
Role of the Immune System in Renal Transplantation, Types of Response, Technical Approaches and Current Challenges


Abstract
:1. Introduction
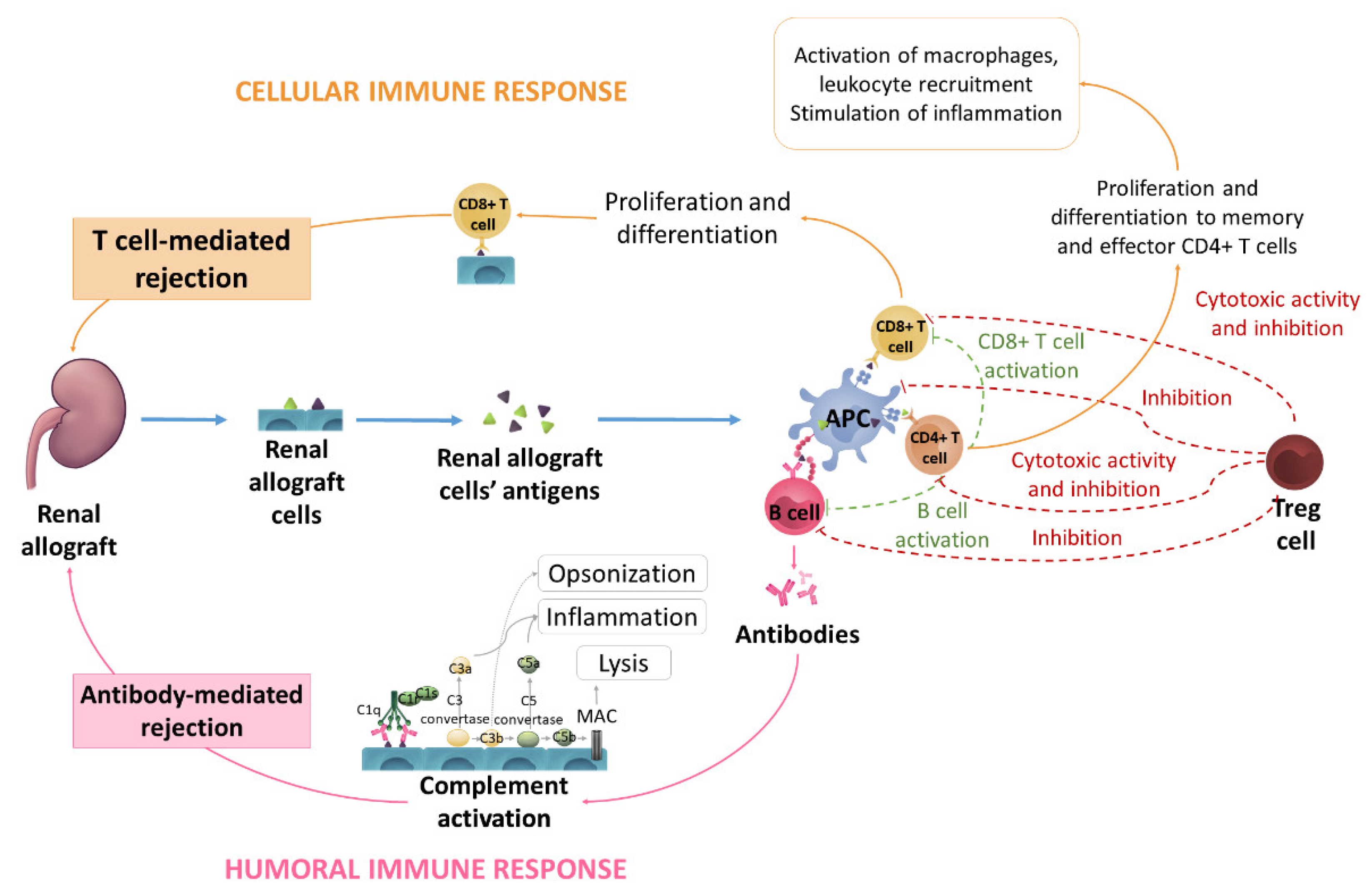
2. Immune Response against the Graft
2.1. HLA Antigens
2.2. Non-HLA Antigens
2.3. Innate Immunity
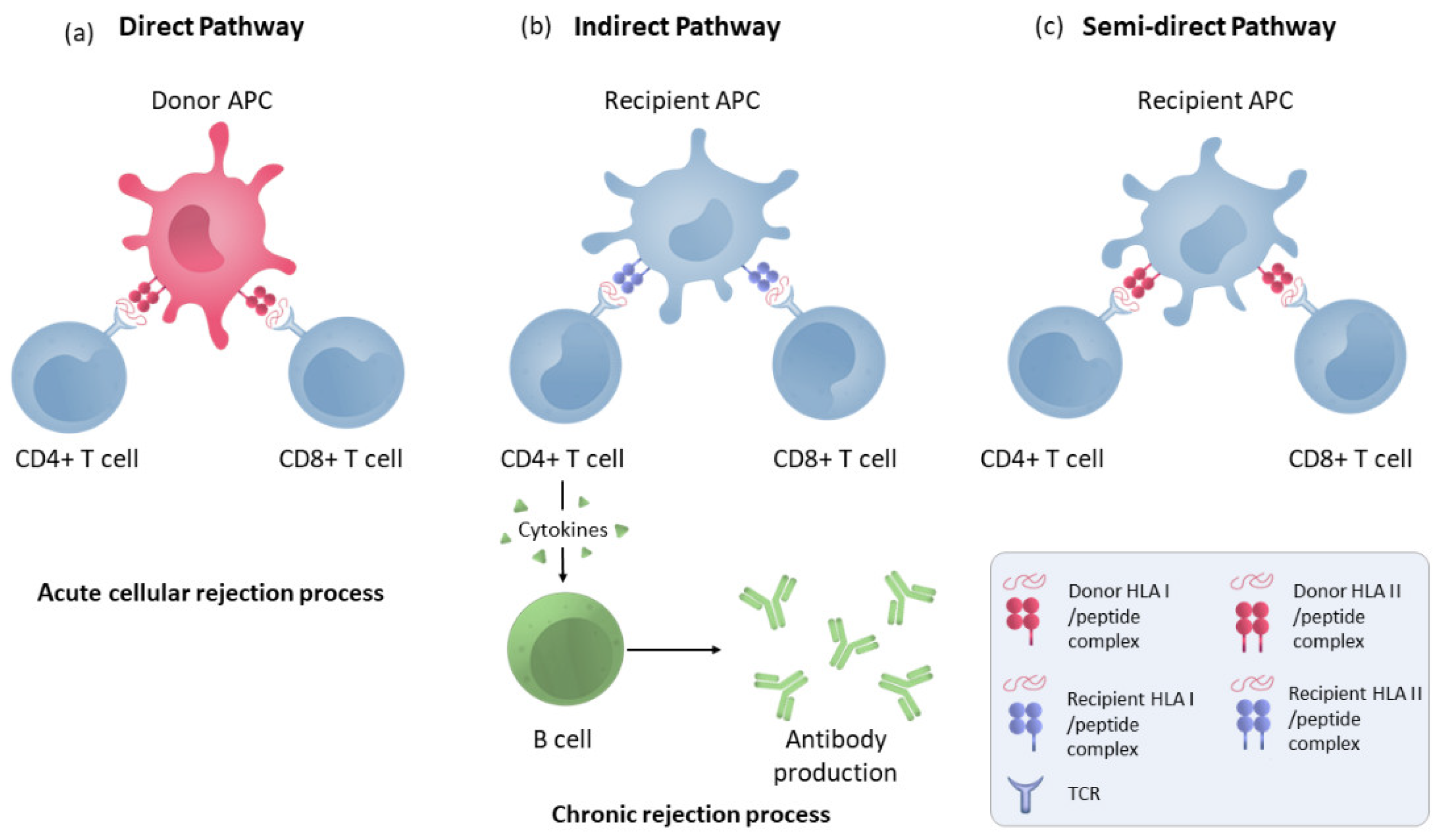
2.4. T-Cell Allorecognition Pathways
2.5. Endothelial Cells
2.6. T-Cells Activation
2.7. B Cells and Humoral Response
3. Biological Drugs Therapies Innovations
4. Antibody Detection Methods
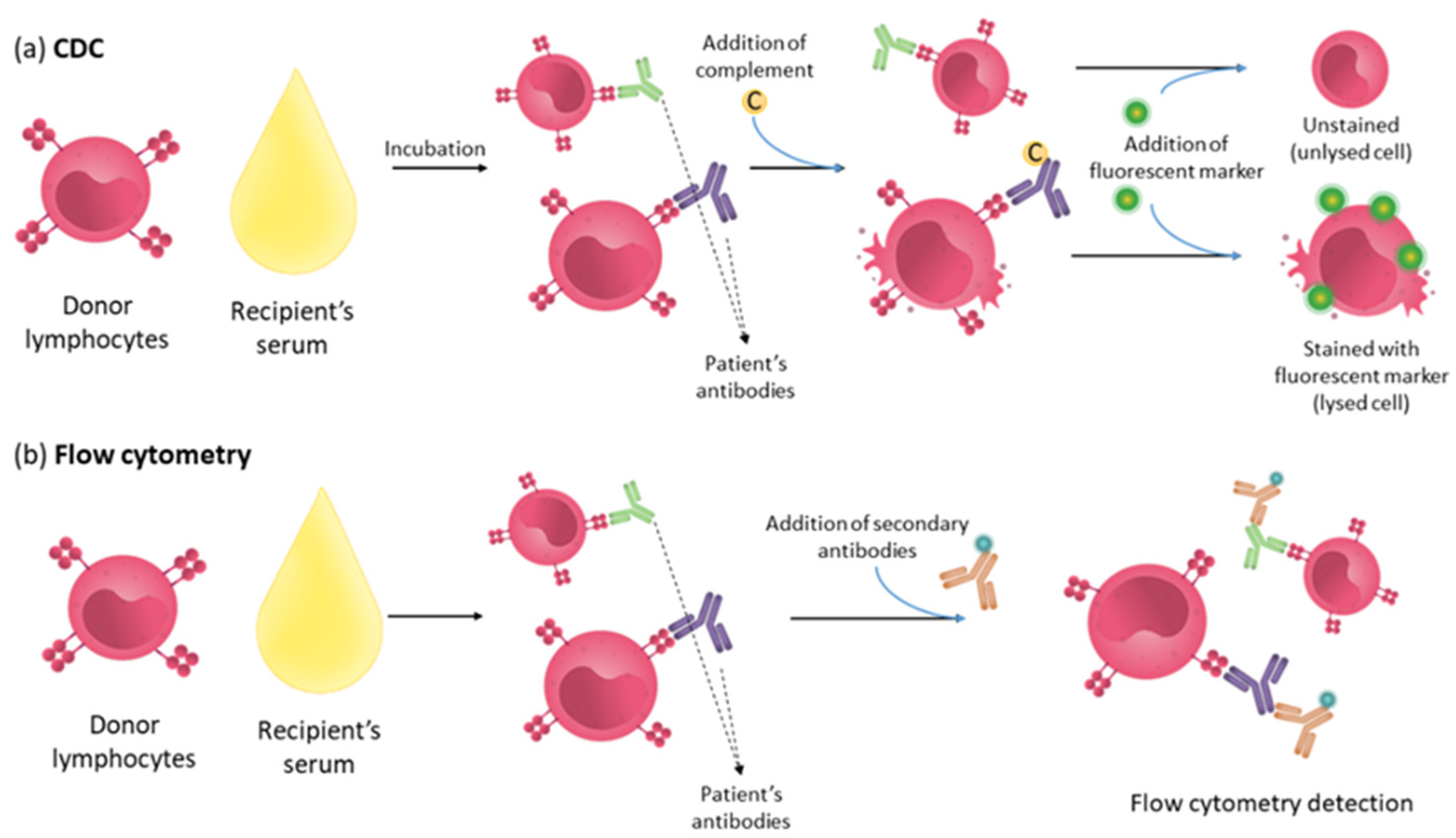
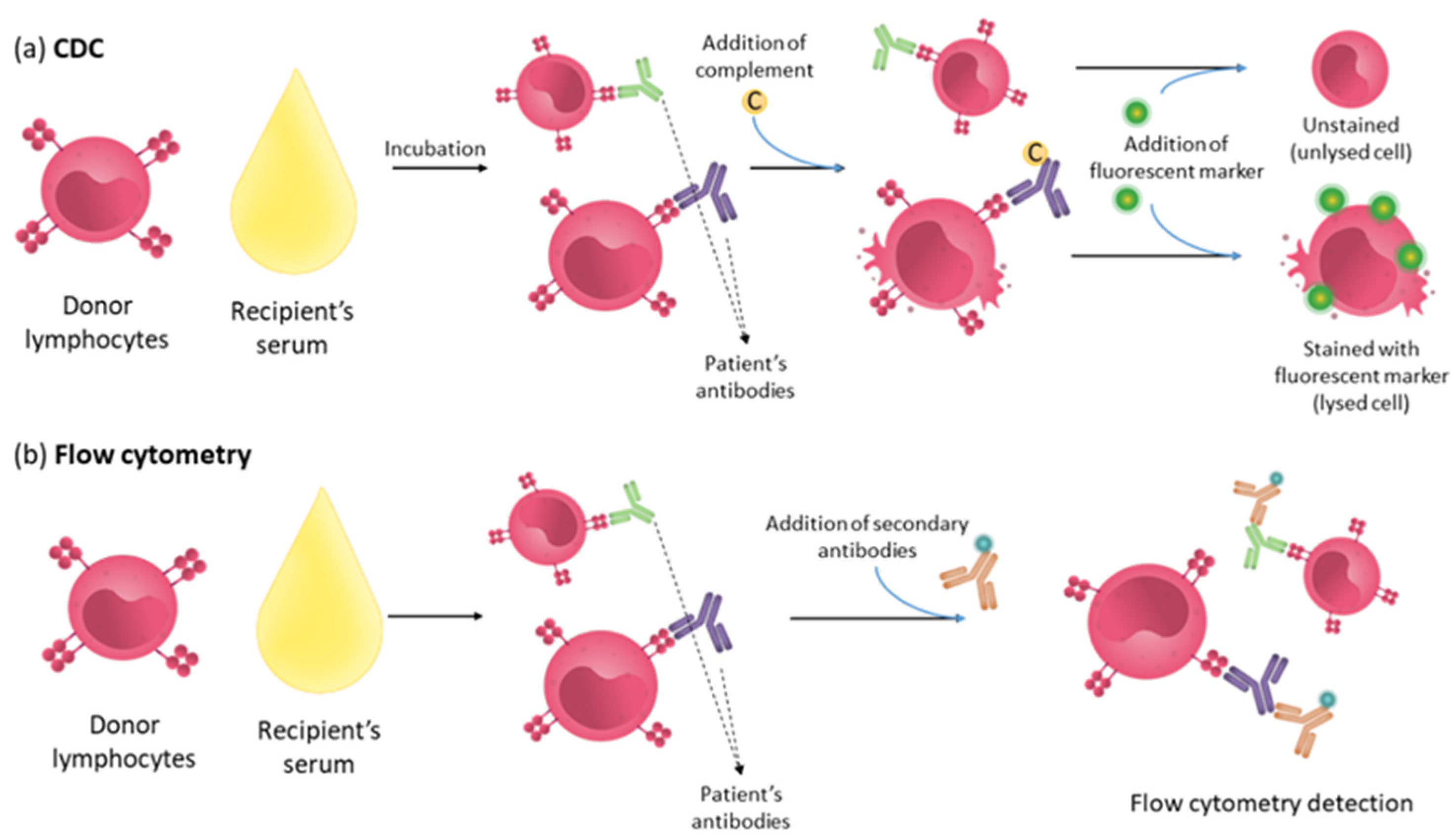
4.1. Complement Dependent Cytotoxicity (CDC) Assay
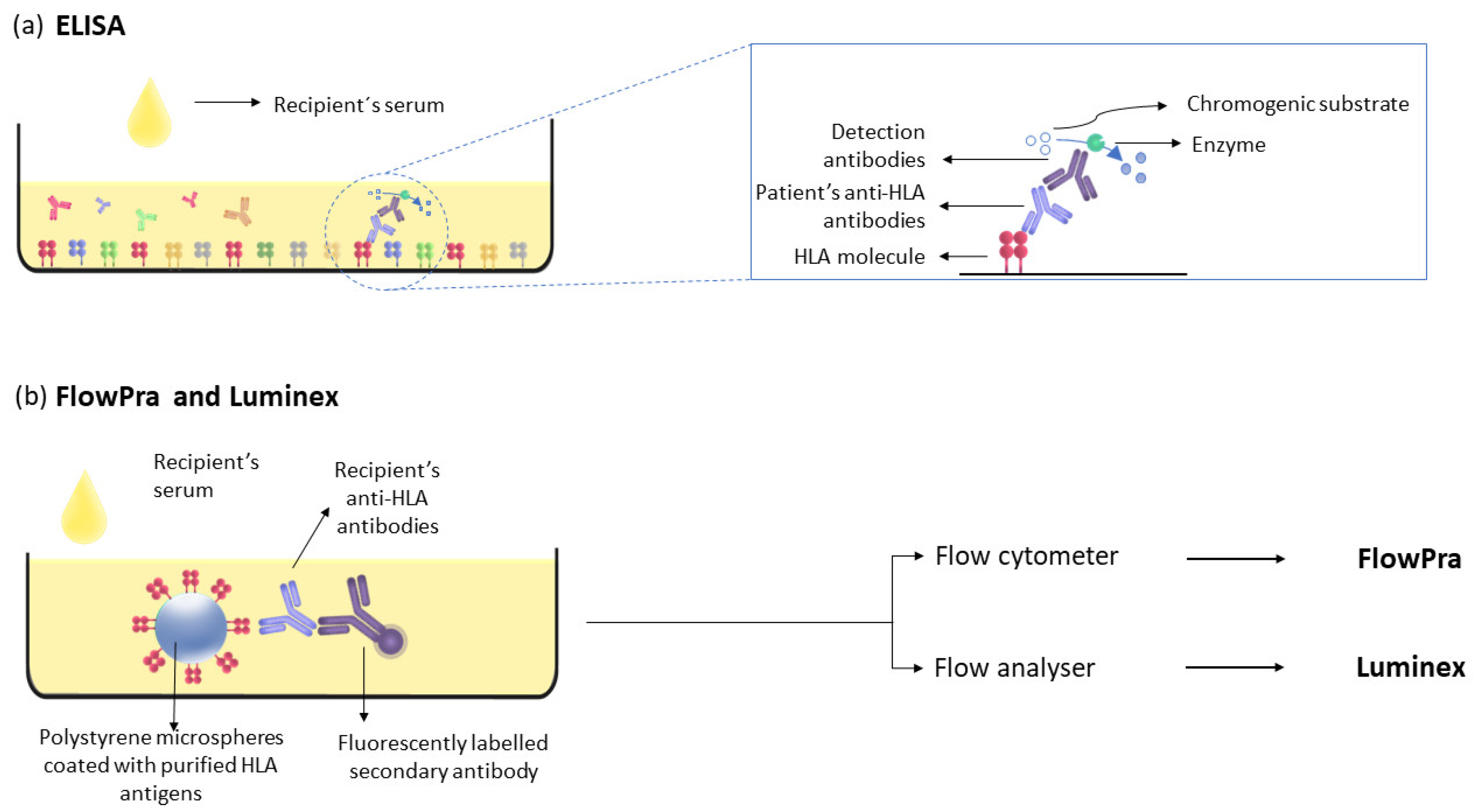
4.2. Luminex
5. Cellular Immunity Detection Methods
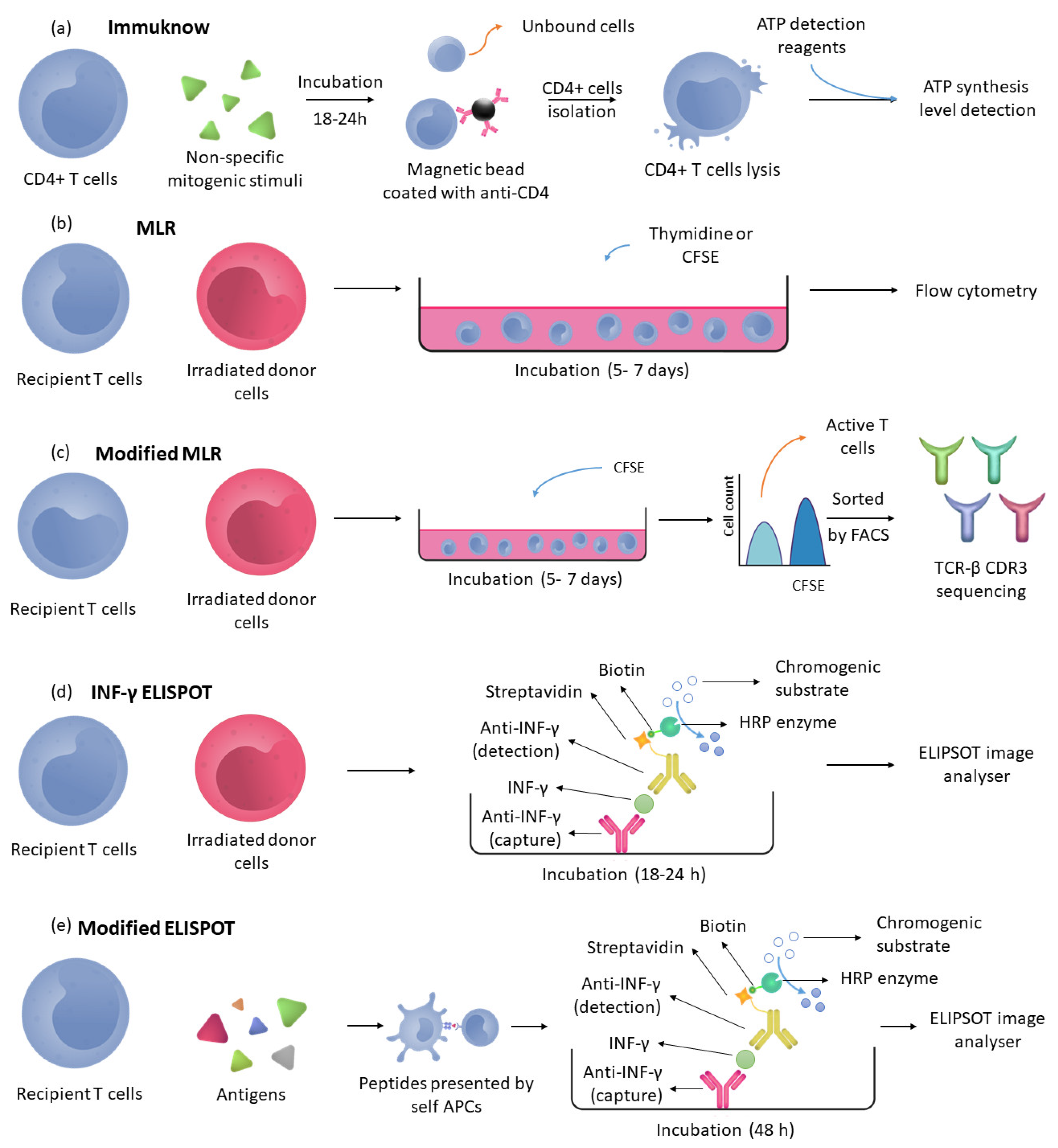
5.1. Immuknow
5.2. Specific Immunity Detection Methods
5.3. Mixed Lymphocytes Reaction (MLR)
5.4. INF-γ ELISPOT
5.5. Memory B Cells Detection Methods
6. Ongoing and Future Challenges
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moreso, F.; Hernández, D. Has the survival of the graft improved after renal transplantation in the era of modern immunosuppression? Nefrologia 2013, 33, 14–26. [Google Scholar] [CrossRef]
- Riella, L.V.; Djamali, A.; Pascual, J. Chronic Allograft Injury: Mechanisms and Potential Treatment Targets; Elsevier B.V.: Amsterdam, The Netherlands, 2017; Volume 31, ISBN 6177325254. [Google Scholar]
- Kissmeyer-Nielsen, F.; Olsen, S.; Petersen, V.P.; Fjeldborg, O. Hyperacute rejection of kidney allografts, associated with pre-existing humoral antibodies against donor cells. Lancet 1966, 288, 662–665. [Google Scholar] [CrossRef]
- Patel, R.; Terasaki, P.I. Significance of The Positive Crossmatch Test in Kidney Transplantation. N. Engl. J. Med. 1969, 280, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Bamoulid, J.; Staeck, O.; Halleck, F.; Khadzhynov, D.; Brakemeier, S.; Dürr, M.; Budde, K. The need for minimization strategies: Current problems of immunosuppression. Transpl. Int. 2015, 28, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Hara, S. Current pathological perspectives on chronic rejection in renal allografts. Clin. Exp. Nephrol. 2017, 21, 943–951. [Google Scholar] [CrossRef]
- Lai, X.; Zheng, X.; Mathew, J.M.; Gallon, L.; Leventhal, J.R.; Zhang, Z.J. Tackling Chronic Kidney Transplant Rejection: Challenges and Promises. Front. Immunol. 2021, 12, 661643. [Google Scholar] [CrossRef] [PubMed]
- Guerra, G.; Srinivas, T.R.; Meier-Kriesche, H.U. Calcineurin inhibitor-free immunosuppression in kidney transplantation. Transpl. Int. 2007, 20, 813–827. [Google Scholar] [CrossRef]
- Josephson, M.A. Monitoring and managing graft health in the kidney transplant recipient. Clin. J. Am. Soc. Nephrol. 2011, 6, 1774–1780. [Google Scholar] [CrossRef] [PubMed]
- Akkina, S.K.; Asrani, S.K.; Peng, Y.; Stock, P.; Kim, W.R.; Israni, A.K. Development of organ-specific donor risk indices. Liver Transplant. 2012, 18, 395–404. [Google Scholar] [CrossRef]
- Williams, W.W.; Taheri, D.; Tolkoff-Rubin, N.; Colvin, R.B. Clinical role of the renal transplant biopsy. Nat. Rev. Nephrol. 2012, 8, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.; Wiebe, C.; Gibson, I.W.; Rush, D.N.; Nickerson, P.W. Immune monitoring of kidney allografts. Am. J. Kidney Dis. 2012, 60, 629–640. [Google Scholar] [CrossRef]
- Lee, P.C.; Ozawa, M.; Hung, C.J.; Lin, Y.J.; Chang, S.S.; Chou, T.C. Eighteen-Year Follow-Up of a Retrospective Study of HLA Antibody on Kidney Graft Survival. Transplant. Proc. 2009, 41, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.; Mistry, K.; McWilliam, H.; Lopez, R.; Parham, P.; Marsh, S.G.E. The IMGT/HLA database. Nucleic Acids Res. 2011, 39, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.A.; Valenzuela, N.M.; Reed, E.F. The perfect storm: HLA antibodies, complement, FcγRs, and endothelium in transplant rejection. Trends Mol. Med. 2015, 21, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Tambur, A.R.; Kosmoliaptsis, V.; Claas, F.H.; Mannon, R.B.; Nickerson, P.; Naesens, M. Significance of HLA-DQ in kidney transplantation: Time to reevaluate human leukocyte antigen matching priorities to improve transplant outcomes? An expert review and recommendations. Kidney Int. 2021, 100, 1012–1022. [Google Scholar] [CrossRef]
- Hernández, D.; Vázquez, T.; Alonso-titos, J.; León, M.; Caballero, A.; Cobo, M.A.; Sola, E.; López, V.; Ruiz-esteban, P.; Cruzado, J.M.; et al. Impact of hla mismatching on early subclinical inflammation in low-immunological-risk kidney transplant recipients. J. Clin. Med. 2021, 10, 1934. [Google Scholar] [CrossRef]
- Collins, A.B.; Chicano, S.L.; Cornell, L.D.; Tolkoff-Rubin, N.; Goes, N.B.; Saidman, S.L.; Farrell, M.L.; Cosimi, A.B.; Colvin, R.B. Putative Antibody-Mediated Rejection With C4d Deposition in HLA-Identical, ABO-Compatible Renal Allografts. Transplant. Proc. 2006, 38, 3427–3429. [Google Scholar] [CrossRef] [PubMed]
- Grafft, C.A.; Cornell, L.D.; Gloor, J.M.; Cosio, F.G.; Gandhi, M.J.; Dean, P.G.; Stegall, M.D.; Amer, H. Antibody-mediated rejection following transplantation from an HLA-identical sibling. Nephrol. Dial. Transplant. 2010, 25, 307–310. [Google Scholar] [CrossRef]
- Zhang, Q.; Reed, E.F. The importance of non-HLA antibodies in transplantation. Nat. Rev. Nephrol. 2016, 12, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Reinsmoen, N.L. Impact and production of Non-HLA-specific antibodies in solid organ transplantation. Int. J. Immunogenet. 2020, 47, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Stallone, G.; Pontrelli, P.; Rascio, F.; Castellano, G.; Gesualdo, L.; Grandaliano, G. Coagulation and Fibrinolysis in Kidney Graft Rejection. Front. Immunol. 2020, 11, 1807. [Google Scholar] [CrossRef] [PubMed]
- Pontrelli, P.; Grandaliano, G.; Van Kooten, C. Editorial: Kidney Transplantation and Innate Immunity. Front. Immunol. 2020, 11, 603982. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Fan, Z.; Koulmanda, M.; Thronley, T.; Strom, T.B. Innate immunity for better or worse govern the allograft response. Curr. Opin. Organ Transplant. 2015, 20, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Goswami, T.K.; Singh, M.; Dhawan, M.; Mitra, S.; Emran, T.B.; Rabaan, A.A.; Mutair, A.A.; Alawi, Z.A.; Alhumaid, S.; Dhama, K. Regulatory T cells (Tregs) and their therapeutic potential against autoimmune disorders—Advances and challenges. Hum. Vaccines Immunother. 2022, 18, 2035117. [Google Scholar] [CrossRef]
- Siu, J.H.Y.; Surendrakumar, V.; Richards, J.A.; Pettigrew, G.J. T cell allorecognition pathways in solid organ transplantation. Front. Immunol. 2018, 9, 2548. [Google Scholar] [CrossRef]
- Al-Lamki, R.S.; Bradley, J.R.; Pober, J.S. Endothelial Cells in Allograft Rejection. Transplantation 2008, 86, 1340–1348. [Google Scholar] [CrossRef]
- Ingulli, E. Mechanism of cellular rejection in transplantation. Pediatr. Nephrol. 2010, 25, 61–74. [Google Scholar] [CrossRef]
- Baker, R.J.; Hernandez-Fuentes, M.P.; Brookes, P.A.; Chaudhry, A.N.; Cook, H.T.; Lechler, R.I. Loss of Direct and Maintenance of Indirect Alloresponses in Renal Allograft Recipients: Implications for the Pathogenesis of Chronic Allograft Nephropathy. J. Immunol. 2001, 167, 7199–7206. [Google Scholar] [CrossRef]
- Conlon, T.M.; Saeb-Parsy, K.; Cole, J.L.; Motallebzadeh, R.; Qureshi, M.S.; Rehakova, S.; Negus, M.C.; Callaghan, C.J.; Bolton, E.M.; Bradley, J.A.; et al. Germinal Center Alloantibody Responses Are Mediated Exclusively by Indirect-Pathway CD4 T Follicular Helper Cells. J. Immunol. 2012, 188, 2643–2652. [Google Scholar] [CrossRef]
- Karahan, G.E.; Claas, F.H.J.; Heidt, S. Pre-existing Alloreactive T and B Cells and Their Possible Relevance for Pre-transplant Risk Estimation in Kidney Transplant Recipients. Front. Med. 2020, 7, 340. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, C.; Wang, J.; Yang, C.; Xu, M.; Rong, R.; Zhu, T.; Zhu, D. Endothelial Cells in Antibody-Mediated Rejection of Kidney Transplantation: Pathogenesis Mechanisms and Therapeutic Implications. J. Immunol. Res. 2017, 2017, 8746303. [Google Scholar] [CrossRef]
- Gavlovsky, P.J.; Tonnerre, P.; Guitton, C.; Charreau, B. Expression of MHC class I-related molecules MICA, HLA-E and EPCR shape endothelial cells with unique functions in innate and adaptive immunity. Hum. Immunol. 2016, 77, 1084–1091. [Google Scholar] [CrossRef]
- Luo, L.; Li, Z.; Wu, W.; Luo, G.; Mei, H.; Sun, Z.; Xu, C. The effect of MICA antigens on kidney transplantation outcomes. Immunol. Lett. 2013, 156, 54–58. [Google Scholar] [CrossRef]
- Stastny, P.; Zou, Y.; Fan, Y.; Qin, Z.; Lavingia, B. The emerging issue of MICA antibodies: Antibodies to MICA and other antigens of endothelial cells. Contrib. Nephrol. 2009, 162, 99–106. [Google Scholar] [CrossRef]
- Thaunat, O.; Graff-Dubois, S.; Fabien, N.; Duthey, A.; Attuil-Audenis, V.; Nicoletti, A.; Patey, N.; Morelon, E. A stepwise breakdown of B-cell tolerance occurs within renal allografts during chronic rejection. Kidney Int. 2012, 81, 207–219. [Google Scholar] [CrossRef]
- Lim, M.A.; Kohli, J.; Bloom, R.D. Immunosuppression for kidney transplantation: Where are we now and where are we going? Transplant. Rev. 2017, 31, 10–17. [Google Scholar] [CrossRef]
- Yang, J.; Brook, M.O.; Carvalho-Gaspar, M.; Zhang, J.; Ramon, H.E.; Sayegh, M.H.; Wood, K.J.; Turka, L.A.; Jones, N.D. Allograft rejection mediated by memory T cells is resistant to regulation. Proc. Natl. Acad. Sci. USA 2007, 104, 19954–19959. [Google Scholar] [CrossRef]
- Adams, A.B.; Williams, M.A.; Jones, T.R.; Shirasugi, N.; Durham, M.M.; Kaech, S.M.; Wherry, E.J.; Onami, T.; Lanier, J.G.; Kokko, K.E.; et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J. Clin. Investig. 2003, 111, 1887–1895. [Google Scholar] [CrossRef]
- Goldfarb, D.A. Immunosuppressive Drugs for Kidney Transplantation. J. Urol. 2005, 173, 2105. [Google Scholar] [CrossRef]
- Tsuji, T.; Iwasaki, S.; Makita, K.; Imamoto, T.; Ishidate, N.; Mitsuke, A.; Fukuzawa, N.; Harada, H.; Fukazawa, Y. Preceding T-Cell-Mediated Rejection Is Associated with the Development of Chronic Active Antibody-Mediated Rejection by de Novo Donor-Specific Antibody. Nephron 2021, 144, 13–17. [Google Scholar] [CrossRef]
- Gupta, G.; Abu Jawdeh, B.G.; Racusen, L.C.; Bhasin, B.; Arend, L.J.; Trollinger, B.; Kraus, E.; Rabb, H.; Zachary, A.A.; Montgomery, R.A.; et al. Late antibody-mediated rejection in renal allografts: Outcome after conventional and novel therapies. Transplantation 2014, 97, 1240–1246. [Google Scholar] [CrossRef]
- Voora, S.; Adey, D.B. Management of Kidney Transplant Recipients by General Nephrologists: Core Curriculum 2019. Am. J. Kidney Dis. 2019, 73, 866–879. [Google Scholar] [CrossRef]
- Noelle, R.J.; Snow, E.C. Cognate interactions between helper T cells and B Cells. Immunol. Today 1990, 11, 361–368. [Google Scholar] [CrossRef]
- Lavinder, J.J.; Wine, Y.; Giesecke, C.; Ippolito, G.C.; Horton, A.P.; Lungu, O.I.; Hoi, K.H.; DeKosky, B.J.; Murrin, E.M.; Wirth, M.M.; et al. Identification and characterization of the constituent human serum antibodies elicited by vaccination. Proc. Natl. Acad. Sci. USA 2014, 111, 2259–2264. [Google Scholar] [CrossRef]
- Bernasconi, N.L.; Traggiai, E.; Lanzavecchia, A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science 2002, 298, 2199–2202. [Google Scholar] [CrossRef]
- Resch, T.; Fabritius, C.; Ebner, S.; Ritschl, P.; Kotsch, K. The Role of Natural Killer Cells in Humoral Rejection. Transplantation 2015, 99, 1335–1340. [Google Scholar] [CrossRef]
- Valenzuela, N.M.; Reed, E.F. Antibodies to HLA Molecules Mimic Agonistic Stimulation to Trigger Vascular Cell Changes and Induce Allograft Injury HHS Public Access. Curr. Transpl. Rep. 2015, 2, 222–232. [Google Scholar] [CrossRef]
- Loupy, A.; Mengel, M.; Haas, M. Thirty years of the International Banff Classification for Allograft Pathology: The past, present, and future of kidney transplant diagnostics. Kidney Int. 2022, 101, 678–691. [Google Scholar] [CrossRef]
- Formica, M.L.; Awde Alfonso, H.G.; Palma, S.D. Biological drug therapy for ocular angiogenesis: Anti-VEGF agents and novel strategies based on nanotechnology. Pharmacol. Res. Perspect. 2021, 9, e00723. [Google Scholar] [CrossRef]
- Sánchez-Magraner, L.; de la Fuente, M.; Evans, C.; Miles, J.; Elexpe, A.; Rodriguez-Astigarraga, M.; Astigarraga, E.; Barreda-Gómez, G. Quantification of PD-1/PD-L1 Interaction between Membranes from PBMCs and Melanoma Samples Using Cell Membrane Microarray and Time-Resolved Förster Resonance Energy Transfer. Analytica 2021, 2, 156–170. [Google Scholar] [CrossRef]
- Aletaha, D.; Smolen, J.S. Diagnosis and Management of Rheumatoid Arthritis: A Review. JAMA-J. Am. Med. Assoc. 2018, 320, 1360–1372. [Google Scholar] [CrossRef]
- Jang, D.I.; Lee, A.H.; Shin, H.Y.; Song, H.R.; Park, J.H.; Kang, T.B.; Lee, S.R.; Yang, S.H. The role of tumor necrosis factor alpha (Tnf-α) in autoimmune disease and current tnf-α inhibitors in therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef]
- Zhao, Y.; Cooper, D.K.C.; Wang, H.; Chen, P.; He, C.; Cai, Z.; Mou, L.; Luan, S.; Gao, H. Potential pathological role of pro-inflammatory cytokines (IL-6, TNF-α, and IL-17) in xenotransplantation. Xenotransplantation 2019, 26, e12502. [Google Scholar] [CrossRef]
- Nagata, Y.; Fujimoto, M.; Nakamura, K.; Isoyama, N.; Matsumura, M.; Fujikawa, K.; Uchiyama, K.; Takaki, E.; Takii, R.; Nakai, A.; et al. Anti-TNF-α agent infliximab and splenectomy are protective against renal ischemia-reperfusion injury. Transplantation 2016, 100, 1675–1682. [Google Scholar] [CrossRef]
- Jordan, S.C.; Choi, J.; Kim, I.; Wu, G.; Toyoda, M.; Shin, B.; Vo, A. Interleukin-6, A cytokine critical to mediation of inflammation, autoimmunity and allograft rejection: Therapeutic implications of IL-6 receptor blockade. Transplantation 2017, 101, 32–44. [Google Scholar] [CrossRef]
- Choi, J.; Aubert, O.; Vo, A.; Loupy, A.; Haas, M.; Puliyanda, D.; Kim, I.; Louie, S.; Kang, A.; Peng, A.; et al. Assesment of Tocilizumab (Anti-IL-6 Receptor Monoclonal) as a Potential Treatment for Chronic Antibody Mediated Rejection and Transplant Glomerulopathy in HLA Sensitized Renal Allograft Recipients. Am. J. Transpl. 2017, 17, 2381–2389. [Google Scholar] [CrossRef] [PubMed]
- Navarra, S.V.; Guzmán, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.M.; Thomas, M.; Kim, H.Y.; León, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Banham, G.D.; Flint, S.M.; Torpey, N.; Lyons, P.A.; Shanahan, D.N.; Gibson, A.; Watson, C.J.E.; O’Sullivan, A.M.; Chadwick, J.A.; Foster, K.E.; et al. Belimumab in kidney transplantation: An experimental medicine, randomised, placebo-controlled phase 2 trial. Lancet 2018, 391, 2619–2630. [Google Scholar] [CrossRef]
- Lundin, J.; Kimby, E.; Björkholm, M.; Broliden, P.A.; Celsing, F.; Hjalmar, V.; Möllgård, L.; Rebello, P.; Hale, G.; Waldmann, H.; et al. Phase II trial of subcutaneous anti-CD52 monoclonal antibody alemtuzumab (Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL). Blood 2002, 100, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Friend, P.J. Alemtuzumab induction therapy in solid organ transplantation. Transplant. Res. 2013, 2, 2–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sureshkumar, K.K.; Hussain, S.M.; Carpenter, B.J.; Sandroni, S.E.; Marcus, R.J. Antibody-mediated rejection following renal transplantation. Expert Opin. Pharmacother. 2007, 8, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Macklin, P.S.; Morris, P.J.; Knight, S.R. A systematic review of the use of rituximab for the treatment of antibody-mediated renal transplant rejection. Transplant. Rev. 2017, 31, 87–95. [Google Scholar] [CrossRef]
- Venetz, J.P.; Pascual, M. New treatments for acute humoral rejection of kidney allografts. Expert Opin. Investig. Drugs 2007, 16, 625–633. [Google Scholar] [CrossRef]
- Grenda, R. Biologics in renal transplantation. Pediatr. Nephrol. 2015, 30, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Cecka, J.M.; Kucheryavaya, A.Y.; Reinsmoen, N.L.; Leffell, M.S. Calculated PRA: Initial results show benefits for sensitized patients and a reduction in positive crossmatches. Am. J. Transplant. 2011, 11, 719–724. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Williams, R.L.; Wong, G.; Lim, W.H. The evolution of hla-matching in kidney transplantation. In Current Issues and Future Direction in Kidney Transplantation; IntechOpen Limited: London, UK, 2013; pp. 273–297. [Google Scholar] [CrossRef]
- Ting, A. The Limfocitotoxic Crossmatch Test in Clinical Renal Transplantation. Transplantation 1983, 35, 403–407. [Google Scholar] [CrossRef]
- Mulley, W.R.; Kanellis, J. Understanding crossmatch testing in organ transplantation: A case-based guide for the general nephrologist. Nephrology 2011, 16, 125–133. [Google Scholar] [CrossRef]
- Claas, F.H.J.; Witvliet, M.D.; Duquesnoy, R.J.; Persijn, G.G.; Doxiadis, I.I.N. The acceptable mismatch program as a fast tool for highly sensitized patients awaiting a cadaveric kidney transplantation: Short waiting time and excellent graft outcome. Transplantation 2004, 78, 190–193. [Google Scholar] [CrossRef]
- Tait, B.D. Solid phase assays for HLA antibody detection in clinical transplantation. Curr. Opin. Immunol. 2009, 21, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Tait, B.D.; Süsal, C.; Gebel, H.M.; Nickerson, P.W.; Zachary, A.A.; Claas, F.H.J.; Reed, E.F.; Bray, R.A.; Campbell, P.; Chapman, J.R.; et al. Consensus guidelines on the testing and clinical management issues associated with HLA and Non-HLA antibodies in transplantation. Transplantation 2013, 95, 19–47. [Google Scholar] [CrossRef]
- Minucci, P.B.; Grimaldi, V.; Casamassimi, A.; Cacciatore, F.; Sommese, L.; Picascia, A.; Resse, M.; Sabia, C.; Russo, A.; Napoli, C. Methodologies for Anti-HLA antibody screening in patients awaiting kidney transplant: A comparative study. Exp. Clin. Transplant. 2011, 9, 381–386. [Google Scholar]
- Rebibou, J.M.; Chabod, J.; Bittencourt, M.C.; Thevénin, C.; Chalopin, J.M.; Hervé, P.; Tiberghien, P. Flow-PRA® evaluation for antibody screening in patients awaiting kidney transplantation. Transpl. Immunol. 2000, 8, 125–128. [Google Scholar] [CrossRef]
- Colombo, M.B.; Haworth, S.E.; Poli, F.; Nocco, A.; Puglisi, G.; Innocente, A.; Serafini, M.; Messa, P.; Scalamogna, M. Luminex technology for anti-HLA antibody screening: Evaluation of performance and of impact on laboratory routine. Cytom. Part B-Clin. Cytom. 2007, 72, 465–471. [Google Scholar] [CrossRef]
- Pei, R.; Wang, G.; Tarsitani, C.; Rojo, S.; Chen, T.; Takemura, S.; Liu, A.; Lee, J. Simultaneous HLA class I and class II antibodies screening with flow cytometry. Hum. Immunol. 1998, 59, 313–322. [Google Scholar] [CrossRef]
- Picascia, A.; Infante, T.; Napoli, C. Luminex and antibody detection in kidney transplantation. Clin. Exp. Nephrol. 2012, 16, 373–381. [Google Scholar] [CrossRef]
- Loupy, A.; Lefaucheur, C.; Vernerey, D.; Prugger, C.; van Huyen, J.-P.D.; Mooney, N.; Suberbielle, C.; Frémeaux-Bacchi, V.; Méjean, A.; Desgrandchamps, F.; et al. Complement-Binding Anti-HLA Antibodies and Kidney-Allograft Survival. N. Engl. J. Med. 2013, 369, 1215–1226. [Google Scholar] [CrossRef]
- Sullivan, H.C.; Gebel, H.M.; Bray, R.A. Understanding solid-phase HLA antibody assays and the value of MFI. Hum. Immunol. 2017, 78, 471–480. [Google Scholar] [CrossRef]
- McCaughan, J.; Xu, Q.; Tinckam, K. Detecting donor-specific antibodies: The importance of sorting the wheat from the chaff. HepatoBiliary Surg. Nutr. 2019, 8, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Süsal, C.; Morath, C. Virtual PRA replaces traditional PRA: Small change but significantly more justice for sensitized patients. Transpl. Int. 2015, 28, 708–709. [Google Scholar] [CrossRef]
- Bingaman, A.W.; Murphey, C.L.; Palma-Vargas, J.; Wright, F. A virtual crossmatch protocol significantly increases access of highly sensitized patients to deceased donor kidney transplantation. Transplantation 2008, 86, 1864–1868. [Google Scholar] [CrossRef]
- Kowalski, R.; Post, D.; Schneider, M.C.; Britz, J.; Thomas, J.; Deierhoi, M.; Lobashevsky, A.; Redfield, R.; Schweitzer, E.; Heredia, A.; et al. Immune cell function testing: An adjunct to therapeutic drug monitoring in transplant patient management. Clin. Transplant. 2003, 17, 77–88. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Li, Y.; Zhang, H.; Wei, X.; Zheng, H.; Xu, C.; Bao, X.; Yuan, X.; Hou, J. Immune function assay (ImmuKnow) as a predictor of allograft rejection and infection in kidney transplantation. Clin. Transplant. 2013, 27, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.T.; Lee, H.M.; Lower, R.R.; Thomas, J.M. Immunological monitoring as a guide to the management of transplant recipients. Surg. Clin. N. Am. 1979, 59, 253–281. [Google Scholar] [CrossRef]
- MacEdo, C.; Orkis, E.A.; Popescu, I.; Elinoff, B.D.; Zeevi, A.; Shapiro, R.; Lakkis, F.G.; Metes, D. Contribution of naïve and memory t-cell populations to the human alloimmune response. Am. J. Transplant. 2009, 9, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.I.; Morris, A.G.; Booth, L.J. Clinical significance of in vitro donor-specific hyporesponsiveness in renal allograft recipients as demonstrated by the MLR. Transpl. Int. 1994, 7, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Crespo, E.; Bestard, O. Biomarkers to assess donor-reactive T-cell responses in kidney transplant patients. Clin. Biochem. 2016, 49, 329–337. [Google Scholar] [CrossRef]
- Emerson, R.O.; Mathew, J.M.; Konieczna, I.M.; Robins, H.S.; Leventhal, J.R. Defining the alloreactive T cell repertoire using high-throughput sequencing of mixed lymphocyte reaction culture. PLoS ONE 2014, 9, e111934. [Google Scholar] [CrossRef]
- Crespo, E.; Lucia, M.; Cruzado, J.M.; Luque, S.; Melilli, E.; Manonelles, A.; Lloberas, N.; Torras, J.; Grinyó, J.M.; Bestard, O. Pre-transplant donor-specific T-cell alloreactivity is strongly associated with early acute cellular rejection in kidney transplant recipients not receiving T-cell depleting induction therapy. PLoS ONE 2015, 10, e117618. [Google Scholar] [CrossRef]
- Nickel, P.; Presber, F.; Bold, G.; Biti, D.; Schönemann, C.; Tullius, S.G.; Volk, H.D.; Reinke, P. Enzyme-linked immunosorbent spot assay for donor-reactive interferon-gamma-producing cells identifies T-cell presensitization and correlates with graft function at 6 and 12 months in renal-transplant recipients. Transplantation 2004, 78, 1640–1646. [Google Scholar] [CrossRef]
- Hricik, D.E.; Augustine, J.; Nickerson, P.; Formica, R.N.; Emilio, D.; Rush, D.; Newell, K.A.; Goebel, J.; Gibson, I.W.; Fairchild, R.L.; et al. Interferon Gamma ELISPOT Testing as a Risk-stratifying Biomarker for Kidney Transplant Injury: Results from the CTOT-01 Multecenter Study. Am. J. Transplant. 2015, 15, 3166–3173. [Google Scholar] [CrossRef] [Green Version]
- Girmanova, E.; Hruba, P.; Viklicky, O.; Slavcev, A. ELISpot assay and prediction of organ transplant rejection. Int. J. Immunogenet. 2022, 49, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Waanders, M.M.; Heidt, S.; Koekkoek, K.M.; Zoet, Y.M.; Doxiadis, I.I.N.; Amir, A.; Heemskerk, M.H.M.; Mulder, A.; Brand, A.; Roelen, D.L.; et al. Monitoring of indirect allorecognition: Wishful thinking or solid data? Tissue Antigens 2008, 71, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zachary, A.A.; Kopchaliiska, D.; Montgomery, R.A.; Leffell, M.S. HLA-specific B cells: I. A method for their detection, quantification, and isolation using HLA tetramers. Transplantation 2007, 83, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Wehmeier, C.; Karahan, G.E.; Heidt, S. HLA-specific memory B-cell detection in kidney transplantation: Insights and future challenges. Int. J. Immunogenet. 2020, 47, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Karahan, G.E.; Claas, F.H.J.; Heidt, S. Detecting the humoral alloimmune response: We need more than serum antibody screening. Transplantation 2015, 99, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Karahan, G.E.; de Vaal, Y.J.H.; Roelen, D.L.; Buchli, R.; Claas, F.H.J.; Heidt, S. Quantification of HLA class II-specific memory B cells in HLA-sensitized individuals. Hum. Immunol. 2015, 76, 129–136. [Google Scholar] [CrossRef]
- Quaglia, M.; Merlotti, G.; Guglielmetti, G.; Castellano, G.; Cantaluppi, V. Recent advances on biomarkers of early and late kidney graft dysfunction. Int. J. Mol. Sci. 2020, 21, 5404. [Google Scholar] [CrossRef]
- Wagner, J.C.; Tang, Q. CAR-Tregs as a Strategy for Inducing Graft Tolerance. Curr. Transpl. Rep. 2020, 7, 205–214. [Google Scholar] [CrossRef]
- Roemhild, A.; Otto, N.M.; Moll, G.; Abou-El-Enein, M.; Kaiser, D.; Bold, G.; Schachtner, T.; Choi, M.; Oellinger, R.; Landwehr-Kenzel, S.; et al. Regulatory T cells for minimising immune suppression in kidney transplantation: Phase I/IIa clinical trial. BMJ 2020, 371, m3734. [Google Scholar] [CrossRef]
- Cravedi, P.; Heeger, P.S. Immunologic monitoring in transplantation revisited. Curr. Opin. Organ Transplant. 2012, 17, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Chopra, B.; Sureshkumar, K.K. Emerging role of cell-free DNA in kidney transplantation. World J. Exp. Med. 2021, 11, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Bloom, R.D.; Bromberg, J.S.; Poggio, E.D.; Bunnapradist, S.; Langone, A.J.; Sood, P.; Matas, A.J.; Mehta, S.; Mannon, R.B.; Sharfuddin, A.; et al. Cell-Free DNA and active rejection in kidney allografts. J. Am. Soc. Nephrol. 2017, 28, 2221–2232. [Google Scholar] [CrossRef] [PubMed]
- Carbone, D.P.; Gandara, D.R.; Antonia, S.J.; Zielinski, C.; Paz-Ares, L. Non-small-cell lung cancer: Role of the immune system and potential for immunotherapy. J. Thorac. Oncol. 2015, 10, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Magraner, L.; Miles, J.; Baker, C.L.; Applebee, C.J.; Lee, D.J.; Elsheikh, S.; Lashin, S.; Withers, K.; Watts, A.G.; Parry, R.; et al. High PD-1/PD-L1 checkpoint interaction infers tumor selection and therapeutic sensitivity to anti-PD-1/PD-L1 treatment. Cancer Res. 2020, 80, 4244–4257. [Google Scholar] [CrossRef]
- Rienda, B.; Elexpe, A.; Tolentino-Cortez, T.; Gulak, M.; Bruzos-Cidón, C.; Torrecilla, M.; Astigarraga, E.; Barreda-Gómez, G. Analysis of Acetylcholinesterase Activity in Cell Membrane Microarrays of Brain Areas as a Screening Tool to Identify Tissue Specific Inhibitors. Analytica 2021, 2, 25–36. [Google Scholar] [CrossRef]
- Fernández, R.; Garate, J.; Tolentino-Cortez, T.; Herraiz, A.; Lombardero, L.; Ducrocq, F.; Rodríguez-Puertas, R.; Trifilieff, P.; Astigarraga, E.; Barreda-Gómez, G.; et al. Microarray and Mass Spectrometry-Based Methodology for Lipid Profiling of Tissues and Cell Cultures. Anal. Chem. 2019, 91, 15967–15973. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Detection of Anti-HLA Antibodies | |||||
| Cell-Based Assays | |||||
| Method | Samples | Requirements | Detection | Diagnosis | |
| Donor | Patient | ||||
| CDC | Lymphocytes or panel of HLA typed lymphocytes | Serum | Complement and fluorescent marker | Complement-binding antibodies | Hyperacute rejection |
| Flow cytometry | Lymphocytes | Serum | Fluorescently labelled secondary antibody | Complement-binding antibodies and non-complement-fixing antibodies | Increased risk of acute rejection |
| Solid phase assays | |||||
| Method | Immobilised | Solid phase | Patient’s sample | Detection method | Sensitivity |
| ELISA | HLA class I and II antigens | Microtiter plate | Serum | Secondary antibody bound to an enzyme and chromogenic substrate | ↑ |
| FlowPra | HLA antigens | Microspheres | Serum | Flow cytometer | ↑↑ |
| Luminex | HLA antigens | Microspheres | Serum | Flow analyser | ↑↑↑ |
| Measure of Cellular Immune Response | ||||||
| T Cell | ||||||
| Method | Samples | Process | Detection | |||
| Patient | Donor | |||||
| Immuknow | Whole blood with nonspecific mitogenic stimuli | - | T cell isolation, addition of lytic agent and ATP synthesis level detection. | Nonspecific T-cell mediated immune responsiveness | ||
| MLR | Lymphocytes | PMBCs with T-cell proliferation blocked | Incubation of both samples for 5–7 days with radioactive thymidine or CFSE and quantification of thymidine or CFSE concentration | Frequency of directly activated recipient T cell | ||
| Modified MLR | Lymphocytes | PMBCs with T-cell proliferation blocked | Incubation of both samples for 5–7 days CFSE, CFSE-low dividing T cells are sorted by FACS, and genomic DNA is isolated for TCR-β CDR3 sequencing. | Alloreactive TCR repertoire | ||
| INF-γ ELISPOT | Lymphocytes | PMBCs with T-cell proliferation blocked | Measurement of secreted INF-γ by cells incubation during 18–24 h on a plate coated with INF-γ capture antibodies | Frequency of memory alloreactive T cells | ||
| Modified ELISPOT | PBMCs | - | Incubation of recipient’s T cells with a source of antigens. | Frequency of indirectly activated recipient T cell | ||
| B cell and memory B cell | ||||||
| Method | Sample | Principle | Analyzer | Detection | ||
| HLA-specific tetramer staining | Peripheral blood B cells | Binding of stained HLA to B cell receptors | Flow cytometry | B cell | ||
| Luminex | Peripheral blood B cells | Detection of anti-HLA antibodies derived from memory B cells | Luminex fluoroanalyser | Memory B cell | ||
| HLA-ELISPOT | 7 days pre-cultured CD2 depleted recipient cells | Binding of anti-HLA antibodies to immobilised anti-IgG antibodies and its detection by binding of labelled HLA to anti-HLA antibodies. | ELISPOT reader | Memory B cell | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Etxebarria, A.; Díez-Martín, E.; Astigarraga, E.; Barreda-Gómez, G. Role of the Immune System in Renal Transplantation, Types of Response, Technical Approaches and Current Challenges. Immuno 2022, 2, 548-570. https://doi.org/10.3390/immuno2040035
Etxebarria A, Díez-Martín E, Astigarraga E, Barreda-Gómez G. Role of the Immune System in Renal Transplantation, Types of Response, Technical Approaches and Current Challenges. Immuno. 2022; 2(4):548-570. https://doi.org/10.3390/immuno2040035
Chicago/Turabian StyleEtxebarria, Aitor, Eguzkiñe Díez-Martín, Egoitz Astigarraga, and Gabriel Barreda-Gómez. 2022. "Role of the Immune System in Renal Transplantation, Types of Response, Technical Approaches and Current Challenges" Immuno 2, no. 4: 548-570. https://doi.org/10.3390/immuno2040035
APA StyleEtxebarria, A., Díez-Martín, E., Astigarraga, E., & Barreda-Gómez, G. (2022). Role of the Immune System in Renal Transplantation, Types of Response, Technical Approaches and Current Challenges. Immuno, 2(4), 548-570. https://doi.org/10.3390/immuno2040035