocytes | [191] | 2021 | |
| -Cognitive impairement in an Alzheimer’s disease model | [178] | 2020 | |
| -Hippocampal neurogenesis in aged mice by polarized MSCs | [190] | 2020 | |
| Lung injury and fibrosis | -MSC-derived EVs attenuate radiation-induced lung injury | [192] | 2021 |
| -Therapeutic efficiency in post-COVID pulmonary fibrosis | [194] | 2021 | |
| Liver regeneration | -Adipose-derived stem cells in hepatobiliary diseases | [195] | 2022 |
| -Reversion of activated to quiescent HeSCs by NDV | [184] | 2009 | |
| -Inhibition of liver fibrosis by mirRNA | [183] | 2014 | |
| -Sustainment of CD8+ T cell memory by IL-15+ HeSCs | [181] | 2022 | |
| Kidney injury | -Sepsis model, remodeling immune homeostasis | [196] | 2023 |
| -Inhibition of ferroptosis via Nrf2-keap1/p53 pathway | [197] | 2023 | |
| -Multi-therapeutic role in diabetic nephropathy | [198] | 2021 | |
| Osteoarthritis | -Amelioration of progression in a rat model | [201] | 2021 |
| -Articular cartilage repair in preclinical studies | [200] | 2020 | |
| Vascular regeneration | -A new conditioning technique to enhance vascular regenerative properties of hMSCs with increases in the expression of endothelial and pericyte markers | [202] | 2023 |
| Autoimmune diseases | -Amelioration of lupus erythematosus | [205] | 2021 |
| -Amelioration of psoriasis-like dermatitis | [208] | 2022 | |
| -Amelioration of type II diabetes mellitus | [207] | 2022 | |
| -Amelioration of experimental autoimmune encephalomyelitis | [191] | 2021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schirrmacher, V. Bone Marrow: The Central Immune System. Immuno 2023, 3, 289-329. https://doi.org/10.3390/immuno3030019
Schirrmacher V. Bone Marrow: The Central Immune System. Immuno. 2023; 3(3):289-329. https://doi.org/10.3390/immuno3030019
Chicago/Turabian StyleSchirrmacher, Volker. 2023. "Bone Marrow: The Central Immune System" Immuno 3, no. 3: 289-329. https://doi.org/10.3390/immuno3030019
APA StyleSchirrmacher, V. (2023). Bone Marrow: The Central Immune System. Immuno, 3(3), 289-329. https://doi.org/10.3390/immuno3030019