
Fisetin Modulates Toll-like Receptor-Mediated Innate Antiviral Response in Chikungunya Virus-Infected Hepatocellular Carcinoma Huh7 Cells

 , , and
, , and 
Abstract
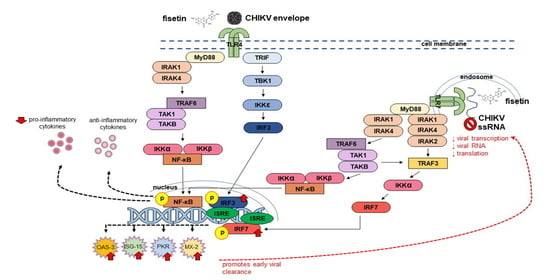
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Virus
2.2. Flavonoid and Agonists
2.3. Virus Plaque Assay
2.4. RNA Extraction, Antiviral Genes, and Viral Yield Reduction Assay
2.5. Cytotoxicity Assay
2.6. Pre-Treatment and Post-Infection Treatment Assay
2.7. Immunofluorescence Assay (IFA)
2.8. Immunoblot Assay
2.9. Cytokine Array
2.10. Statistical Analysis
3. Results
3.1. Fisetin Reduced CHIKV Replication in a Dose-Dependent Manner
3.2. CHIKV Inhibitory Potential of Fisetin
3.3. Fisetin Reduced CHIKV-Induced Cytopathic Effects (CPE) in Huh7 Cells
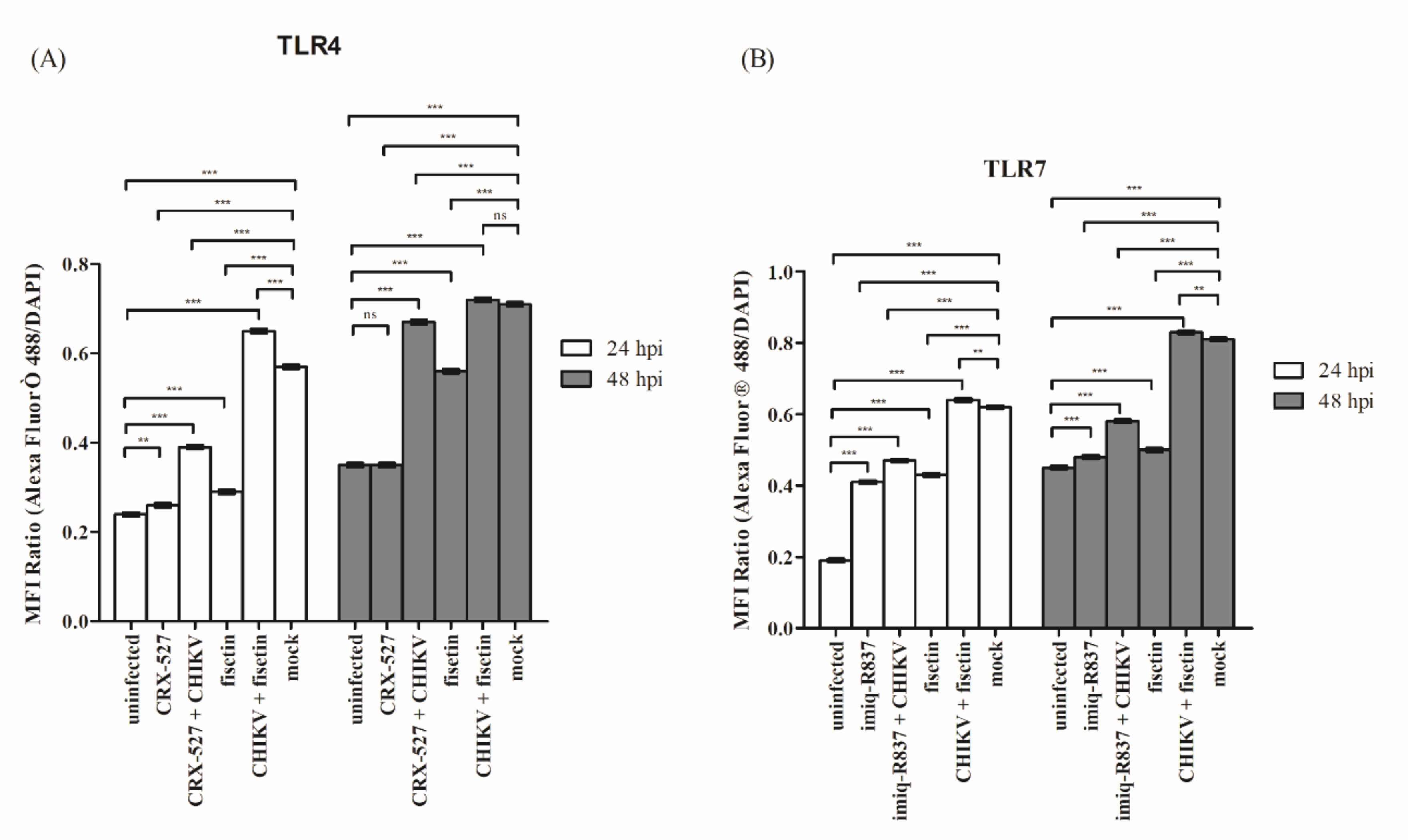
3.4. Fisetin Treatment and CHIKV Infection Induced Endogenous TLR4 and TLR7 Protein Expression
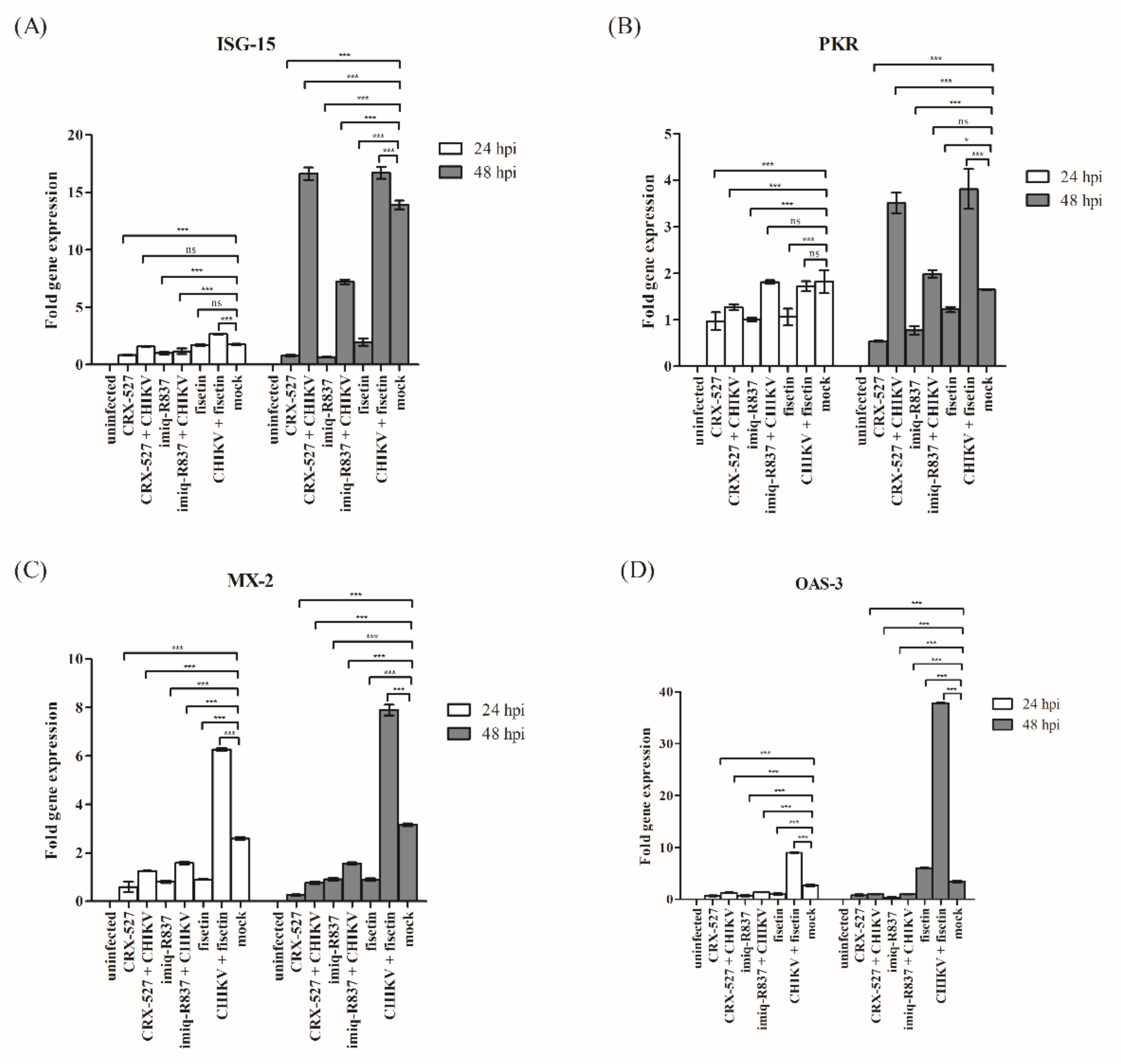
3.5. Induction of Endogenous Antiviral Genes in Huh7 Cells
3.6. Fisetin Reversed the Regulation of Pro- and Anti-Inflammatory Cytokines Evoked by CHIKV Infection
3.7. Effect of Fisetin on the Expression of Endogenous IRF3, IRF7 and Their Respective Phosphorylated Form in CHIKV-Infected Huh7 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef] [PubMed]
- Jose, J.; Snyder, J.E.; Kuhn, R.J. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 2009, 4, 837–856. [Google Scholar] [CrossRef] [PubMed]
- Poo, Y.S.; Nakaya, H.; Gardner, J.; Larcher, T.; Schroder, W.A.; Le, T.T.; Major, L.D.; Suhrbier, A. CCR2 deficiency promotes exacerbated chronic erosive neutrophil-dominated chikungunya virus arthritis. J. Virol. 2014, 88, 6862–6872. [Google Scholar] [CrossRef] [PubMed]
- Javelle, E.; Ribera, A.; Degasne, I.; Gaüzère, B.A.; Marimoutou, C.; Simon, F. Specific management of post-chikungunya rheumatic disorders: A retrospective study of 159 cases in Reunion Island from 2006–2012. PLoS Negl. Trop. Dis. 2015, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Caglioti, C.; Lalle, E.; Castilletti, C.; Carletti, F.; Capobianchi, M.R.; Bordi, L. Chikungunya virus infection: An overview. New Microbiol. 2013, 36, 211–227. [Google Scholar]
- Silva, L.A.; Dermody, T.S. Chikungunya virus: Epidemiology, replication, disease mechanisms, and prospective intervention strategies. J. Clin. Invest. 2017, 127, 737–749. [Google Scholar] [CrossRef]
- Volk, S.M.; Chen, R.; Tsetsarkin, K.A.; Adams, A.P.; Garcia, T.I.; Sall, A.A.; Nasar, F.; Schuh, A.J.; Holmes, E.C.; Higgs, S.; et al. Genome-scale phylogenetic analyses of chikungunya virus reveal independent emergences of recent epidemics and various evolutionary rates. J. Virol. 2010, 84, 6497–6504. [Google Scholar] [CrossRef]
- da Cunha, R.V.; Trinta, K.S. Chikungunya virus: Clinical aspects and treatment-A Review. Mem. Inst. Oswaldo Cruz. 2017, 112, 523–531. [Google Scholar] [CrossRef]
- Lani, R.; Agharbaoui, F.E.; Hassandarvish, P.; Teoh, B.T.; Sam, S.S.; Zandi, K.; Rahman, N.A.; AbuBakar, S. In silico studies of fisetin and silymarin as novel chikungunya virus nonstructural proteins inhibitors. Future Virol. 2021, 16, 167–180. [Google Scholar] [CrossRef]
- Lani, R.; Hassandarvish, P.; Shu, M.H.; Phoon, W.H.; Chu, J.J.H.; Higgs, S.; Vanlandingham, D.; Bakar, S.A.; Zandi, K. Antiviral activity of selected flavonoids against Chikungunya virus. Antiviral Res. 2016, 133, 50–61. [Google Scholar] [CrossRef]
- Willits, M.G.; Giovanni, M.; Prata, R.T.; Kramer, C.M.; De Luca, V.; Steffens, J.C.; Graser, G. Bio-fermentation of modified flavonoids: An example of in vivo diversification of secondary metabolites. Phytochemistry 2004, 65, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Peer, W.A.; Murphy, A.S. Flavonoids and auxin transport: Modulators or regulators? Trends Plant Sci. 2007, 12, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Maleki, S.J.; Crespo, J.F.; Cabanillas, B. Anti-inflammatory effects of flavonoids. Food Chem. 2019, 299, 1–11. [Google Scholar] [CrossRef]
- Nigusse, T.; Zhang, L.; Wang, R.; Wang, X.N.; Li, J.; Liu, C. Flavonoids in a crude extract of Catha edulis inhibit rat intestinal contraction via blocking Ca2+ channels. Neurogastroenterol Motil. 2019, 31, 7. [Google Scholar] [CrossRef]
- Gazola, A.C.; Costa, G.M.; Zucolotto, S.M.; Castellanos, L.; Ramos, F.A.; de Lima, T.C.M.; Schenkel, E.P. The sedative activity of flavonoids from Passiflora quadrangularis is mediated through the GABAergic pathway. Biomed Pharmacother. 2018, 100, 388–393. [Google Scholar] [CrossRef]
- Lipkovskaya, N.A.; Barvinchenko, V.N.; Fedyanina, T.V. Physicochemical properties of quercetin and rutin in aqueous solutions of decamethoxin antiseptic drug. Russ. J. Appl. Chem. 2014, 87, 36–41. [Google Scholar] [CrossRef]
- Vinayagam, R.; Xu, B. Antidiabetic properties of dietary flavonoids: A cellular mechanism review. Nutr. Metab. 2015, 12, 1–20. [Google Scholar] [CrossRef]
- Abdel-Salam, N.A.; Ghazy, N.M.; Sallam, S.M.; Radwan, M.M.; Wanas, A.S.; ElSohly, M.A.; El-Demellawy, M.A.; Abdel-Rahman, N.M.; Piacente, S.; Shenouda, M.L. Flavonoids of Alcea rosea L. and their immune stimulant, antioxidant and cytotoxic activities on hepatocellular carcinoma HepG-2 cell line. Nat. Prod. Res. 2018, 32, 702–706. [Google Scholar] [CrossRef]
- He, Y.; Xia, Z.; Yu, D.; Wang, J.; Jin, L.; Huang, D.; Ye, X.; Li, X.; Zhang, B. Hepatoprotective effects and structure-activity relationship of five flavonoids against lipopolysaccharide/d-galactosamine induced acute liver failure in mice. Int. Immunopharmacol. 2019, 68, 171–178. [Google Scholar] [CrossRef]
- Sarbu, L.G.; Bahrin, L.G.; Babii, C.; Stefan, M.; Birsa, M.L. Synthetic flavonoids with antimicrobial activity: A review. J. Appl. Microbiol. 2019, 127, 1282–1290. [Google Scholar] [CrossRef]
- Ninfali, P.; Antonelli, A.; Magnani, M.; Scarpa, E.S. Antiviral properties of flavonoids and delivery strategies. Nutrients 2020, 12, 2534. [Google Scholar] [CrossRef] [PubMed]
- Althunibat, O.Y.; Al Hroob, A.M.; Abukhalil, M.H.; Germoush, M.O.; Bin-Jumah, M.; Mahmoud, A.M. Fisetin ameliorates oxidative stress, inflammation and apoptosis in diabetic cardiomyopathy. Life Sci. 2019, 221, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Choi, N.E.; Lee, J.H.; Kang, H.M.; Yu, S.B.; Kim, H.J.; Kang, H.K.; Kim, I.R. Crosstalk between fisetin-induced apoptosis and autophagy in human oral squamous cell carcinoma. J. Cancer 2019, 10, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Xu, X.; Zhou, S.; Chen, Y.; Ding, G.; Cao, L. Fisetin induces autophagy in pancreatic cancer cells via endoplasmic reticulum stress-and mitochondrial stress-dependent pathways. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Sabroe, I.; Parker, L.C.; Dower, S.K.; Whyte, M.K.B. The role of TLR activation in inflammation. J. Pathol. 2008, 214, 126–135. [Google Scholar] [CrossRef]
- Delgado, M.A.; Elmaoued, R.A.; Davis, A.S.; Kyei, G.; Deretic, V. Toll-like receptors control autophagy. EMBO J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef]
- Salaun, B.; Romero, P.; Lebecque, S. Toll-like receptors’ two-edged sword: When immunity meets apoptosis. Eur. J. Immunol. 2007, 37, 3311–3318. [Google Scholar] [CrossRef]
- Lavieri, R.; Piccioli, P.; Carta, S.; Delfino, L.; Castellani, P.; Rubartelli, A. TLR costimulation causes oxidative stress with unbalance of proinflammatory and anti-inflammatory cytokine production. J. Immunol. 2014, 192, 5373–5381. [Google Scholar] [CrossRef]
- Hertzog, P.J.; O’Neill, L.A.; Hamilton, J.A. The interferon in TLR signaling: More than just antiviral. Trends Immunol. 2003, 24, 534–539. [Google Scholar] [CrossRef]
- Kaczanowska, S.; Joseph, A.M.; Davila, E. TLR agonists: Our best frenemy in cancer immunotherapy. J. Leukoc. Biol. 2013, 93, 847–863. [Google Scholar] [CrossRef]
- Watts, C.; West, M.A.; Zaru, R. TLR signalling regulated antigen presentation in dendritic cells. Curr. Opin. Immunol. 2010, 22, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Hua, Z.; Hou, B. TLR signaling in B-cell development and activation. Cell. Mol. Immunol. 2013, 10, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.A.; Tsuji, M.; Suzuki, K.; Meek, B.; Yasuda, N.; Kaisho, T.; Fagarasan, S. Regulation of B1 cell migration by signals through Toll-like receptors. J. Exp. Med. 2006, 203, 2541–2550. [Google Scholar] [CrossRef] [PubMed]
- Van Duin, D.; Medzhitov, R.; Shaw, A.C. Triggering TLR signaling in vaccination. Trends Immunol. 2006, 27, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhang, L.; Zhao, Y. Modulation of immune responses through direct activation of Toll-like receptors to T cells. Clin. Exp. Immunol. 2010, 160, 168–175. [Google Scholar] [CrossRef]
- Kovuru, N.; Raghuwanshi, S.; Sangeeth, A.; Malleswarapu, M.; Sharma, D.S.; Dahariya, S.; Pallepati, A.; Gutti, R.K. Co-stimulatory effect of TLR2 and TLR4 stimulation on megakaryocytic development is mediated through PI3K/NF-ĸB and XBP-1 loop. Cell Signal. 2021, 80, 109924. [Google Scholar] [CrossRef]
- Hovland, A.; Jonasson, L.; Garred, P.; Yndestad, A.; Aukrust, P.; Lappegård, K.T.; Espevik, T.; Mollnes, T.E. The complement system and toll-like receptors as integrated players in the pathophysiology of atherosclerosis. Atherosclerosis 2015, 241, 480–494. [Google Scholar] [CrossRef]
- Priya, R.; Dhanwani, R.; Patro, I.K.; Rao, P.V.L.; Parida, M.M. Differential regulation of TLR mediated innate immune response of mouse neuronal cells following infection with novel ECSA genotype of Chikungunya virus with and without E1: A226V mutation. Infect Genet. Evol. 2013, 20, 396–406. [Google Scholar] [CrossRef]
- Dutta, S.K.; Tripathi, A. Association of toll-like receptor polymorphisms with susceptibility to chikungunya virus infection. Virology 2017, 511, 207–213. [Google Scholar] [CrossRef]
- Pathak, H.; Mohan, M.C.; Ravindran, V. Chikungunya arthritis. Clin. Med. 2019, 19, 381–385. [Google Scholar] [CrossRef]
- Messaoudi, I.; Vomaske, J.; Totonchy, T.; Kreklywich, C.N.; Haberthur, K.; Springgay, L.; Brien, J.D.; Diamond, M.S.; DeFilippis, V.R.; Streblow, D.N. Chikungunya virus infection results in higher and persistent viral replication in aged rhesus macaques due to defects in anti-viral immunity. PLoS Negl. Trop. Dis. 2013, 7, e2343. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.G.; Siripanyaphinyo, U.; Tumkosit, U.; Noranate, N.; Nuegoonpipat, A.; Pan, Y.; Kameoka, M.; Kurosu, T.; Ikuta, K.; Takeda, N.; et al. Poly I: C, an agonist of toll like receptor- 3, inhibits replication of the Chikungunya virus in BEAS-2B cells. Virol. J. 2012, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Wauquier, N.; Becquart, P.; Nkoghe, D.; Padilla, C.; Ndjoyi, M.A.; Leroy, E.M. The acute phase of chikungunya virus infection in humans is associated with strong innate immunity and T CD8 cell activation. J. Infect. Dis. 2011, 204, 115–123. [Google Scholar] [CrossRef]
- Thangamani, S.; Higgs, S.; Ziegler, S.; Vanlandingham, D.; Tesh, R. Host immune response to mosquito-transmitted Chikungunya virus differs from that elicited by needle inoculated virus. PLoS ONE. 2010, 5, e12137. [Google Scholar] [CrossRef] [PubMed]
- Hiroki, C.H.; Toller-Kawahisa, J.E.; Fumagalli, M.J.; Colon, D.F.; Figueiredo, L.; Fonseca, B.A.; Franca, R.F.; Cunha, F.Q. Neutrophil extracellular traps effectively control acute chikungunya virus infection. Front. Immunol. 2020, 10, 3108. [Google Scholar] [CrossRef]
- Farrell, P.J.; Broeze, R.J.; Lengyel, P. Accumulation of an mRNA and protein in interferon-treated Ehrlich ascites tumour cells. Nature 1979, 279, 523–525. [Google Scholar] [CrossRef] [PubMed]
- Korant, B.D.; Blomstrom, D.C.; Jonak, G.J.; Knight Jr, E. Interferon-induced proteins. Purification and characterization of a 15,000-dalton protein from human and bovine cells induced by interferon. J. Biol. Chem. 1984, 259, 14835–14839. [Google Scholar] [CrossRef]
- Haas, A.L.; Ahrens, P.; Bright, P.M.; Ankel, H. Interferon induces a 15-kilodalton protein exhibiting marked homology to ubiquitin. J. Biol. Chem. 1987, 262, 11315–11323. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, D.E. Interferon-stimulated gene 15 and the protein ISGylation system. J. Interferon Cytokine Res. 2011, 31, 119–130. [Google Scholar] [CrossRef]
- Hsu, L.C.; Park, J.M.; Zhang, K.; Luo, J.L.; Maeda, S.; Kaufman, R.J.; Karin, M. The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature 2004, 428, 341–345. [Google Scholar] [CrossRef]
- White, L.K.; Sali, T.; Alvarado, D.; Gatti, E.; Pierre, P.; Streblow, D.; DeFilippis, V.R. Chikungunya virus induces IPS-1-dependent innate immune activation and protein kinase R-independent translational shutoff. J. Virol. 2011, 85, 606–620. [Google Scholar] [CrossRef] [PubMed]
- Turan, K.; Mibayashi, M.; Sugiyama, K.; Saito, S.; Numajiri, A.; Nagata, K. Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic Acids Res. 2004, 32, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.H. Viral encounters with 2′, 5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 2007, 81, 12720–12729. [Google Scholar] [CrossRef] [PubMed]
- Roques, P.; Thiberville, S.D.; Dupuis-Maguiraga, L.; Lum, F.M.; Labadie, K.; Martinon, F.; Gras, G.; Lebon, P.; Ng, L.F.; De Lamballerie, X.; et al. Paradoxical effect of chloroquine treatment in enhancing chikungunya virus infection. Viruses. 2018, 10, 268. [Google Scholar] [CrossRef]
- Ninla-Aesong, P.; Mitarnun, W.; Noipha, K. Proinflammatory cytokines and chemokines as biomarkers of persistent arthralgia and severe disease after chikungunya virus infection: A 5-year follow-up study in Southern Thailand. Viral Immunol. 2019, 32, 442–452. [Google Scholar] [CrossRef]
- Ng, L.F.; Chow, A.; Sun, Y.J.; Kwek, D.J.; Lim, P.L.; Dimatatac, F.; Ng, L.C.; Ooi, E.E.; Choo, K.H.; Her, Z.; et al. IL-1β, IL-6, and RANTES as biomarkers of Chikungunya severity. PLoS ONE 2019, 4, e4261. [Google Scholar] [CrossRef]
- Simarmata, D.; Ng, D.C.E.; Kam, Y.W.; Lee, B.; Sum, M.S.H.; Her, Z.; Chow, A.; Leo, Y.S.; Cardosa, J.; Perera, D.; et al. Early clearance of Chikungunya virus in children is associated with a strong innate immune response. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Kashyap, R.S.; Morey, S.; Bhullar, S.; Baheti, N.; Chandak, N.; Purohit, H.; Taori, G.; Daginawala, H. Determination of Toll-like receptor-induced cytokine profiles in the blood and cerebrospinal fluid of Chikungunya patients. Neuroimmunomodulation 2014, 21, 338–346. [Google Scholar] [CrossRef]
- Chaaithanya, I.K.; Muruganandam, N.; Sundaram, S.G.; Kawalekar, O.; Sugunan, A.P.; Manimunda, S.P.; Ghosal, S.R.; Muthumani, K.; Vijayachari, P. Role of proinflammatory cytokines and chemokines in chronic arthropathy in CHIKV infection. Viral Immunol. 2011, 24, 265–271. [Google Scholar] [CrossRef]
- Chang, A.Y.; Tritsch, S.; Reid, S.P.; Martins, K.; Encinales, L.; Pacheco, N.; Amdur, R.L.; Porras-Ramirez, A.; Rico-Mendoza, A.; Li, G.; et al. The cytokine profile in acute chikungunya infection is predictive of chronic arthritis 20 months post infection. Diseases 2018, 6, 95. [Google Scholar] [CrossRef]
- Kulkarni, S.P.; Ganu, M.; Jayawant, P.; Thanapati, S.; Ganu, A.; Tripathy, A.S. Regulatory T cells and IL-10 as modulators of chikungunya disease outcome: A preliminary study. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2475–2481. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Kapus, A.; Marsden, P.A.; Li, Y.H.; Oreopoulos, G.; Marshall, J.C.; Frantz, S.; Kelly, R.A.; Medzhitov, R.; Rotstein, O.D. Regulation of Toll-like receptor 4 expression in the lung following hemorrhagic shock and lipopolysaccharide. J. Immunol. 2002, 168, 5252–5259. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Kodys, K.; Szabo, G. Impaired expression and function of toll-like receptor 7 in hepatitis C virus infection in human hepatoma cells. Hepatology 2010, 51, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lani, R.; Teoh, B.-T.; Sam, S.-S.; AbuBakar, S.; Hassandarvish, P. Fisetin Modulates Toll-like Receptor-Mediated Innate Antiviral Response in Chikungunya Virus-Infected Hepatocellular Carcinoma Huh7 Cells. Immuno 2022, 2, 703-719. https://doi.org/10.3390/immuno2040043
Lani R, Teoh B-T, Sam S-S, AbuBakar S, Hassandarvish P. Fisetin Modulates Toll-like Receptor-Mediated Innate Antiviral Response in Chikungunya Virus-Infected Hepatocellular Carcinoma Huh7 Cells. Immuno. 2022; 2(4):703-719. https://doi.org/10.3390/immuno2040043
Chicago/Turabian StyleLani, Rafidah, Boon-Teong Teoh, Sing-Sin Sam, Sazaly AbuBakar, and Pouya Hassandarvish. 2022. "Fisetin Modulates Toll-like Receptor-Mediated Innate Antiviral Response in Chikungunya Virus-Infected Hepatocellular Carcinoma Huh7 Cells" Immuno 2, no. 4: 703-719. https://doi.org/10.3390/immuno2040043
APA StyleLani, R., Teoh, B.-T., Sam, S.-S., AbuBakar, S., & Hassandarvish, P. (2022). Fisetin Modulates Toll-like Receptor-Mediated Innate Antiviral Response in Chikungunya Virus-Infected Hepatocellular Carcinoma Huh7 Cells. Immuno, 2(4), 703-719. https://doi.org/10.3390/immuno2040043