
Immunotherapeutic Development of a Tri-Specific NK Cell Engager Recognizing BCMA



Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
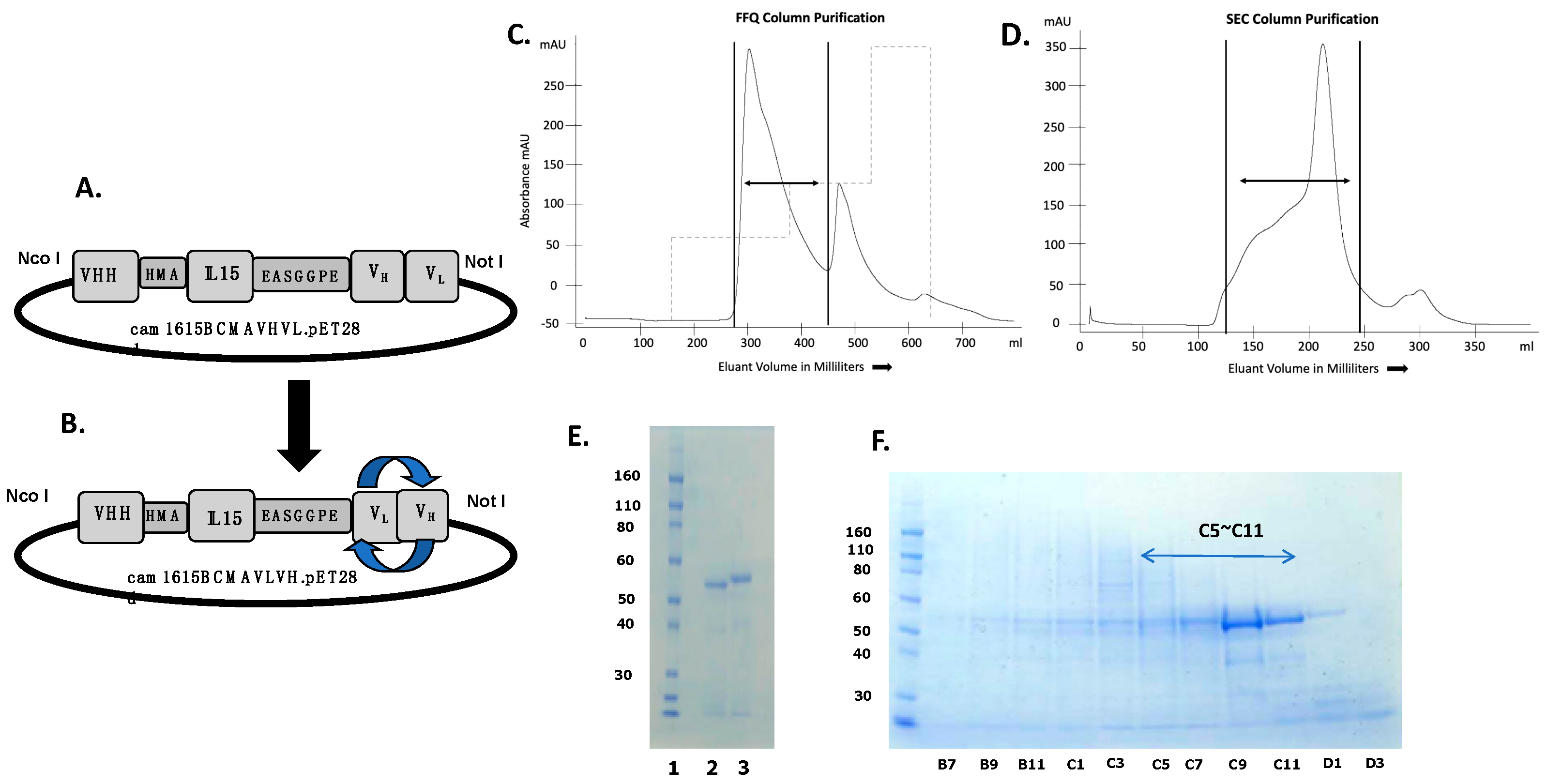
2.1. Construction and Purification of cam1615BCMA
2.2. Cell Lines and Blood and Human Blood
2.3. Evaluation of NK Cell Activity
2.4. NK Cell Expansion via IL-15 Stimulation
2.5. In Vivo Mouse Study and Imaging
2.6. Statistical Analysis
3. Results
3.1. Construction of cam1615BCMA TriKE
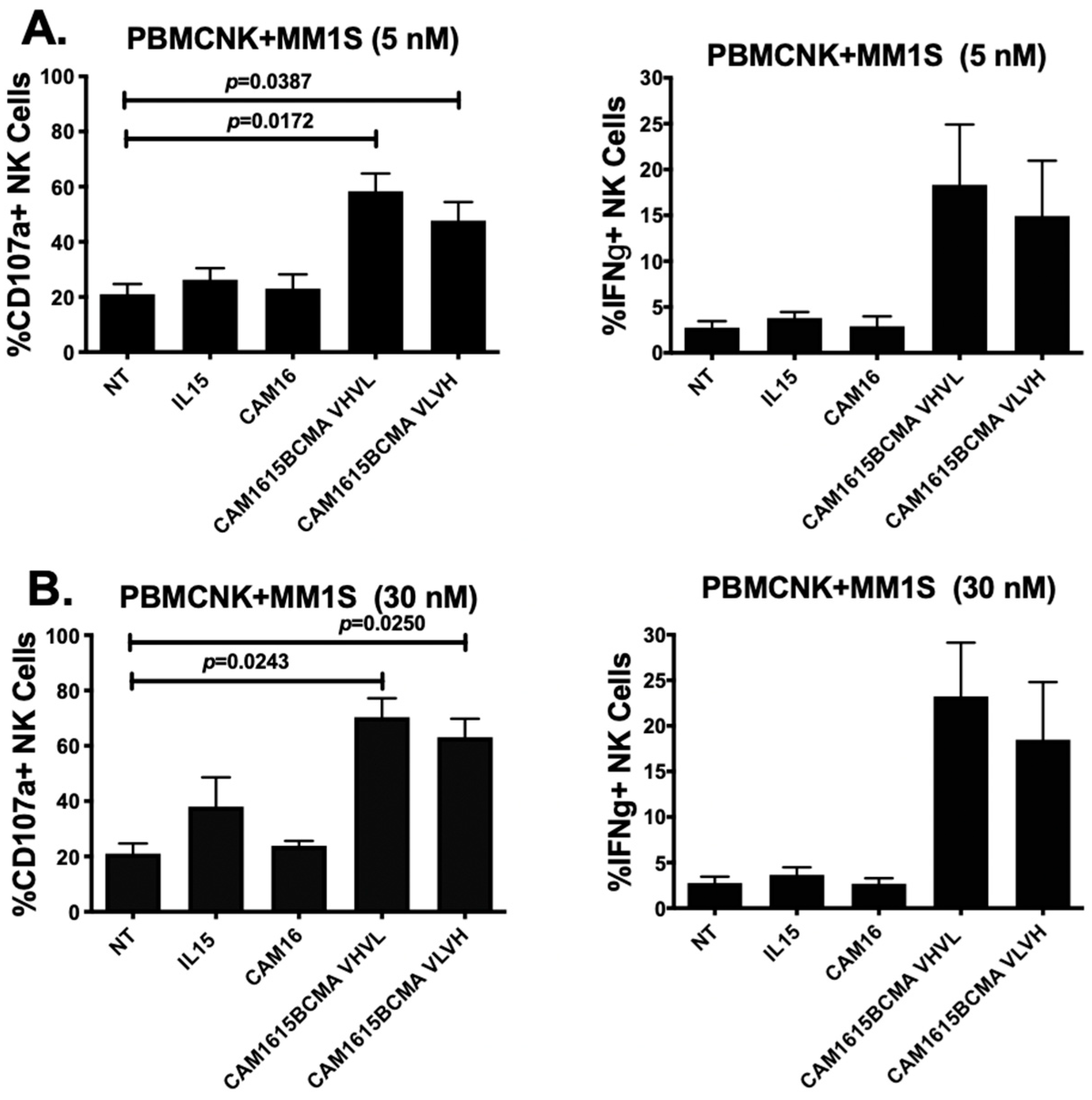
3.2. cam1615BCMA TriKE Cytotoxicity Is Dose Dependent
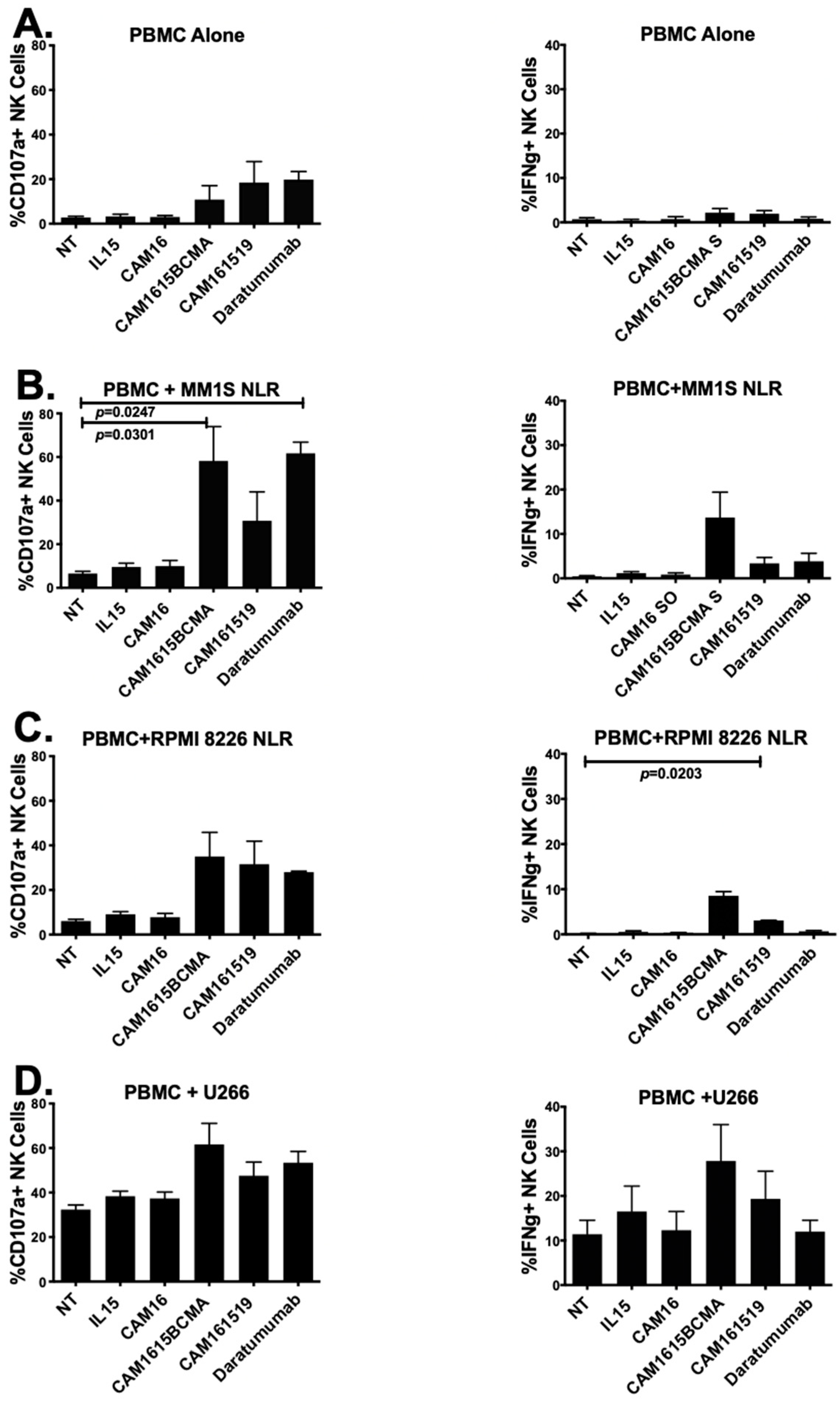
3.3. cam1615BCMA TriKE Is Cytotoxic against Various Multiple Myeloma Targets
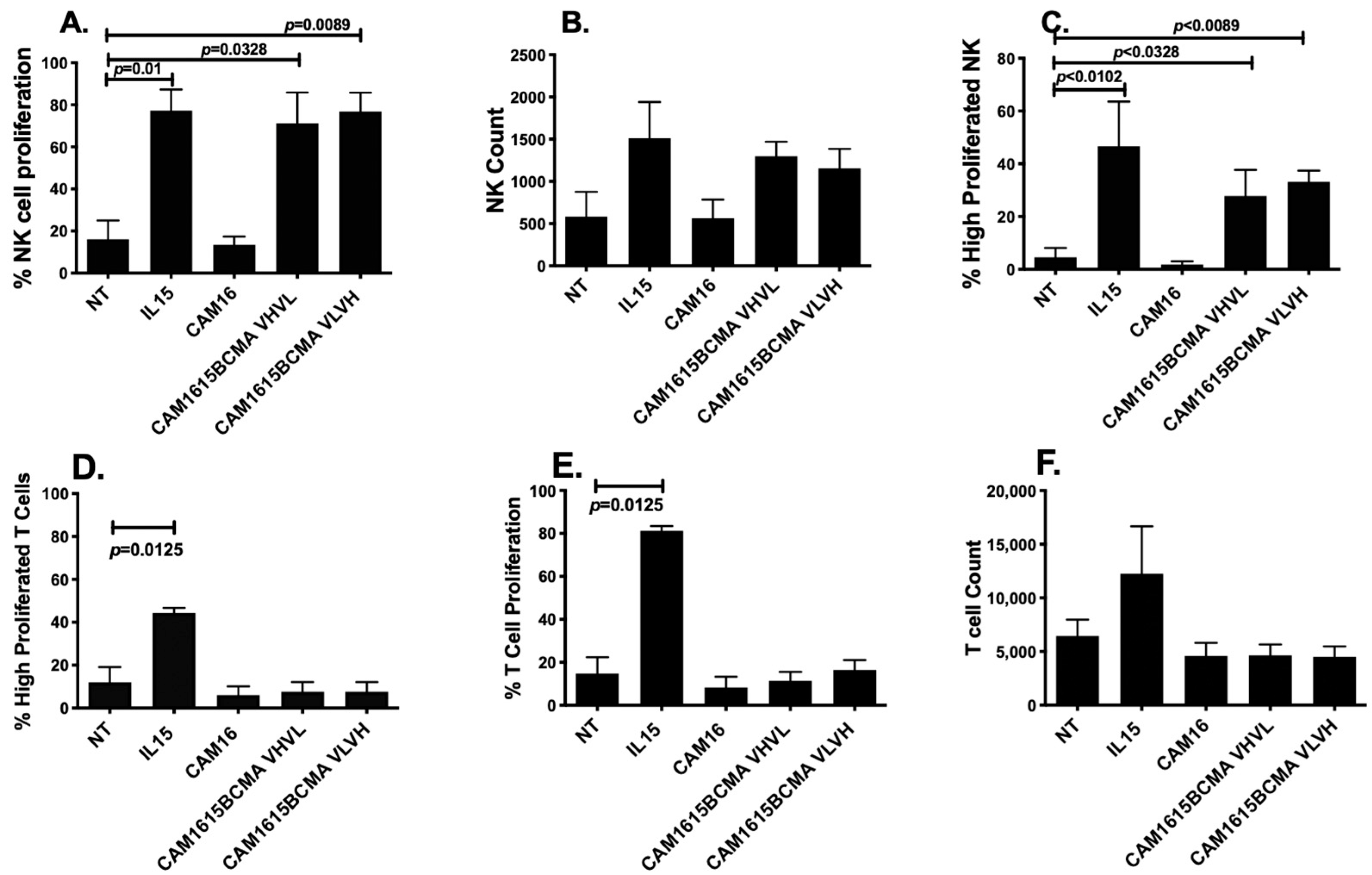
3.4. cam1615BCMA TriKE Induces Specific NK Cell Proliferation
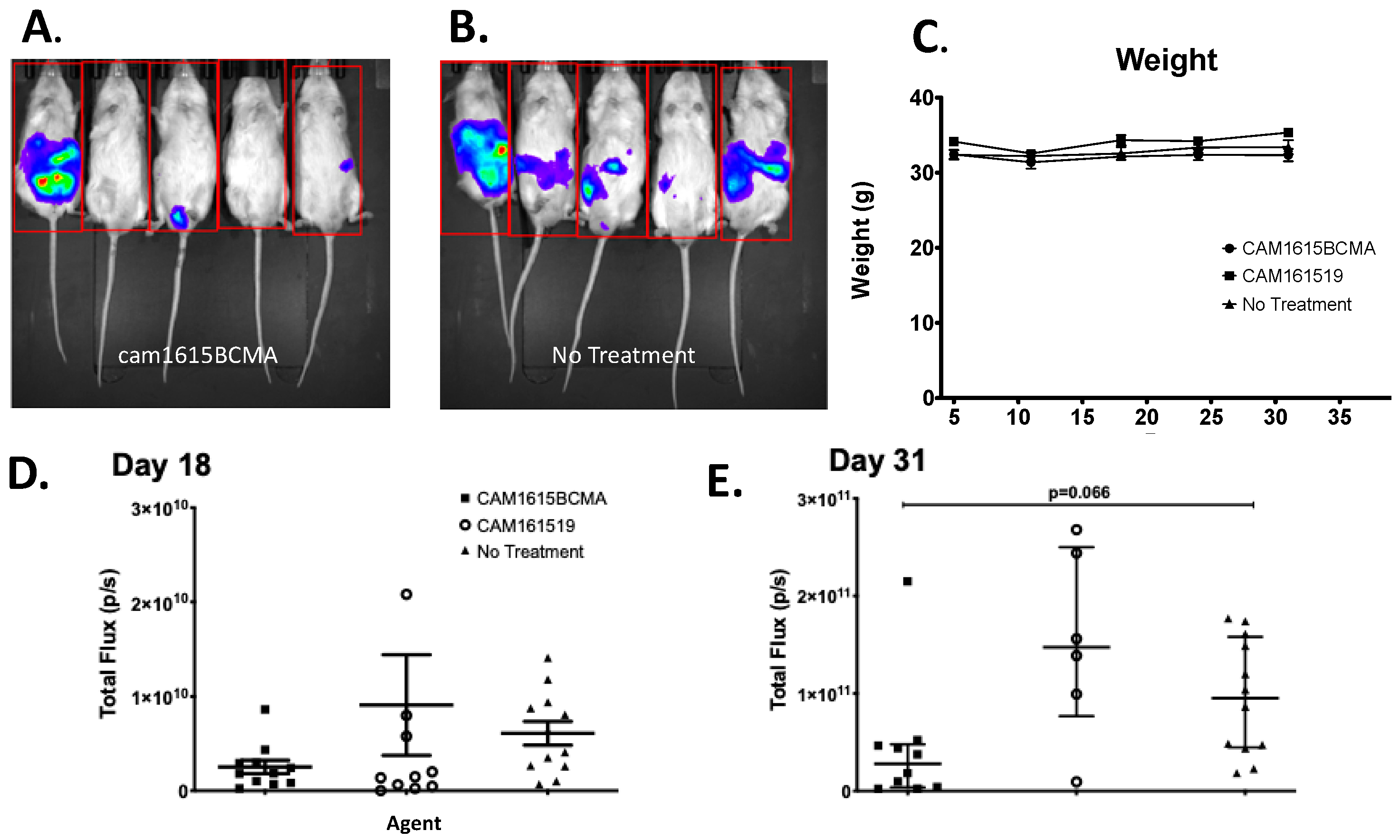
3.5. In Vivo Efficacy of cam1615BCMA TriKE
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cowan, A.J.; Green, D.J.; Kwok, M.; Lee, S.; Coffey, D.G.; Holmberg, L.A.; Tuazon, S.; Gopal, A.K.; Libby, E.N. Diagnosis and Management of Multiple Myeloma: A Review. JAMA 2022, 327, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Mian, H.; Eisfeld, C.; Venner, C.P.; Masih-Khan, E.; Kardjadj, M.; Jimenez-Zepeda, V.H.; Khandanpour, C.; Lenz, G.; McCurdy, A.; Sebag, M.; et al. Efficacy of Daratumumab-Containing Regimens Among Patients With Multiple Myeloma Progressing on Lenalidomide Maintenance: Retrospective Analysis. Front. Oncol. 2022, 12, 826342. [Google Scholar] [CrossRef]
- Swan, D.; Routledge, D.; Harrison, S. The evolving status of immunotherapies in multiple myeloma: The future role of bispecific antibodies. Br. J. Haematol. 2022, 196, 488–506. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, H.T.; Singh, M.; Milce, J.; Haidar, M.; Rieth, A.; Lebioda, A.; Kohnke, J. Management of Patients with Relapsed and/or Refractory Multiple Myeloma Treated with Novel Combination Therapies in Routine Clinical Practice in Germany. Adv. Ther. 2022, 39, 1247–1266. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Dimopoulos, M.A.; Kastritis, E.; Terpos, E.; Nahi, H.; Goldschmidt, H.; Hillengass, J.; Leleu, X.; Beksac, M.; Alsina, M.; et al. Natural history of relapsed myeloma, refractory to immunomodulatory drugs and proteasome inhibitors: A multicenter IMWG study. Leukemia 2017, 31, 2443–2448. [Google Scholar] [CrossRef]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef]
- Lee, L.; Bounds, D.; Paterson, J.; Herledan, G.; Sully, K.; Seestaller-Wehr, L.M.; Fieles, W.E.; Tunstead, J.; McCahon, L.; Germaschewski, F.M.; et al. Evaluation of B cell maturation antigen as a target for antibody drug conjugate mediated cytotoxicity in multiple myeloma. Br. J. Haematol. 2016, 174, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Eckhert, E.; Hewitt, R.; Liedtke, M. B-cell maturation antigen directed monoclonal antibody therapies for multiple myeloma. Immunotherapy 2019, 11, 801–811. [Google Scholar] [CrossRef]
- Tai, Y.T.; Acharya, C.; An, G.; Moschetta, M.; Zhong, M.Y.; Feng, X.; Cea, M.; Cagnetta, A.; Wen, K.; van Eenennaam, H.; et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016, 127, 3225–3236. [Google Scholar] [CrossRef]
- Darce, J.R.; Arendt, B.K.; Wu, X.; Jelinek, D.F. Regulated expression of BAFF- binding receptors during human B cell differentiation. J. Immunol. 2007, 179, 7276–7286. [Google Scholar] [CrossRef]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef]
- Teachey, D.T.; Rheingold, S.R.; Maude, S.L.; Zugmaier, G.; Barrett, D.M.; Seif, A.E.; Nichols, K.E.; Suppa, E.K.; Kalos, M.; Berg, R.A.; et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood 2013, 121, 5154–5157. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Boll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef]
- Lanier, L.L.; Ruitenberg, J.J.; Phillips, J.H. Functional and biochemical analysis of CD16 antigen on natural killer cells and granulocytes. J. Immunol. 1988, 141, 3478–3485. [Google Scholar] [CrossRef]
- Yokoyama, W.M.; Plougastel, B.F. Immune functions encoded by the natural killer gene complex. Nat. Rev. Immunol. 2003, 3, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. Natural killer cell receptor signaling. Curr. Opin. Immunol. 2003, 15, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- Felices, M.; Lenvik, T.; Kodal, B.; Lenvik, A.; Hinderlie, P.; Bendzick, L.; Schirm, D.; Kaminski, M.; McElmurry, R.; Geller, M.; et al. Potent Cytolytic Activity and Specific IL15 Delivery in a Second-Generation Trispecific Killer Engager. Cancer Immunol. Res. 2020, 8, 1139–1149. [Google Scholar] [CrossRef]
- Vallera, D.A.; Oh, F.; Kodal, B.; Hinderlie, P.; Geller, M.A.; Miller, J.S.; Felices, M. A HER2 Tri-Specific NK Cell Engager Mediates Efficient Targeting of Human Ovarian Cancer. Cancers 2021, 8, 3994. [Google Scholar] [CrossRef]
- Vallera, D.A.; Ferrone, S.; Kodal, B.; Hinderlie, P.; Bendzick, L.; Ettestad, B.; Hallstrom, C.; Zorko, N.A.; Rao, A.; Fujioka, N.; et al. NK-Cell-Mediated Targeting of Various Solid Tumors Using a B7-H3 Tri-Specific Killer Engager In Vitro and In Vivo. Cancers 2020, 12, 2659. [Google Scholar] [CrossRef]
- Schmohl, J.U.; Felices, M.; Taras, E.; Miller, J.S.; Vallera, D.A. Enhanced ADCC and NK Cell Activation of an Anticarcinoma Bispecific Antibody by Genetic Insertion of a Modified IL-15 Cross-linker. Mol. Ther. 2016, 24, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.L.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef]
- Steel, J.C.; Waldmann, T.A.; Morris, J.C. Interleukin-15 biology and its therapeutic implications in cancer. Trends Pharmacol. Sci. 2012, 33, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Carson, W.E.; Giri, J.G.; Lindemann, M.J.; Linett, M.L.; Ahdieh, M.; Paxton, R.; Anderson, D.; Eisenmann, J.; Grabstein, K.; Caligiuri, M.A. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J. Exp. Med. 1994, 180, 1395–1403. [Google Scholar] [CrossRef]
- Zhang, Y.; Wallace, D.L.; De Lara, C.M.; Ghattas, H.; Asquith, B.; Worth, A.; Griffin, G.E.; Taylor, G.P.; Tough, D.F.; Beverley, P.C.L.; et al. In vivo kinetics of human natural killer cells: The effects of aging and acute and chronic viral infection. Immunology 2007, 121, 258–265. [Google Scholar] [CrossRef]
- Berger, S.C.; Berger, M.; Hackman, R.C.; Gough, M.; Elliott, C.; Jensen, M.C.; Riddell, S.R. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood 2009, 114, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Munger, W.; Dejoy, S.Q.; Jeyaseelan, R.; Torley, L.W.; Grabstein, K.H.; Eisenmann, J.; Paxton, R.; Cox, T.; Wick, M.M.; Kerwar, S.S. Studies evaluating the antitumor activity and toxicity of interleukin-15, a new T cell growth factor: Comparison with interleukin-2. Cell. Immunol. 1995, 165, 289–293. [Google Scholar] [CrossRef]
- Behar, G.; Siberil, S.; Groulet, A.; Chames, P.; Pugniere, M.; Boix, C.; Sautes-Fridman, C.; Teillaud, J.L.; Baty, D. Isolation and characterization of anti-FcgammaRIII (CD16) llama single-domain antibodies that activate natural killer cells. Protein Eng. Des. Sel. 2008, 21, 1–10. [Google Scholar] [CrossRef]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Todhunter, D.; Kuroki, D.W.; Shu, Y.; Sicheneder, A.; Panoskaltsis-Mortari, A.; Vallera, V.D.; Chen, H. Molecular modification of a recombinant, bivalent anti-human CD3 immunotoxin (Bic3) results in reduced in vivo toxicity in mice. Leuk. Res. 2005, 29, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol. Asp. Med. 2006, 27, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Zaritskaya, L.; Shurin, M.R.; Sayers, T.J.; Malyguine, A.M. New flow cytometric assays for monitoring cell-mediated cytotoxicity. Expert Rev. Vaccines 2010, 9, 601–616. [Google Scholar] [CrossRef]
- Lowdell, M.W.; Lamb, L.; Hoyle, C.; Velardi, A.; Prentice, H.G. Non-MHC-restricted cytotoxic cells: Their roles in the control and treatment of leukaemias. Br. J. Haematol. 2001, 114, 11–24. [Google Scholar] [CrossRef]
- Hamilton, S.; Odili, J.; Gundogdu, O.; Wilson, G.D.; Kupsch, J.M. Improved production by domain inversion of single-chain Fv antibody fragment against high molecular weight proteoglycan for the radioimmunotargeting of melanoma. Hybrid. Hybridomics 2001, 20, 351–360. [Google Scholar] [CrossRef]
- Vallera, D.A.; Chen, H.; Sicheneder, A.R.; Panoskaltsis-Mortari, A.; Taras, E.P. Genetic alteration of a bispecific ligand-directed toxin targeting human CD19 and CD22 receptors resulting in improved efficacy against systemic B cell malignancy. Leuk. Res. 2009, 33, 1233–1242. [Google Scholar] [CrossRef]
- Romee, R.; Foley, B.; Lenvik, T.; Wang, Y.; Zhang, B.; Ankarlo, D.; Luo, X.; Cooley, S.; Verneris, M.; Walcheck, B.; et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 2013, 121, 2599–3608. [Google Scholar] [CrossRef]
- Wiernik, A.; Foley, B.; Zhang, B.; Verneris, M.R.; Warlick, E.; Gleason, M.K.; Ross, J.A.; Luo, X.; Weisdorf, D.J.; Walcheck, B.; et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16×33 bispecific killer cell engager and ADAM17 inhibition. Clin. Cancer Res. 2013, 19, 3844–3855. [Google Scholar] [CrossRef]
- Ali, A.K.; Nandagopal, N.; Lee, S.H. IL-15-PI3K-AKT-mTOR: A critical pathway in the life journey of Natural Killer cells. Front. Immunol. 2015, 6, 355. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, D.; Brandt, L.; Felices, M.; Guldevall, K.; Lenvik, T.; Hinderlie, P.; Curtsinger, J.; Warlick, E.; Spellman, S.R.; Blazar, B.R.; et al. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv. 2018, 2, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A. The biology of interleukin-2 and interleukin-15: Implications for cancer therapy and vaccine design. Nat. Rev. Immunol. 2006, 6, 595–601. [Google Scholar] [CrossRef]
- Sneller, M.C.; Kopp, W.C.; Engelke, K.J.; Yovandich, J.L.; Creekmore, S.P.; Waldmann, T.A.; Lane, H.C. IL-15 administered by continuous infusion to rhesus macaques induces massive expansion of CD8+ T effector memory population in peripheral blood. Blood 2011, 118, 6845–6848. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Morishima, C.; McNeel, D.G.; Patel, M.R.; Kohrt, H.E.K.; Thompson, J.A.; Sondel, P.M.; Wakelee, H.A.; Disis, M.L.; Kaiser, J.C.; et al. A first-in-human phase I study of subcutaneous outpatient recombinant human IL15 (rhIL15) in adults with advanced solid tumors. Clin. Cancer Res. 2018, 24, 1525–1535. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Effector: Target Ratio | ||
|---|---|---|---|
| 20:1 | 6.6:1 | 2.2:1 | |
| no treatment | 0.0 | 0.2 | 0.0 |
| cam1615BCMA | 38.7 | 20.0 | 10.3 |
| IL15 alone | 0.1 | 0.0 | 0.0 |
| anti-cam16 alone | 0.0 | 0.0 | 0.0 |
| anti-BCMA alone | 0.2 | 0.0 | 0.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, F.; Felices, M.; Kodal, B.; Miller, J.S.; Vallera, D.A. Immunotherapeutic Development of a Tri-Specific NK Cell Engager Recognizing BCMA. Immuno 2023, 3, 237-249. https://doi.org/10.3390/immuno3020016
Oh F, Felices M, Kodal B, Miller JS, Vallera DA. Immunotherapeutic Development of a Tri-Specific NK Cell Engager Recognizing BCMA. Immuno. 2023; 3(2):237-249. https://doi.org/10.3390/immuno3020016
Chicago/Turabian StyleOh, Felix, Martin Felices, Behiye Kodal, Jeffrey S. Miller, and Daniel A. Vallera. 2023. "Immunotherapeutic Development of a Tri-Specific NK Cell Engager Recognizing BCMA" Immuno 3, no. 2: 237-249. https://doi.org/10.3390/immuno3020016
APA StyleOh, F., Felices, M., Kodal, B., Miller, J. S., & Vallera, D. A. (2023). Immunotherapeutic Development of a Tri-Specific NK Cell Engager Recognizing BCMA. Immuno, 3(2), 237-249. https://doi.org/10.3390/immuno3020016