Abstract
Advances over the last decades have made renal transplantation an important therapy for patients with end-stage renal disease, as the incidences of acute rejection and short-term transplant loss have been significantly reduced. However, long-term transplant survival remains a challenge in the renal transplantation community. The main causes of long-term graft loss are acute and chronic rejection, as well as the complications related to immunosuppression therapy. In spite of the breakthroughs achieved in recent years, histology is the gold standard technique to confirm the activation of the immune system against the graft with all the ensuing problems that taking biopsies brings to immunosuppressed patients. For this reason, several assays have been developed to try to monitor the immune function, but they show serious constraints owing to the fact that they require substantial laboratory work, they are not clinically available and they provide controversial results, so the combination of multiple assays is often needed to obtain a reliable diagnosis. Thus, the aim of this review is to perform a retrospective study of the immune system in renal transplantation, with special emphasis on the cutting-edge technological developments for monitoring, classification and early detection of rejection episodes in order to contribute to a better adjustment of immunosuppressive therapies and, hence, to a more personalized medicine that improves the quality of life of patients.
Keywords:
graft rejection; immune response; cellular response; kidney transplantation; HLA; Immunoassay; ELISPOT; Luminex; ELISA; DSA 1. Introduction
Kidney transplantation is the better choice for patients with end stage kidney disease (ESKD), since it improves the quality of life of the recipients and reduces the associated cost of dialysis [1]. Over the last few decades, important advances made in the understanding of transplantation immunity, surgical techniques and patient immunosuppression have improved transplant survival, especially in the short term. The incidence of acute rejection of kidney transplantation is now below 15%, and graft survival during the first year exceeds 90% [1]. This has led to transplantation being considered an acceptable medical treatment. However, these results have not been transferred to the long term. Indeed, more than 50% of transplants are lost after 10 years, and the annual transplant loss rate after the first year has remained constant for the last 25 years, being 3–5% for transplants from optimal deceased donors and 2–3% for living donors [2].
The major limitation facing transplantation medicine is rejection by the recipient’s immune system, which differentiates between self and non-self, resulting in immune damage to the graft and loss of the graft. This rejection can be hyperacute, taking place within minutes after transplantation; acute, within days to months after transplantation or chronic, occurring years after transplantation.
Hyperacute rejection is a severe immune system reaction resulting in graft loss that is caused by preformed donor-specific antibodies (DSA), due to prior sensitisation of the patient from blood transfusions, pregnancy or a previous transplant. This relationship was established in the 1960s [3], and the detection of these preformed antibodies in patient sera prior to transplantation by the complement-dependent cytotoxicity crossmatch assay (CDC) was a milestone in organ allocation [4]. Hyperacute rejection is now an extremely rare event due to the universal adoption of pretransplant crossmatching techniques.
Improvements in immunosuppression therapies have led to major advances in transplant survival. The introduction of calcineurin inhibitors, cyclosporine in the late 1980s and tacrolimus in the 1990s, together with other immunosuppressors such as mycophenolate, the mammalian target of rapamycin (mTor) inhibitors and interleukin-2 receptor (IL-2R) antibodies have improved acute rejection rates from 40–50% and transplant survival during the first year from 80–85% to the current rejection and transplant survival rates [5].
Currently, long-term transplant loss is due to multifactorial causes, including immunological and nonimmunological causes. On the one hand, immunological causes include mainly acute rejection and chronic rejection. The current immunosuppressive therapies have significantly reduced acute rejection, making chronic rejection the main cause of poor graft outcomes [6]. Chronic rejection is characterised by a persistent allogeneic immune response to which risk factors such as early ischaemia reperfusion injury, acute rejection episodes or infectious diseases of the transplant may contribute [7].
On the other hand, nonimmunological causes include nephrotoxicity due to long-term exposure to calcineurin inhibitors, as well as viral infections or cancer due to overimmunosuppression [5]. While a reduction in immunosuppressors would ameliorate nonimmunological causes of transplant loss, it would increase the likelihood of immune rejection of the graft, so it is necessary to achieve an ideal therapeutic immunosuppression sufficient to prevent rejection but below the levels that lead to toxicity, opportunistic infections or cancer [8]. The adoption of individualised immunosuppressive treatment based on the risk level of each recipient is recognised as a promising strategy to improve long-term rejection. This requires the early stratification of high and low immunological risk recipients in order to establish appropriate immunosuppression guidelines for each case. Ongoing monitoring of the transplant is necessary as a part of post-transplantation management for the correct detection of graft failure, leading to the correct treatment to improve transplant survival [9].
Some combinations of donor and recipient characteristics are associated with poorer transplant outcomes. These characteristics include donor age, cause of death, weight or race, and classification systems based on the risk of graft failure have been developed using various combinations of donor and recipient characteristics, such as the kidney donor risk index (KDRI), to assist clinicians in better transplant allocation [10].
Post-transplant monitoring of the graft function is performed with measurements of the serum creatinine levels and glomerular filtration rate (GFR). However, these markers are not specific for rejection and represent a late event in the rejection process, so that, by the time they are detected, the tissue damage may be irreversible. Biopsy remains the gold standard for the diagnosis of transplant failure, as well as a prognosis indicator [11]. However, a biopsy is an invasive and costly technique that can lead to surgical complications.
Screening for anti-HLA antibodies has become the standard clinical practice. There is overwhelming evidence that the presence of antibodies to both donor HLA and non-HLA antigens both pre-transplant and de novo generated post-transplant has a negative impact on transplant survival. Furthermore, longitudinal studies revealed that the appearance of these antibodies in serum occurs long before the rise in blood creatinine in rejection episodes, making antibodies a good predictor of graft rejection [12].
An integrated diagnostic system is needed, with different techniques and approaches supporting the current gold standard methodology [11]. In this line, important advances have been made in the search for new non-invasive markers, as well as the development of new technologies that allow a correct diagnosis and reduce the need for biopsies. However, most of these techniques are still awaiting clinical validation.
2. Immune Response against the Graft
2.1. HLA Antigens
The main antigens recognised by the recipient’s immune system are the components of the highly polymorphic HLA system of the donor [13]. There are two classes of HLA molecules, HLA-I and HLA-II, which are involved in antigen presentation. Moreover, there are three types of HLA class I molecules (HLA-A, -B and -C) and three types of HLA class II molecules (HLA-DP, -DQ and -DR). To date, more than 6000 class I proteins and more than 2000 class II proteins have been found [14]. HLA-I molecules are expressed on all nucleated cells in the body and present endogenous antigens to cytotoxic CD8+ T cells. HLA-II molecules are primarily expressed on antigen-presenting cells (APCs), which are primarily dendritic cells, macrophages and B cells that are involved in the presentation of exogenous antigens to CD 4+ lymphocytes. However, other cell types such as endothelial cells express HLA class II in response to stress or inflammation in response to TNF-α. The host immune system recognises these alloantigens and generates an immune response by generating specific antibodies against the donor HLA system [15].
For organ allocation, a correct HLA donor–recipient match is desirable, as the extent of mismatches at these loci is associated with poorer graft performance. The consequence of an HLA mismatch is the potential generation of antibodies against the mismatched HLA antigen. There is a strong correlation between HLA mismatches at the HLA-A, -B and -DR loci and poor graft outcomes. Compatibility at these loci is still considered the gold standard by the majority of allocation agencies [16].
Although, in the new era of potent immunosuppressants, HLA mismatches seem to have become less important in short-term transplant failure, some studies link the increased number of mismatches to episodes of early subclinical inflammation that would result in poorer long-term transplant outcomes [17].
2.2. Non-HLA Antigens
There is a growing acknowledgment of the role of other non-HLA antigens in the acute and chronic rejection process. The strongest evidence for the involvement of non-HLA antibodies in graft rejection was provided by reports of antibody-mediated rejection (AMR) in HLA-identical grafts [18,19]. These non-HLA antigens can be polymorphic, such as MICA, or non-polymorphic. The latter are graft-specific antigens that are exposed to the recipient’s immune system in situations of tissue damage and inflammation, such as those caused by ischaemia reperfusion and transplant rejection, and can lead to the formation of autoantibodies [20].
In recent years, an increasing number of non-HLA antigens have been reported to be associated with graft rejection and transplant loss, including angiotensin II type 1 receptor (AT1R), collagen V, perlecan, K-α-tubulin, vimentin and endothelial cell antigens [21].
2.3. Innate Immunity
Over the past decades, evidence has accumulated, indicating that innate immunity contributes significantly to amplifying and modulating the antigen-specific adaptive immune response against the graft. This makes innate immunity a good therapeutic target to limit the effects of adaptive immunity in transplant rejection [22].
The innate immune system is comprised of cellular components, such as macrophages, neutrophils, dendritic cells and natural killer cells, and blood proteins such as the complement and blood coagulation systems. These three components of innate immunity are closely interrelated and can activate each other. In the early post-transplant period, activation and proinflammatory crosstalk between elements of the innate immune system occur in response to the release of danger-associated molecular patterns (DAMPs) and tissue factor expression in graft cells because of tissue damage and hypoxia following brain death and ischaemia reperfusion. As a result, there will be a potent secretion of proinflammatory cytokines such as IL-6 and IL-21, as well as the activation and recruitment of innate immune cells into the graft [23].
The presence of proinflammatory cytokines in the microenvironment in which T cells recognise the antigen will direct their phenotypic differentiation into tissue-destroying effector T-helper cells (Th1, Th2 and Th17) and limit the differentiation of naïve T cells into tissue-protective regulatory T cells (Tregs) [24]. The latter are immunosuppressive cells that play a central role in immune tolerance and homeostasis. The function of Tregs is involved in the prevention of autoimmune diseases and in the generation of graft tolerance by suppressing the function and proliferation of other CD4+ T cells, CD8+ T cells, B cells, NK cells, macrophages or dendritic cells. Thus, Tregs modulate both the adaptive and innate immunity and are key regulators of inflammation. In organ transplantation, the explosion of proinflammatory cytokines generated by innate immunity leads to an aggressive adaptive response against the graft due to events such as an imbalance between effector T cells and reg T cells [25].
2.4. T-Cell Allorecognition Pathways
Rejection is caused by an activation of T cells that will develop a coordinated immune response between the adaptive immune response and the innate immune response against the graft. The T cell recognises donor antigens via antigen-presenting cells (APCs) through interactions between the T-cell receptor (TCR) and HLA molecules expressed on APC membranes. In transplantation immunology, both donor and recipient APCs can present antigens to T cells in what is called the direct and indirect allorecognition pathways, respectively [26].
Brain death and ischaemia reperfusion processes produce a secretion of cytokines and DAMPs [27] that will lead to the recruitment and maturation of donor macrophages and dendritic cells in the kidneys, which can then leave to target the recipient’s secondary lymphoid organs and activate the recipient’s T cells via the direct pathway. This direct activation (Figure 1) is a process unique to alloimmunity and occurs by the recognition of intact HLA molecules from donor APCs by recipient’s CD4+ and CD8+ lymphocytes. This pathway is considered to be short-lived due to the short lifespan of the donor APCs, produces a more potent alloresponse than conventional T-cell activation and is considered to be the predominant pathway in the acute cellular rejection process [28].
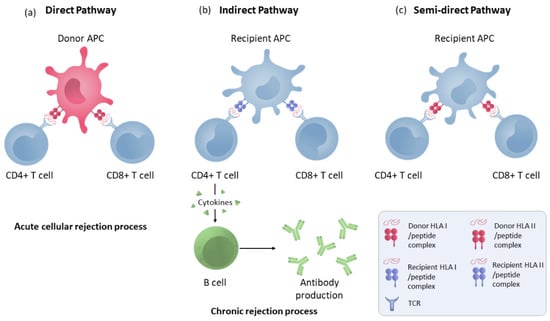
Figure 1.
Direct, indirect and semi-direct allorecognition pathways. (a) T-lymphocyte direct pathway activation by HLA molecule presentation by donor APCs to the recipient’s T cells. (b) Indirect pathway recognition by T CD4 and CD8 cells in which the alloantigens presented as peptides processed by recipient APCs through self-HLA molecules. CD4+ lymphocytes activate B cells to make antibodies. (c) T-cell semi-direct pathway activation: T cells recognise intact HLA molecules from the donor, but their own APC presents them.
In the indirect pathway (Figure 1), T CD4 cells recognise the alloantigens presented as peptides processed by recipient APCs trough self-HLA II molecules. The indirect pathway is considered to be long-lasting and predominant in chronic rejection [29]. Indirectly activated CD4 T cells can activate cytotoxic CD8 lymphocytes and are the only cells that can activate alloreactive B cells [30]. Moreover, a semi-direct pathway (Figure 1) has also been described in which T cells recognise intact HLA molecules from the donor, similar to direct recognition, but on their own APC membranes that have acquired these HLA molecules from the donor via various pathways such as cell–cell contacts or exosomes, suggesting that both direct and indirect recognition play a role in chronic rejection [31].
2.5. Endothelial Cells
The endothelial cells (EC) of the graft will play a fundamental role in the rejection process, as they will be directly exposed to the bloodstream and therefore represent the first barrier between the recipient’s immune system and the graft. EC-expressed antigens are the main targets of the antibodies generated by the recipient’s immune system against the graft. In addition, EC are semi-professional presenting cells that can activate both CD4 and CD8 T-lymphocytes [32].
Under conditions of stress or inflammation, such as those occurring in a transplanted organ, endothelial cells increase the expression of both class I and class II HLA molecules and may be recognised by circulating T lymphocytes. In addition, under these inflammatory conditions, ECs express other minor histocompatibility complex molecules, such as HLA-E and HLA-G, as well as Major Histocompatibility Complex Class I-Related Chain A Antigens (MICA) [33]. MICA is a highly polymorphic protein that has been associated with rejection and late renal failure [34]. Studies have reported that both pre-existing and de novo generated anti-MICA antibodies are associated with acute and chronic rejection [35]. However, MICA is not expressed on resting T or B cells, and therefore, the CDC test is not valid for detecting anti-MICA antibodies. The MICA polymorphism is in addition to the HLA polymorphism, so it is important to perform donor–recipient matching on the MICA genes, which is currently not done. In addition, MIC molecules function as ligands for NKG2D and can stimulate γδ T and CD8+T cells.
Furthermore, in transplantation, the immune system may react against nonpolymorphic graft antigens, predominantly from endothelial cells, generated as self-antigens in a process involving a loss of B-cell tolerance associated with chronic rejection. In rejection episodes, there is a release of self-antigens in the noncanonical tertiary lymphoid tissues formed in the graft. The cytokine microenvironment of these tissues, enriched in IL-17 and the B-lymphocyte stimulator (BLyS), together with a low concentration of Treg lymphocytes, causes autoreactive B cells to miss deletion signals and differentiate into autoantibody-producing plasma cells [36].
2.6. T-Cells Activation
Three signals between the T cell and the APC are required for successful T-cell activation. The interaction between the TCR of the lymphocyte and the HLA II molecule with the peptide antigen of the APC results in the first activation signal. It is activated through the calcineurin pathway, which promotes the expression of several cytokines and receptors that are important for lymphocyte proliferation. Lymphocyte activation requires a non-antigen-specific costimulatory signal that, if absent, leads the lymphocyte into a state of anergy and apoptosis. This costimulatory signal requires the interaction of CD28 expressed on the lymphocyte membrane with its ligands, B7.1 (CD80) and B7.2 (CD86), expressed on APCs [37]. Once the T cell has received the activation signal and the costimulatory signal, it will receive further instructions in the form of cytokines, which will induce cell proliferation through IL-2, and define the T-cell subtype into which it will differentiate.
Once activated, a fraction of T cells will differentiate into long-lived memory T cells, which upon re-encounter with the antigen produce an immune response faster and with less need for co-stimulation than naive T cells, and are less susceptible to immunosuppressive treatments [38]. In addition, it is known that memory T cells initially primed by various infectious agents or environmental antigens can cross-react with foreign HLA molecules without having had prior contact with them through a mechanism known as heterologous immunity [39].
The process of T-lymphocyte activation is a common target of the currently used immunosuppressive agents. Immunosuppressive agents such as anti-IL-2 receptor antibodies and the mammalian target of rapamycin (mTor) act by inhibiting the effects of IL-2 [40]. Both cyclosporine and tacrolimus inhibit the calcineurin pathway by binding to their cytoplasmic receptors. Although calcineurin inhibitors (CNIs) have produced an important advance in terms of transplant survival, they produce important side effects due to their nephrotoxicity, and in the long term, they can produce interstitial fibrosis [37].
At the time of transplantation, patients are usually treated with induction therapies directed against T-lymphocytes, such as a T-lymphocyte-depleting agent (thymoglobulin) or IL-2 inhibitors (basilixumab). This has led to a significant reduction in cellular rejection, which is more likely to occur in the first weeks to months post-transplant. It can also occur later, especially in association with reduced immunosuppression [6]. Chronic cellular rejection can present as tubulointerstitial or vascular and is characterised by an infiltration of T-lymphocytes, monocytes and plasma cells into the interstitium, causing tubulitis, or into the subendothelium and intima of the graft arteries, respectively. It is common for cellular and antibody-mediated rejection to coexist in a graft injury. In fact, episodes of early cellular rejection may result in the production of de novo anti-DSA antibodies, preceding AMR [41]. However, modern immunosuppressive treatments do not sufficiently address the humoral immune response of the recipient [42]. AMR is responsible for 30–50% of acute rejection cases and 60% of late graft failure [43].
2.7. B Cells and Humoral Response
B cells play a very important role in the immune response against transplantation, as they are the cells that will produce antibodies against donor-specific antigens that will result in acute or chronic AMR. Once they recognise the antigen via the BCR receptor, B cells internalise the antigen by receptor-mediated endocytosis and migrate to the lymphoid organ, where they present the antigen as a processed peptide via their own HLA II to a cognate Tfh lymphocyte with the involvement of the CD40–CD40L interaction [44]. A fraction of B cells will give rise to short-lived IgM-producing plasma cells, while another fraction of B cells develops in germinal centres, undergoing immunoglobulin class switching, somatic hypermutation and affinity maturation. Once activated, B cells leave the germinal centres as memory B cells or plasma cells. While activated B cells with the highest affinity for antigen differentiate into antibody-producing plasma cells, B cells with less affinity are selected to differentiate into memory B cells. This less stringent selection of memory B cells results in a more diverse repertoire of memory B cells than of antibodies in serum [45]. Plasma cells migrate and settle in the bone marrow or mucosal tissues. These cells are differentiated, nondividing cells that are the main source of antibodies in the serum. Memory B cells remain in quiescent mode and circulate between secondary lymphoid organs and peripheral blood until they encounter the antigen again. After re-encountering the antigen, the memory B cell generates a rapid, enhanced response and differentiates into an antibody-producing cell. Importantly, memory B cells can also be activated in a non-antigen-specific manner through the cytokine environment via bystander activation [46]. In addition, activated B cells are efficient APCs that can interact with naïve T cells and activate them through the HLA-II they express. This makes B cells also mediate and regulate cellular rejection.
Anti-donor antibodies produced by mature B cells are responsible for AMR and can cause a graft injury through several mechanisms. Complement-binding antibodies, the IgG1 and IgG3 subclasses, are capable of activating the complement system, leading to the C5b–C9 membrane attack complex, causing lysis of the target cell. This pathway is detected by the presence in biopsies of C4d, a by-product of the classical complement pathway. In addition, the crystalline fragment (Fc) of both complement-binding and non-complement-binding antibodies acts as a stimulus for FcγR-expressing cells of innate immunity, such as macrophages, dendritic cells and especially natural killer cells, leading to antibody-dependent cell-mediated cytotoxicity [47]. Lastly, the binding of antibodies to HLA molecules on endothelial cells results in a series of endothelial and smooth muscle cell modifications, such as altered cytoskeleton, cell proliferation and the expression of leucocyte adhesion molecules, which generate the histological features of chronic rejection [48].
Acute AMR occurs from the first weeks to years after transplantation. It is characterised by cell death, loss of vascular integrity and tissue damage. Chronic AMR is characterised by a long and progressive process of loss of graft function whose histological features include transplant glomerulopathy, the deposition of fibrillar material in the subendothelium and C4d deposition. For a proper diagnosis of both acute and chronic AMR, according to the criteria of Banff classification, it is necessary to observe by biopsy the pathophysiology associated with each rejection and to detect donor-specific antibodies in the serum. Since the 2013 Banff classification meeting, the presence of C4d in biopsies is not a prerequisite for the diagnosis of AMR but rather an indication of a recent interaction between the antibody and the microvasculature of the endothelium [49]. The treatment of AMR is less effective, and unfortunately, there are no standardised treatments against antibody production.
3. Biological Drugs Therapies Innovations
In recent years, some therapies initially designed for the treatment of cancer or autoimmune diseases have been adapted for the treatment of chronic transplant rejection due to the similarities shared by these fields [7]. Indeed, in transplant rejection, autoimmune disease and cancer, protein–protein interactions trigger signalling events that ultimately lead to disease development. These interactions are usually cell–cell contacts or cytokine–receptor interactions, which result in signal transduction in immune system cells that can lead to their chronic activation and cause pathology.
Immunotherapies using biological drugs (BD), monoclonal antibodies (mAb) obtained from living organisms [50], can block these protein–protein interactions that generate the pathophysiology of the disease. Therefore, the current immunotherapies using BDs have revolutionised the treatment and improved clinical outcomes for patients with these pathologies [51]. Indeed, BDs have demonstrated better efficacy and safety profiles than synthetic drugs [52]. Some of the BDs used for the treatment of transplant rejection are mAbs designed to block the signalling of specific cytokines.
Infliximab is a chimeric monoclonal antibody that specifically binds to TNF-α and blocks its interaction with both its soluble and membrane receptors. TNF-α is a cytokine with a central role in the inflammatory response and is a major pathological component in autoimmune diseases. Hence, the use of TNF-α inhibitor mAbs (infliximab, golimumab, adalimumab, etanercept and certolizumab pegol) is a common strategy in the treatment of these pathologies [53].
TNF-α induces inflammation and activates immune cells, promoting the activation of the immune response against the transplant and aggravating rejection. Consequently, blocking TNF-α or other cytokines such as IL-1β is also helpful in reducing the immune response in allotransplantation [54]. Infliximab treatment has been shown to reduce cytokine expression, macrophage recruitment and improve renal function following ischaemia reperfusion injury [55].
Tocilizumab is an anti–interleukin 6 receptor mAb. Interleukin 6 (IL-6) is a pleiotropic cytokine involved in the generation of donor-specific antibodies and graft rejection. IL-6 affects the ratio of effector-to-regulatory T cells by promoting the maturation of Th17 cells while favouring the maturation of B cells to plasma cells [56]. Tocilizumab has been used for the treatment of rheumatoid arthritis [52] and, in recent years, has been proposed to treat chronic antibody-mediated rejection, showing an improvement in the graft outcomes in sensitised patients who do not respond to the standard desensitisation protocol [57].
Belimumab is a humanized anti-B-lymphocyte stimulator (BLyS) antibody with successful therapeutic results in the treatment of Lupus erythematosus. BLyS is a cytokine that enhances B-cell and plasma cell survival and is overexpressed in lupus erythematosus and other autoimmune diseases, causing B-cell hyperreactivity [58]. Belimumab has recently been proposed for use in transplantation to treat rejection as a modulator of B-cell alloreactivity. In a phase II clinical trial, patients with a standard level of immunological risk are treated by administering belimumab together with the standard of care immunosuppression for 24 weeks. The results show a reduction of both preformed HLA antibodies and non-HLA autoantibodies, as well as a reduction of activated memory B cells, without an increase in infections compared to the control group treated with the standard of care immunosuppression. This same study showed a rebound in the serum BLyS concentration after the cessation of belimumab administration, raising the question of whether belimumab should be administered longer after transplantation, with a gradual reduction and monitoring of the serum BLyS levels [59].
In terms of cancer treatments that can be translated into therapies that prevent graft rejection, the first type of drugs is those used for some tumours of haematopoietic and lymphoid tissues. This is because the recipient’s immune system is activated to attack the graft during the rejection. Consequently, there is an interest in decreasing the immune system’s activity, specifically depleting T and B cells [28]. Cell-depleting immunotherapies used for the treatment of transplant rejection include alemtuzumab and rituximab.
Alemtuzumab is a humanised anti-CD52 monoclonal antibody originally used for the treatment of lymphocytic leukaemia [60]. This antibody causes cell lysis via the complement system or antibody-dependent cellular cytotoxicity to CD52-expressing cells via its crystallisable fraction Fc. As CD52 is expressed on all peripheral blood lymphocytes, alemtuzumab induces potent immunosuppression by depleting both T cells and B cells. Alemtuzumab has shown good results as an induction agent in solid organ transplantation compared to other induction agents such as IL2R antibodies in preventing rejection in the first months after transplantation, without any significant increase in malignancy or generation of autoimmunity compared to other induction regimens [61].
Rituximab is a chimeric monoclonal antibody with a mechanism of action similar to alemtuzumab that acts by depleting CD20-expressing B cells. In addition, as B cells are antigen-presenting cells, they indirectly inhibit T-cell activity. Therefore, this antibody acts by blocking the production of antibodies. In 80% of patients, it depletes the circulating antibody levels for a period of six to twelve months [62]. Rituximab has been used to treat patients with relapsed low-grade or follicular NHL, as well as for the treatment of autoimmune diseases. More recently, it has been incorporated as a rescue therapy in the treatment of antibody-mediated rejection. Some studies suggest that rituximab has benefits in the treatment of acute antibody-mediated rejection; however, the data show less effectiveness in the treatment of chronic AMR [63]. Recently, acute AMR has been treated using a combination of plasmapheresis, tacrolimus mycophenolate mofetil and/or intravenous immunoglobulin, together with rituximab, allowing 70–80% of the affected grafts to be rescued. However, there is no consensus on the optimal protocol for the treatment of acute AMR [64]. Rituximab is currently part of the standard protocol for the desensitisation of patients at high immunological risk combined with apheresis, conventional triple immunosuppression (CNI, mycophenolic acid and steroids) and intravenous polyvalent immunoglobulin (IVIg).
In general, BDs are potent immunosuppressants that interfere with the normal immune response, which can lead to adverse side effects. The safety profile of BDs can be dose-dependent and may be associated with certain combinations with other drugs. Therefore, the risk–benefit of each BD treatment and its dosage should be assessed on a case-by-case basis [65].
4. Antibody Detection Methods
The detection of anti-HLA antibodies both pre-transplant and post-transplant is a common clinical practice in transplantation. It is included in the Banff classification criteria for the diagnosis of AMR and is essential for a correct transplant allocation and for correct transplant management [49].
DSA detection tests (Table 1) have evolved from early cell-based methods (CDC) to modern solid-phase methods, such as the single antigen bead (SAB), which allows the detection of individual HLA specificities. The increased sensitivity of anti-DSA antibody detection methods has allowed the generation of increasingly accurate immune risk level assignment tools, such as calculated PRA and virtual crossmatching [66], which has led to a marked improvement in organ allocation [67].

Table 1.
Methods to detect anti-HLA antibodies.
4.1. Complement Dependent Cytotoxicity (CDC) Assay
CDC (Figure 2) is a cell-based antibody detection method in which antibody detection is performed by incubating the patient’s serum with donor lymphocytes expressing HLA I and HLA II molecules [68]. Upon the addition of a complement, the presence of reactive antibodies results in cell lysis. Then, the lysed cells are stained with a fluorescent marker, usually a cocktail of ethidium bromide and acrylamide orange, that discriminates positive from negative results. This method detects complement-binding antibodies present in high titres in the recipient’s serum that trigger hyperacute rejection and represent a contraindication for transplantation [69]. To overcome the limitation of donor cells, the serum is incubated against a randomly selected panel of HLA-typed lymphocytes that are representative of the donor population. The result is expressed as a percentage of reactivity against the panel of lymphocytes against which the serum has reacted, known as the panel reactive antigen (PRA). This percentage reflects the patient’s probability of detecting antibodies against the donor [70].
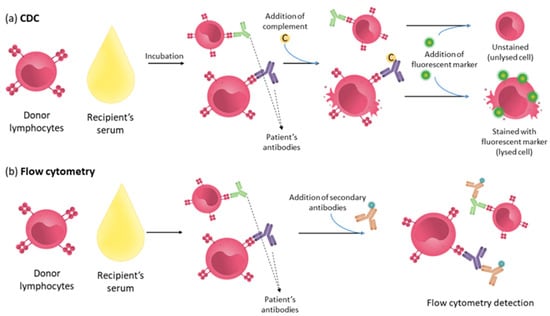
Figure 2.
Assays detecting anti-HLA antibodies. (a) CDC: Recipient’s serum is incubated with donor’s lymphocytes; after this, the incubation complement is added, triggering cell lysis on cells bound to complement-binding antibodies. Detection is made using a fluorescent marker that stains lysed cells. (b) Flow cytometry: Recipient’s serum is incubated with donor’s lymphocytes. In this assay, after this incubation, secondary antibodies fluorescently labelled are added to samples to detect cells with the patient’s antibodies bound.
This method is simple to perform and reflects the in vivo situation, where the HLA molecules are in a native conformation, making it a very useful clinical information tool. Numerous studies have associated the antibodies detected by this technique with hyperacute rejection. Currently, a positive CDC result is still considered a contraindication for transplantation. However, it has a number of limitations. The CDC assay detects any antibody that binds to cells and fixes the complement; therefore, it does not detect non-complement-binding antibodies and does not discriminate between HLA and non-HLA antibodies. The assignment of antibody specificity is performed by comparing the reactivity of the test with DNA typing of the target cells. However, this assignment is not very accurate using this technique, especially for HLA-II molecules, because B cells express both HLA-I and HLA II, and these cells are found in low frequencies in the peripheral blood. Additionally, as HLA molecules are co-dominantly expressed and some alleles of close loci show linkage disequilibrium, the assignment of antibody specificity is done on the basis of probability calculations [71]. Some cytolytic therapies interfere with the assay, and finally, it is a low-sensitivity assay [72]. To improve the sensitivity of the CDC technique, flow cytometry was introduced for cell analysis (Figure 2). In this case, the patient serum is incubated with lymphocytes, and the detection of antibodies is performed by incubation with a fluorescently labelled secondary antibody. In addition, this technique can distinguish between complement-fixing and non-complement-fixing antibodies using the secondary antibodies specific to each IgG subtype.
The higher sensitivity of this technique allows the detection of antibodies that are not detected by CDC and whose effect on the graft outcome is not entirely clear. However, transplants with a negative CDC crossmatch and a positive flow cytometry crossmatch have been shown to have an increased risk of acute rejection and early graft loss [67]. Nevertheless, the main limitation of this technique is the need for viable cells. To overcome this limitation, solid-phase assays were introduced, which use purified HLA molecules and have a higher level of sensitivity than cell-based assays.
There are three types of solid-phase assays: enzyme-linked immunosorbent assay (ELISA), FlowPra and Luminex, each with a higher level of sensitivity than the previous one [73].
The identification of anti-HLA antibodies by ELISA (Figure 3) is performed by immobilising HLA antigens, recombinant or purified from human platelets, on the surface of a microtiter plate with a subsequent incubation with the recipient’s serum. The antibodies are detected by binding to an enzyme-linked secondary antibody, and then, a chromogenic substrate is added. The ELISA results are expressed as the optical density.
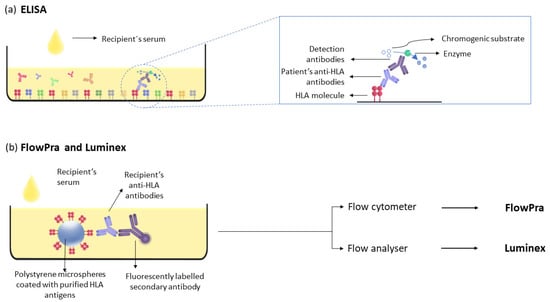
Figure 3.
Solid-phase assays for detecting anti-HLA antibodies. (a) ELISA assay: HLA antigens are immobilised on the microtiter plate, and anti-HLA antibodies are detected after incubation with the recipient’s serum. (b) FlowPra and Luminex assays: anti-HLA antibodies from the recipient’s serum are detected by incubating the serum with polystyrene microspheres coated with HLA antigens and secondary antibodies.
The next step in the evolution of antibody detection methods is based on the detection of antibodies on polystyrene microspheres coated with purified HLA antigens. Their detection can be performed either with a flow cytometer (FlowPRA) [74] or a flow analyser (Luminex) (Figure 3). The latter is the gold standard method for the detection of anti-donor antibodies due to its sensitivity and specificity.
4.2. Luminex
The Luminex technique is based on the detection of anti-DSA antibodies on polystyrene beads coated with purified HLA molecules. The beads have different ratios of two different fluorophores, allowing each bead to be identified individually. The patient serum is incubated with the beads, and antibody detection is performed using a fluorescently labelled secondary antibody [75]. The Luminex fluoroanalyser has two lasers: one laser excites the fluorophore of the beads, and the other detects the fluorophore of the secondary antibody, allowing the identification of antibody specificity. The results are reported as the mean fluorescence intensity (MFI). Solid-phase assays allow three levels of resolution, depending on the composition of HLA molecules with which the beads are coated. In the first level of detection, known as the panel pool, the beads are coated with several HLA I or HLA II molecules for screening [76]. In the next level of detection, called the phenotypic panel, the beads are coated with HLA I or HLA II molecules obtained from an individual patient cell line. At the highest resolution level, called a single antigen bead, the different populations of beads are coated with a single recombinant HLA allele [77]. This assay allows the precise identification of the specificity of the detected antibodies. The Luminex assay has a high sensitivity and specificity, allowing the detection of both allele specificity and epitope specificity of each antibody [72]. In addition, improvements to this technique have been developed that allow the detection of complement-binding antibodies through the detection of C1q to potentially identify more injurious antibodies [78].
These new technologies have made possible the detection of antibodies that were not detectable with the CDC technique. However, the clinical relevance of these low-affinity antibodies is not entirely clear, and the presence of these antibodies is not always a contraindication. The increased sensitivity of solid-phase assays has led to an increase in the number of highly sensitised patients. This has the potential drawback of refusing transplants for patients based on the presence of clinically irrelevant antibodies, leading to an increase in the waiting list time. This makes it vitally important to discriminate among the antibodies detected; those antibodies that are effectively unacceptable. The main value used for this purpose is MFI obtained for each antibody, establishing a cut-off value. However, the MFI value gives a semi-quantitative measure that does not necessarily determine the pathogenicity of the antibody [79]. The Luminex assay is subject to a number of limitations that may result in false positives, due to the presence of neoepitopes in recombinant HLA molecules, or false negatives due to serum-interfering substances or antibodies with shared epitopes that spread among several antigens, resulting in lower MFI values [80]. Therefore, MFI should not be taken as an absolute determinant of whether a patient receives or refuses a transplant but rather as a guide, together with HLA typing, the patient’s history of alloimmunisation and the specificity of the antibody epitope, which helps to discriminate between acceptable and unacceptable antigens.
To improve the transplant allocation in these highly immunised patients, the traditional PRA was replaced by the calculated PRA (cPRA) [81], which is calculated based on the phenotypic frequency of unacceptable HLA alleles in the donor population. Organ allocation using the virtual crossmatch, which predicts a negative crossmatch based on unacceptable antigens, has improved organ allocation to highly immunised patients [82].
For the detection of non-HLA antibodies, EC are used as the target for anti-endothelial cell antibodies (AECA), as non-HLA antigens are normally expressed on EC in the graft. Different approaches have been used utilizing endothelial cells for the detection of AECAs, including CDC, flow cytometry and immunofluorescence [72]. However, the detection of these antibodies is a limitation of these techniques, as non-HLA antigens are not always constitutively expressed on the surfaces of endothelial cells, and their identities are usually unknown.
The use of surrogate endothelial cells as a sensor for non-HLA antigens could be useful for the detection of non-polymorphic antigens but has the disadvantage that they do not constitutively express MICA.
Currently, the most feasible option for the detection of non-HLA antibodies, such as anti-MICA antibodies or autoantibodies, is the use of purified peptides in solid-phase assays and the Luminex platform.
5. Cellular Immunity Detection Methods
Although the detection of donor-specific antibodies has proven to be an effective tool for an important advancement in transplant allocation and management, it does not give complete information on the immune system and its responsiveness to the donor.
The search for new non-invasive methods and biomarkers that provide useful information to clinicians and allow a good diagnosis continues to be an important field in transplantation research. Some of these methods are focused on measuring the cellular immune response, which, together with the detection of antibodies, allows for a broader view of the immune response (Table 2). There are several methods to measure the cellular immune response; some of them, such as the Immuknow assay, quantify the nonspecific immune response, and others, such as the mixed lymphocyte reaction (MLR) or the ELISPOT assay, quantify the specific immune response.
Table 2.
Methods to measure the cellular immune response.
5.1. Immuknow
The Immuknow assay (Figure 4a) is a test approved by the Food and Drug Administration (FDA) for monitoring immunosuppression on an individualised basis. Immuknow provides a measure of the overall cellular immune response. The test measures the level of ATP synthesis of the patient’s CD4+ T cells after incubation of the patient’s blood with nonspecific mitogenic stimuli such as phytohetoglutin-L for 15–18 h [83]. The test uses whole blood to maintain the presence of immunosuppressants during incubation. After incubation, the T cells are isolated from the blood using a magnetically coated anti-CD4 monoclonal antibody, allowing magnetic separation. A lytic agent is added for cell lysis and ATP release, which is then labelled with a luciferin luciferase mixture and measured in a luminometer at a maximum emission of 562 nm.
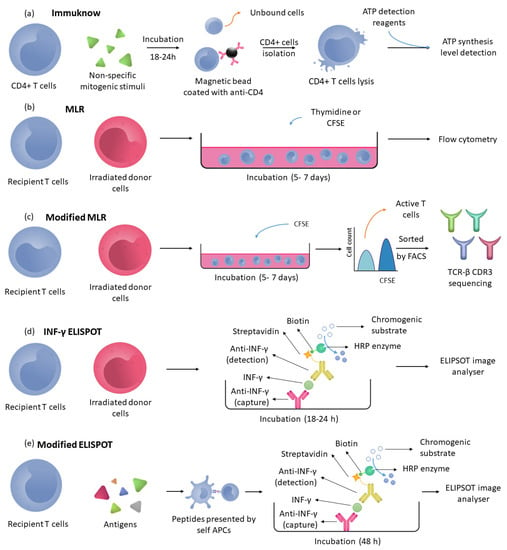
Figure 4.
Cellular immunity detection method. (a) Immuknow: Recipient’s T cells are incubated with nonspecific mitogens; then, CD4+ T cells are isolated using a magnetic bead coated with anti-CD4 antibodies, and finally, these CD4+ cells are lysed to measure the ATP levels. (b) MLR: Recipient’s T cells are incubated with irradiated donor cells and thymidine or CFSE to quantify the cell proliferation by measuring the thymidine or CFSE concentration in cells using flowcytometry. (c) Modified MLR follows the same process as the MLR assay, but the cells are incubated only with CFSE to quantify CFSE and isolate CFSE-low cells (active T cells) by fluorescence-activated cell sorting (FACS), and finally, the TCR-β CDR3 receptors are sequenced. (d) INF-γ ELISPOT: Recipient’s T cells are incubated with irradiated T cells in microtiter wells. In these wells, anti-INF-γ is immobilised, and after incubation with detection antibodies, INF-γ cells can be detected to quantify memory T cells. (e) Modified ELISPOT used the recipient’s T cells and processed peptides (antigens) to quantify the frequency of indirectly activated recipient T cells measuring INF-γ.
The ATP levels obtained in this assay indicate nonspecific T-cell-mediated immune responsiveness [84]. Early studies linked high levels of ATP production with the risk of rejection and low levels of ATP with the risk of opportunistic infections due to overimmunosuppression. However, later studies reported that the technique has little value as an indicator of rejection. Low ATP levels detected by Immuknow would be useful for predicting infection risks and adjusting the patient’s immunosuppressive dose if a serial follow-up is performed, as some studies have shown a single measurement of ATP synthesis with Immuknow has a poor predictive value for diagnosing rejection or infection.
5.2. Specific Immunity Detection Methods
Other approaches proposed for immunological risk assessments include quantification of the immune response of donor-specific T cells by MLR, as well as detection of the frequency and specificity of the repertoire of alloreactive memory B and T cells.
5.3. Mixed Lymphocytes Reaction (MLR)
One of the first in vitro methods to quantify alloreactivity was the mixed lymphocytes reaction (MLR) (Figure 4b) [85]. This method quantifies the level of cellular alloreactivity by measuring the cell proliferation of recipient T cells after reacting with donor PBMCs. MLR consists of incubating lymphocytes from the patient with donor lymphocytes for 5–7 days. The donor T-cell proliferation is blocked, and it is usually carried out by previously irradiating the donor PBMCs or by adding a CD3 T-cell-depleting agent. The resulting cell proliferation is due to the direct allorecognition pathway activation of recipient CD4 and CD8 T cells and involves both memory T cells and naive T cells [86]. This approach is interesting as the direct pathway of recognition is strongly associated with acute rejection that occurs in the first weeks post-transplantation.
To quantify cell proliferation, the cells are labelled with radioactive thymidine or carboxyfluorescein succimidyl ester (CFSE), which is diluted with each cell replication so that the diluted CFSE cell population can be detected by flow cytometry.
Some studies have linked high levels of cell proliferation in this assay with a risk of acute rejection and low levels of cell proliferation with a good transplant outcome within the first year of transplantation [87]. However, other studies have shown that this assay has a low predictive value. This technique has limited clinical use in kidney transplantation [88].
An interesting MLR-based approach (Figure 4c) is to sequence the TCR-beta CDR3 of the patient’s reactive T-cell population to generate a catalogue of the size and diversity of the reactive T-cell repertoire before and after transplantation. Using CFSE, rather than radioactive thymidine, the reactive T-cell population can be monitored as CFSE is diluted during each cell replication. This allows alloreactive cells to be separated by FACS and their TCR to be sequenced [89].
5.4. INF-γ ELISPOT
The IFN-γ ELISPOT assay (Figure 4d) is an MLR-based assay used to assess the frequency of memory alloreactive T cells. These cells are formed in the individual through exposure to environmental antigens. Some of these memory cells may cross-react with donor alloantigens without having anti-donor antibodies in the serum. Therefore, detection of the patient’s population of memory immune cells may provide additional information to that obtained with the current methods of detecting DSA antibodies in the patient’s serum [90].
Memory T cells react faster than naïve T cells to alloantigens, allowing their frequency to be measured after 18–24 h of MLR by detecting the IFN-γ they secrete upon activation. The ELISPOT assay involves the detection of INF-γ in a plate with wells coated with INF-γ capture antibodies, where patient PBMCs are incubated with donor PBMCs in MLR. Detection is performed with a biotinylated anti-INF-γ detection antibody and HRP streptavidin [91]. The resulting spots are measured with an ELISPOT image analyser, and the result is expressed as INF-γ spots per 300,000 PBMCs.
This approach has been used as a screening tool to try to predict and diagnose acute rejection. To overcome the limitation of having donor cells, the panel of reactive T cells (PRT) was introduced, consisting of a representative sample of the donor population in a manner analogous to the PRA. However, the results obtained against the TRP have shown a lower predictive value for acute rejection than those obtained against the donor.
Regarding the predictive value of this test, the results obtained are contradictory. The assay is able to predict the immunological risk but is influenced by T-cell suppressor therapies [92].
Although some studies have linked high frequencies of pre-transplant reactive T cells obtained by the ELISPOT assay to an increased risk of acute rejection in the first weeks post-transplant, others have found this relationship only with those patients who have not received induction therapy [93].
Another ELISPOT assay (Figure 4e) modality exists to measure the alloreactivity of indirectly activated recipient T cells, although there is no consensus for an assay to monitor indirect T-cell activation [94]. This approach is interesting, as indirectly activated T cells play a predominant role in chronic rejection and are the only ones that can activate B cells. The ELISPOT assay for the indirect pathway is performed by stimulating recipient T cells with processed peptides presented by self APCs via their HLA II molecules. This implies that the recipient PBMCs have to be incubated with a source of antigens that are processed and presented to the T cells by the recipient APCs. Different approaches have been tested as a source of antigens to measure the alloreactivity of activated T cells, including processed membranes, synthetic peptides and HLA monomers [31]. However, the use of these antigen sources presents certain difficulties and limitations that prevent the establishment of a protocol for monitoring the frequency of indirectly activated T cells. The use of fragmented membranes has the advantage of providing the full antigenic profile of the donor but has the disadvantage of not providing the specificity of the alloreactive T cell. Furthermore, the use of this antigen source does not ensure the absence of intact HLA molecules, leading to direct or semi-direct presentation and interfering with the measurements and therefore requires appropriate controls. This is a problem that can be solved by using synthetic peptides; however, the use of synthetic peptides has been shown to generate neoepitopes that interfere with the specificity of the assay. These problems, coupled with the low frequency of indirectly activated T cells, make this assay currently unfeasible for clinical use [94].
5.5. Memory B Cells Detection Methods
A number of techniques for the detection of memory B cells have emerged in recent years. The current antibody detection methods are based on the detection of antibodies in serum, so having information on the specificity and frequency of the memory B-cell repertoire gives a broader view of the humoral response. Due to the low frequency of B cells in peripheral blood, highly sensitive assays are needed to detect them.
Early approaches to detect memory B cells were based on the ability of B cells to bind to fluorescently labelled HLA tetramers. This allows flow cytometric detection and the separation of HLA tetramer-binding B cells. However, this method requires a subsequent B-cell culture step to confirm the production of antibodies from the tetramer-positive cells. One of the limitations of this method is that, due to the significant loss of cell viability inherent in the cytometer, the detection of the memory alloantibody repertoire is incomplete [95].
An analysis of the specificity and frequency of memory B cells requires their ex vivo stimulation by polyclonal activation to produce antibodies. This is performed via CD40 activation by incubating B cells with a fibroblast line expressing CD40L, along with a cocktail of B-cell cytokines and growth factors. It is important that memory B-cell activation is carried out without inducing an isotype switching in naïve B cells. The presence of memory antibodies against the donor in the culture supernatant can be tested by the Luminex technique [96]. This method allows the identification of the repertoire of alloreactive memory antibodies, providing qualitative information but does not indicate the frequency of HLA-specific memory B cells [97]. The memory B-cell frequency can be estimated by the detection of anti-HLA antibodies in ELISPOT plates modified for the detection of anti-HLA antibodies. The high sensitivity of the ELISPOT assay makes this technique particularly useful for measuring the low frequency of HLA-specific B cells.
Polyclonally activated B cells from a 6 to 7-day preculture are transferred to an ELISPOT plate coated with anti-IgG antibodies to capture all antibodies of this isotype produced. Visualisation is performed by the addition of biotinylated synthetic HLA class I or II molecules and a subsequent incubation with alkaline phosphatase-conjugated streptavidin. Detection is carried out by a chromogenic substrate, each spot representative of a single HLA-specific antibody-producing cell [98].
6. Ongoing and Future Challenges
One of the great challenges in the renal transplantation field is the search for new approaches and the generation of techniques that allow for early diagnosis, individualised immune monitoring and follow-up of the efficacy and dosage of each patient’s therapy [7], replacing renal biopsy as the gold standard technique, which, in addition to its invasiveness, presents certain drawbacks, such as low sensitivity and specificity [99]. Recent advances in high-throughput cellular and molecular methods are enabling major breakthroughs in each of these fields, as well as expanding our understanding of the immunological mechanisms underlying the rejection process (Figure 5). These advances are in the direction of personalised medicine as the best strategy to improve the long-term outcomes of transplantation and enhance patients’ quality of life by providing clinicians with the potential to adjust the current standards of immunosuppression for each patient, reducing the adverse effects [5]. As Tregs are key regulators of immune homeostasis, there are several strategies to reduce immunosuppression based on Treg cell therapy. Tregs therapies involve the culture and isolation of autologous polyclonal Tregs from the patient’s PBMCs extracted prior to transplantation and their subsequent administration to the patient to restore the immune balance and modulate the patient’s immune response against the graft, allowing the reduction or withdrawal of immunosuppression [25]. Another Tregs-based strategy is the development of genetically engineered antigen-specific Tregs by the transduction of chimeric antigen receptors (CARs). By this technique, CAR-Tregs against graft-specific antigens can be achieved, with a more potent in vitro suppressive capacity than polyclonal Tregs when stimulated by that antigen [100].
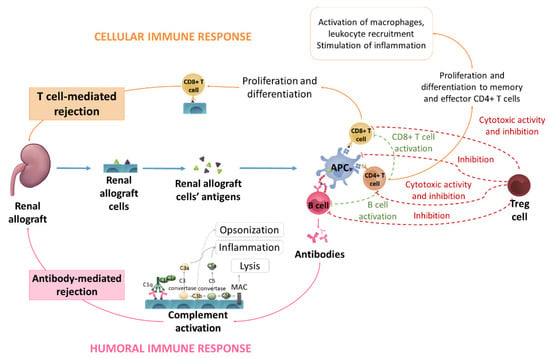
Figure 5.
Schematic representation of different allograft rejection mechanisms. Renal allograft cells’ antigens are processed by APC cells that present the antigens to T and B cells. Activated B cells produce antibodies, which activate the complement system, leading to an antibody-mediated rejection via opsonization, inflammation and lysis processes. Activated CD4+ T cells start their proliferation and differentiation to memory and effector CD4+ T cells. These effector CD4+ T cells activate macrophages and leukocyte recruitment and stimulate inflammation. Moreover, CD4+ cells activate B and CD8+ T cells. CD8+ T cells can also be triggered by APC cells; once CD8+ T cells are activated, they proliferate and differentiate, leading to allograft T-cell-mediated rejection. Treg cells participate in this mechanism by inhibiting B cells and APC cell activity and releasing granzymes and perforins that destroy T cells.
Despite the lack of clinical trials of CAR Treg therapies, phase I and phase I/IIa clinical trials with polyclonal Tregs therapy have shown the potential of these therapies. Patients receiving therapy 7 days after transplantation were monitored for 3 years, with a subsequent reduction of triple immunosuppression at low doses of tacrolimus. The results indicated a high degree of efficacy in reducing immunosuppression, as well as safety and feasibility, supporting that Tregs therapies could become a good strategy in transplantation and autoimmune diseases [101].
A hot line of research is the search for non-invasive or minimally invasive markers that relate to graft rejection and allow therapeutic evaluation for patient care. Scientific effort has focused on identifying such markers primarily in serum and urine involving mRNA markers and protein markers such as CXCL9 and CXCL10, but further research is required [102].
Donor-derived cell-free DNA (dd-cfDNA) has become one of the most widely used rejection markers in recent years [103]. Donor cfDNA are fragments of donor DNA that circulate in the recipient’s bloodstream, indicating tissue graft injury. The measurement and quantification of dd-cfDNA is based on the detection of differences between the recipient and donor in highly homozygous single-nucleotide polymorphisms (SNPs). Using this method, donor and recipient DNA can be discriminated without prior knowledge of their genotypes. The result of this method is expressed as a percentage of dd-cfDNA with respect to the total circulating cell-free DNA in the recipient’s plasma. The DART multicentre study comparing rejection diagnoses made by biopsy with the percentage of dd-cfDNA reported that the percentage of dd-cell-free DNA discriminates between rejection and non-rejection with a cut-off of 1% [104].
Another approach to monitor the immune system in transplant patients is the study of specific protein–protein interactions between cells involved in immune system response and graft rejection, such as HLA-I/CD8+ T cells and HLA-II/CD4+ T cells for immune activation, cytotoxic T lymphocyte antigen-4 (CTLA-4) and CD80/86 for therapy dose regulation and PD-1/PD-L1 for immune inhibition. Programmed death receptor 1 (PD-1) and its complimentary ligand, programmed death ligand 1 (PD-L1), are crucial players in developing the adaptive immune resistance, as this interaction leads to repressing the activity of several intracellular molecules involved in propagating signals downstream of the T-cell receptor (TCR), reducing the cytokine release, cytotoxic activity and halted T-cell proliferation [105,106]. To quantify these interactions between functional states of biomarkers, the two-site amplified time-resolved Förster Resonance Energy has been successfully used using pairs of complementary antibodies against PD-1/PD-L1 interactions not only in living cells but also in cell membranes [51]. In this sense, an iFRET platform (commercially known as QF-Pro®) was used to perform a quantitative readout of PD-1/PD-L1 ligand–receptor interactions using model membrane systems derived from cell lines and tumour samples [107]. The study of specific interactions between membrane proteins of PBMCs and membrane proteins of the membrane microarray gives relevant information on the state of the immune system [51] as a promising approach that can be useful in the field of transplantation for immune system monitoring or for therapy dose surveillance. Membrane microarray technology uses cell membrane homogenates printed onto glass slides, which overcomes the limitation of having viable cells and maintains the functionality of receptors, enzymes and other membrane proteins. Due to its characteristics as a high-throughput, miniaturised and parallelisable analysis tool, cell membrane microarray technology has been used as a powerful tool for drug and antibody screening [108,109] and is a promising approach to implement in the field of transplantation [51] not only for PD-1/PD-L1 interactions but also for other ligand–receptor interactions involved in immune activation and therapy dose regulation.
Funding
This research was funded by the Spanish Ministry of Science and Innovation Center for Technological and Industrial Development (CDTI) (exp. PTQ2019-010650) the Basque Government (BIKAINTEK program, exp. 006-B2/2021) and the University of Basque Country (exp. GEB11 and PIFIND21/02).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Moreso, F.; Hernández, D. Has the survival of the graft improved after renal transplantation in the era of modern immunosuppression? Nefrologia 2013, 33, 14–26. [Google Scholar] [CrossRef]
- Riella, L.V.; Djamali, A.; Pascual, J. Chronic Allograft Injury: Mechanisms and Potential Treatment Targets; Elsevier B.V.: Amsterdam, The Netherlands, 2017; Volume 31, ISBN 6177325254. [Google Scholar]
- Kissmeyer-Nielsen, F.; Olsen, S.; Petersen, V.P.; Fjeldborg, O. Hyperacute rejection of kidney allografts, associated with pre-existing humoral antibodies against donor cells. Lancet 1966, 288, 662–665. [Google Scholar] [CrossRef]
- Patel, R.; Terasaki, P.I. Significance of The Positive Crossmatch Test in Kidney Transplantation. N. Engl. J. Med. 1969, 280, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Bamoulid, J.; Staeck, O.; Halleck, F.; Khadzhynov, D.; Brakemeier, S.; Dürr, M.; Budde, K. The need for minimization strategies: Current problems of immunosuppression. Transpl. Int. 2015, 28, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Hara, S. Current pathological perspectives on chronic rejection in renal allografts. Clin. Exp. Nephrol. 2017, 21, 943–951. [Google Scholar] [CrossRef]
- Lai, X.; Zheng, X.; Mathew, J.M.; Gallon, L.; Leventhal, J.R.; Zhang, Z.J. Tackling Chronic Kidney Transplant Rejection: Challenges and Promises. Front. Immunol. 2021, 12, 661643. [Google Scholar] [CrossRef] [PubMed]
- Guerra, G.; Srinivas, T.R.; Meier-Kriesche, H.U. Calcineurin inhibitor-free immunosuppression in kidney transplantation. Transpl. Int. 2007, 20, 813–827. [Google Scholar] [CrossRef]
- Josephson, M.A. Monitoring and managing graft health in the kidney transplant recipient. Clin. J. Am. Soc. Nephrol. 2011, 6, 1774–1780. [Google Scholar] [CrossRef] [PubMed]
- Akkina, S.K.; Asrani, S.K.; Peng, Y.; Stock, P.; Kim, W.R.; Israni, A.K. Development of organ-specific donor risk indices. Liver Transplant. 2012, 18, 395–404. [Google Scholar] [CrossRef]
- Williams, W.W.; Taheri, D.; Tolkoff-Rubin, N.; Colvin, R.B. Clinical role of the renal transplant biopsy. Nat. Rev. Nephrol. 2012, 8, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Wiebe, C.; Gibson, I.W.; Rush, D.N.; Nickerson, P.W. Immune monitoring of kidney allografts. Am. J. Kidney Dis. 2012, 60, 629–640. [Google Scholar] [CrossRef]
- Lee, P.C.; Ozawa, M.; Hung, C.J.; Lin, Y.J.; Chang, S.S.; Chou, T.C. Eighteen-Year Follow-Up of a Retrospective Study of HLA Antibody on Kidney Graft Survival. Transplant. Proc. 2009, 41, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.; Mistry, K.; McWilliam, H.; Lopez, R.; Parham, P.; Marsh, S.G.E. The IMGT/HLA database. Nucleic Acids Res. 2011, 39, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.A.; Valenzuela, N.M.; Reed, E.F. The perfect storm: HLA antibodies, complement, FcγRs, and endothelium in transplant rejection. Trends Mol. Med. 2015, 21, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Tambur, A.R.; Kosmoliaptsis, V.; Claas, F.H.; Mannon, R.B.; Nickerson, P.; Naesens, M. Significance of HLA-DQ in kidney transplantation: Time to reevaluate human leukocyte antigen matching priorities to improve transplant outcomes? An expert review and recommendations. Kidney Int. 2021, 100, 1012–1022. [Google Scholar] [CrossRef]
- Hernández, D.; Vázquez, T.; Alonso-titos, J.; León, M.; Caballero, A.; Cobo, M.A.; Sola, E.; López, V.; Ruiz-esteban, P.; Cruzado, J.M.; et al. Impact of hla mismatching on early subclinical inflammation in low-immunological-risk kidney transplant recipients. J. Clin. Med. 2021, 10, 1934. [Google Scholar] [CrossRef]
- Collins, A.B.; Chicano, S.L.; Cornell, L.D.; Tolkoff-Rubin, N.; Goes, N.B.; Saidman, S.L.; Farrell, M.L.; Cosimi, A.B.; Colvin, R.B. Putative Antibody-Mediated Rejection With C4d Deposition in HLA-Identical, ABO-Compatible Renal Allografts. Transplant. Proc. 2006, 38, 3427–3429. [Google Scholar] [CrossRef] [PubMed]
- Grafft, C.A.; Cornell, L.D.; Gloor, J.M.; Cosio, F.G.; Gandhi, M.J.; Dean, P.G.; Stegall, M.D.; Amer, H. Antibody-mediated rejection following transplantation from an HLA-identical sibling. Nephrol. Dial. Transplant. 2010, 25, 307–310. [Google Scholar] [CrossRef]
- Zhang, Q.; Reed, E.F. The importance of non-HLA antibodies in transplantation. Nat. Rev. Nephrol. 2016, 12, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Reinsmoen, N.L. Impact and production of Non-HLA-specific antibodies in solid organ transplantation. Int. J. Immunogenet. 2020, 47, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Stallone, G.; Pontrelli, P.; Rascio, F.; Castellano, G.; Gesualdo, L.; Grandaliano, G. Coagulation and Fibrinolysis in Kidney Graft Rejection. Front. Immunol. 2020, 11, 1807. [Google Scholar] [CrossRef] [PubMed]
- Pontrelli, P.; Grandaliano, G.; Van Kooten, C. Editorial: Kidney Transplantation and Innate Immunity. Front. Immunol. 2020, 11, 603982. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Fan, Z.; Koulmanda, M.; Thronley, T.; Strom, T.B. Innate immunity for better or worse govern the allograft response. Curr. Opin. Organ Transplant. 2015, 20, 8–12. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Goswami, T.K.; Singh, M.; Dhawan, M.; Mitra, S.; Emran, T.B.; Rabaan, A.A.; Mutair, A.A.; Alawi, Z.A.; Alhumaid, S.; Dhama, K. Regulatory T cells (Tregs) and their therapeutic potential against autoimmune disorders—Advances and challenges. Hum. Vaccines Immunother. 2022, 18, 2035117. [Google Scholar] [CrossRef]
- Siu, J.H.Y.; Surendrakumar, V.; Richards, J.A.; Pettigrew, G.J. T cell allorecognition pathways in solid organ transplantation. Front. Immunol. 2018, 9, 2548. [Google Scholar] [CrossRef]
- Al-Lamki, R.S.; Bradley, J.R.; Pober, J.S. Endothelial Cells in Allograft Rejection. Transplantation 2008, 86, 1340–1348. [Google Scholar] [CrossRef]
- Ingulli, E. Mechanism of cellular rejection in transplantation. Pediatr. Nephrol. 2010, 25, 61–74. [Google Scholar] [CrossRef]
- Baker, R.J.; Hernandez-Fuentes, M.P.; Brookes, P.A.; Chaudhry, A.N.; Cook, H.T.; Lechler, R.I. Loss of Direct and Maintenance of Indirect Alloresponses in Renal Allograft Recipients: Implications for the Pathogenesis of Chronic Allograft Nephropathy. J. Immunol. 2001, 167, 7199–7206. [Google Scholar] [CrossRef]
- Conlon, T.M.; Saeb-Parsy, K.; Cole, J.L.; Motallebzadeh, R.; Qureshi, M.S.; Rehakova, S.; Negus, M.C.; Callaghan, C.J.; Bolton, E.M.; Bradley, J.A.; et al. Germinal Center Alloantibody Responses Are Mediated Exclusively by Indirect-Pathway CD4 T Follicular Helper Cells. J. Immunol. 2012, 188, 2643–2652. [Google Scholar] [CrossRef]
- Karahan, G.E.; Claas, F.H.J.; Heidt, S. Pre-existing Alloreactive T and B Cells and Their Possible Relevance for Pre-transplant Risk Estimation in Kidney Transplant Recipients. Front. Med. 2020, 7, 340. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, C.; Wang, J.; Yang, C.; Xu, M.; Rong, R.; Zhu, T.; Zhu, D. Endothelial Cells in Antibody-Mediated Rejection of Kidney Transplantation: Pathogenesis Mechanisms and Therapeutic Implications. J. Immunol. Res. 2017, 2017, 8746303. [Google Scholar] [CrossRef]
- Gavlovsky, P.J.; Tonnerre, P.; Guitton, C.; Charreau, B. Expression of MHC class I-related molecules MICA, HLA-E and EPCR shape endothelial cells with unique functions in innate and adaptive immunity. Hum. Immunol. 2016, 77, 1084–1091. [Google Scholar] [CrossRef]
- Luo, L.; Li, Z.; Wu, W.; Luo, G.; Mei, H.; Sun, Z.; Xu, C. The effect of MICA antigens on kidney transplantation outcomes. Immunol. Lett. 2013, 156, 54–58. [Google Scholar] [CrossRef]
- Stastny, P.; Zou, Y.; Fan, Y.; Qin, Z.; Lavingia, B. The emerging issue of MICA antibodies: Antibodies to MICA and other antigens of endothelial cells. Contrib. Nephrol. 2009, 162, 99–106. [Google Scholar] [CrossRef]
- Thaunat, O.; Graff-Dubois, S.; Fabien, N.; Duthey, A.; Attuil-Audenis, V.; Nicoletti, A.; Patey, N.; Morelon, E. A stepwise breakdown of B-cell tolerance occurs within renal allografts during chronic rejection. Kidney Int. 2012, 81, 207–219. [Google Scholar] [CrossRef]
- Lim, M.A.; Kohli, J.; Bloom, R.D. Immunosuppression for kidney transplantation: Where are we now and where are we going? Transplant. Rev. 2017, 31, 10–17. [Google Scholar] [CrossRef]
- Yang, J.; Brook, M.O.; Carvalho-Gaspar, M.; Zhang, J.; Ramon, H.E.; Sayegh, M.H.; Wood, K.J.; Turka, L.A.; Jones, N.D. Allograft rejection mediated by memory T cells is resistant to regulation. Proc. Natl. Acad. Sci. USA 2007, 104, 19954–19959. [Google Scholar] [CrossRef]
- Adams, A.B.; Williams, M.A.; Jones, T.R.; Shirasugi, N.; Durham, M.M.; Kaech, S.M.; Wherry, E.J.; Onami, T.; Lanier, J.G.; Kokko, K.E.; et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J. Clin. Investig. 2003, 111, 1887–1895. [Google Scholar] [CrossRef]
- Goldfarb, D.A. Immunosuppressive Drugs for Kidney Transplantation. J. Urol. 2005, 173, 2105. [Google Scholar] [CrossRef]
- Tsuji, T.; Iwasaki, S.; Makita, K.; Imamoto, T.; Ishidate, N.; Mitsuke, A.; Fukuzawa, N.; Harada, H.; Fukazawa, Y. Preceding T-Cell-Mediated Rejection Is Associated with the Development of Chronic Active Antibody-Mediated Rejection by de Novo Donor-Specific Antibody. Nephron 2021, 144, 13–17. [Google Scholar] [CrossRef]
- Gupta, G.; Abu Jawdeh, B.G.; Racusen, L.C.; Bhasin, B.; Arend, L.J.; Trollinger, B.; Kraus, E.; Rabb, H.; Zachary, A.A.; Montgomery, R.A.; et al. Late antibody-mediated rejection in renal allografts: Outcome after conventional and novel therapies. Transplantation 2014, 97, 1240–1246. [Google Scholar] [CrossRef]
- Voora, S.; Adey, D.B. Management of Kidney Transplant Recipients by General Nephrologists: Core Curriculum 2019. Am. J. Kidney Dis. 2019, 73, 866–879. [Google Scholar] [CrossRef]
- Noelle, R.J.; Snow, E.C. Cognate interactions between helper T cells and B Cells. Immunol. Today 1990, 11, 361–368. [Google Scholar] [CrossRef]
- Lavinder, J.J.; Wine, Y.; Giesecke, C.; Ippolito, G.C.; Horton, A.P.; Lungu, O.I.; Hoi, K.H.; DeKosky, B.J.; Murrin, E.M.; Wirth, M.M.; et al. Identification and characterization of the constituent human serum antibodies elicited by vaccination. Proc. Natl. Acad. Sci. USA 2014, 111, 2259–2264. [Google Scholar] [CrossRef]
- Bernasconi, N.L.; Traggiai, E.; Lanzavecchia, A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science 2002, 298, 2199–2202. [Google Scholar] [CrossRef]
- Resch, T.; Fabritius, C.; Ebner, S.; Ritschl, P.; Kotsch, K. The Role of Natural Killer Cells in Humoral Rejection. Transplantation 2015, 99, 1335–1340. [Google Scholar] [CrossRef]
- Valenzuela, N.M.; Reed, E.F. Antibodies to HLA Molecules Mimic Agonistic Stimulation to Trigger Vascular Cell Changes and Induce Allograft Injury HHS Public Access. Curr. Transpl. Rep. 2015, 2, 222–232. [Google Scholar] [CrossRef][Green Version]
- Loupy, A.; Mengel, M.; Haas, M. Thirty years of the International Banff Classification for Allograft Pathology: The past, present, and future of kidney transplant diagnostics. Kidney Int. 2022, 101, 678–691. [Google Scholar] [CrossRef]
- Formica, M.L.; Awde Alfonso, H.G.; Palma, S.D. Biological drug therapy for ocular angiogenesis: Anti-VEGF agents and novel strategies based on nanotechnology. Pharmacol. Res. Perspect. 2021, 9, e00723. [Google Scholar] [CrossRef]
- Sánchez-Magraner, L.; de la Fuente, M.; Evans, C.; Miles, J.; Elexpe, A.; Rodriguez-Astigarraga, M.; Astigarraga, E.; Barreda-Gómez, G. Quantification of PD-1/PD-L1 Interaction between Membranes from PBMCs and Melanoma Samples Using Cell Membrane Microarray and Time-Resolved Förster Resonance Energy Transfer. Analytica 2021, 2, 156–170. [Google Scholar] [CrossRef]
- Aletaha, D.; Smolen, J.S. Diagnosis and Management of Rheumatoid Arthritis: A Review. JAMA-J. Am. Med. Assoc. 2018, 320, 1360–1372. [Google Scholar] [CrossRef]
- Jang, D.I.; Lee, A.H.; Shin, H.Y.; Song, H.R.; Park, J.H.; Kang, T.B.; Lee, S.R.; Yang, S.H. The role of tumor necrosis factor alpha (Tnf-α) in autoimmune disease and current tnf-α inhibitors in therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef]
- Zhao, Y.; Cooper, D.K.C.; Wang, H.; Chen, P.; He, C.; Cai, Z.; Mou, L.; Luan, S.; Gao, H. Potential pathological role of pro-inflammatory cytokines (IL-6, TNF-α, and IL-17) in xenotransplantation. Xenotransplantation 2019, 26, e12502. [Google Scholar] [CrossRef]
- Nagata, Y.; Fujimoto, M.; Nakamura, K.; Isoyama, N.; Matsumura, M.; Fujikawa, K.; Uchiyama, K.; Takaki, E.; Takii, R.; Nakai, A.; et al. Anti-TNF-α agent infliximab and splenectomy are protective against renal ischemia-reperfusion injury. Transplantation 2016, 100, 1675–1682. [Google Scholar] [CrossRef]
- Jordan, S.C.; Choi, J.; Kim, I.; Wu, G.; Toyoda, M.; Shin, B.; Vo, A. Interleukin-6, A cytokine critical to mediation of inflammation, autoimmunity and allograft rejection: Therapeutic implications of IL-6 receptor blockade. Transplantation 2017, 101, 32–44. [Google Scholar] [CrossRef]
- Choi, J.; Aubert, O.; Vo, A.; Loupy, A.; Haas, M.; Puliyanda, D.; Kim, I.; Louie, S.; Kang, A.; Peng, A.; et al. Assesment of Tocilizumab (Anti-IL-6 Receptor Monoclonal) as a Potential Treatment for Chronic Antibody Mediated Rejection and Transplant Glomerulopathy in HLA Sensitized Renal Allograft Recipients. Am. J. Transpl. 2017, 17, 2381–2389. [Google Scholar] [CrossRef] [PubMed]
- Navarra, S.V.; Guzmán, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.M.; Thomas, M.; Kim, H.Y.; León, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Banham, G.D.; Flint, S.M.; Torpey, N.; Lyons, P.A.; Shanahan, D.N.; Gibson, A.; Watson, C.J.E.; O’Sullivan, A.M.; Chadwick, J.A.; Foster, K.E.; et al. Belimumab in kidney transplantation: An experimental medicine, randomised, placebo-controlled phase 2 trial. Lancet 2018, 391, 2619–2630. [Google Scholar] [CrossRef]
- Lundin, J.; Kimby, E.; Björkholm, M.; Broliden, P.A.; Celsing, F.; Hjalmar, V.; Möllgård, L.; Rebello, P.; Hale, G.; Waldmann, H.; et al. Phase II trial of subcutaneous anti-CD52 monoclonal antibody alemtuzumab (Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL). Blood 2002, 100, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Friend, P.J. Alemtuzumab induction therapy in solid organ transplantation. Transplant. Res. 2013, 2, 2–5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sureshkumar, K.K.; Hussain, S.M.; Carpenter, B.J.; Sandroni, S.E.; Marcus, R.J. Antibody-mediated rejection following renal transplantation. Expert Opin. Pharmacother. 2007, 8, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Macklin, P.S.; Morris, P.J.; Knight, S.R. A systematic review of the use of rituximab for the treatment of antibody-mediated renal transplant rejection. Transplant. Rev. 2017, 31, 87–95. [Google Scholar] [CrossRef]
- Venetz, J.P.; Pascual, M. New treatments for acute humoral rejection of kidney allografts. Expert Opin. Investig. Drugs 2007, 16, 625–633. [Google Scholar] [CrossRef]
- Grenda, R. Biologics in renal transplantation. Pediatr. Nephrol. 2015, 30, 1087–1098. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cecka, J.M.; Kucheryavaya, A.Y.; Reinsmoen, N.L.; Leffell, M.S. Calculated PRA: Initial results show benefits for sensitized patients and a reduction in positive crossmatches. Am. J. Transplant. 2011, 11, 719–724. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Williams, R.L.; Wong, G.; Lim, W.H. The evolution of hla-matching in kidney transplantation. In Current Issues and Future Direction in Kidney Transplantation; IntechOpen Limited: London, UK, 2013; pp. 273–297. [Google Scholar] [CrossRef]
- Ting, A. The Limfocitotoxic Crossmatch Test in Clinical Renal Transplantation. Transplantation 1983, 35, 403–407. [Google Scholar] [CrossRef]
- Mulley, W.R.; Kanellis, J. Understanding crossmatch testing in organ transplantation: A case-based guide for the general nephrologist. Nephrology 2011, 16, 125–133. [Google Scholar] [CrossRef]
- Claas, F.H.J.; Witvliet, M.D.; Duquesnoy, R.J.; Persijn, G.G.; Doxiadis, I.I.N. The acceptable mismatch program as a fast tool for highly sensitized patients awaiting a cadaveric kidney transplantation: Short waiting time and excellent graft outcome. Transplantation 2004, 78, 190–193. [Google Scholar] [CrossRef]
- Tait, B.D. Solid phase assays for HLA antibody detection in clinical transplantation. Curr. Opin. Immunol. 2009, 21, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Tait, B.D.; Süsal, C.; Gebel, H.M.; Nickerson, P.W.; Zachary, A.A.; Claas, F.H.J.; Reed, E.F.; Bray, R.A.; Campbell, P.; Chapman, J.R.; et al. Consensus guidelines on the testing and clinical management issues associated with HLA and Non-HLA antibodies in transplantation. Transplantation 2013, 95, 19–47. [Google Scholar] [CrossRef]
- Minucci, P.B.; Grimaldi, V.; Casamassimi, A.; Cacciatore, F.; Sommese, L.; Picascia, A.; Resse, M.; Sabia, C.; Russo, A.; Napoli, C. Methodologies for Anti-HLA antibody screening in patients awaiting kidney transplant: A comparative study. Exp. Clin. Transplant. 2011, 9, 381–386. [Google Scholar]
- Rebibou, J.M.; Chabod, J.; Bittencourt, M.C.; Thevénin, C.; Chalopin, J.M.; Hervé, P.; Tiberghien, P. Flow-PRA® evaluation for antibody screening in patients awaiting kidney transplantation. Transpl. Immunol. 2000, 8, 125–128. [Google Scholar] [CrossRef]
- Colombo, M.B.; Haworth, S.E.; Poli, F.; Nocco, A.; Puglisi, G.; Innocente, A.; Serafini, M.; Messa, P.; Scalamogna, M. Luminex technology for anti-HLA antibody screening: Evaluation of performance and of impact on laboratory routine. Cytom. Part B-Clin. Cytom. 2007, 72, 465–471. [Google Scholar] [CrossRef]
- Pei, R.; Wang, G.; Tarsitani, C.; Rojo, S.; Chen, T.; Takemura, S.; Liu, A.; Lee, J. Simultaneous HLA class I and class II antibodies screening with flow cytometry. Hum. Immunol. 1998, 59, 313–322. [Google Scholar] [CrossRef]
- Picascia, A.; Infante, T.; Napoli, C. Luminex and antibody detection in kidney transplantation. Clin. Exp. Nephrol. 2012, 16, 373–381. [Google Scholar] [CrossRef]
- Loupy, A.; Lefaucheur, C.; Vernerey, D.; Prugger, C.; van Huyen, J.-P.D.; Mooney, N.; Suberbielle, C.; Frémeaux-Bacchi, V.; Méjean, A.; Desgrandchamps, F.; et al. Complement-Binding Anti-HLA Antibodies and Kidney-Allograft Survival. N. Engl. J. Med. 2013, 369, 1215–1226. [Google Scholar] [CrossRef]
- Sullivan, H.C.; Gebel, H.M.; Bray, R.A. Understanding solid-phase HLA antibody assays and the value of MFI. Hum. Immunol. 2017, 78, 471–480. [Google Scholar] [CrossRef]
- McCaughan, J.; Xu, Q.; Tinckam, K. Detecting donor-specific antibodies: The importance of sorting the wheat from the chaff. HepatoBiliary Surg. Nutr. 2019, 8, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Süsal, C.; Morath, C. Virtual PRA replaces traditional PRA: Small change but significantly more justice for sensitized patients. Transpl. Int. 2015, 28, 708–709. [Google Scholar] [CrossRef]
- Bingaman, A.W.; Murphey, C.L.; Palma-Vargas, J.; Wright, F. A virtual crossmatch protocol significantly increases access of highly sensitized patients to deceased donor kidney transplantation. Transplantation 2008, 86, 1864–1868. [Google Scholar] [CrossRef]
- Kowalski, R.; Post, D.; Schneider, M.C.; Britz, J.; Thomas, J.; Deierhoi, M.; Lobashevsky, A.; Redfield, R.; Schweitzer, E.; Heredia, A.; et al. Immune cell function testing: An adjunct to therapeutic drug monitoring in transplant patient management. Clin. Transplant. 2003, 17, 77–88. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Li, Y.; Zhang, H.; Wei, X.; Zheng, H.; Xu, C.; Bao, X.; Yuan, X.; Hou, J. Immune function assay (ImmuKnow) as a predictor of allograft rejection and infection in kidney transplantation. Clin. Transplant. 2013, 27, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.T.; Lee, H.M.; Lower, R.R.; Thomas, J.M. Immunological monitoring as a guide to the management of transplant recipients. Surg. Clin. N. Am. 1979, 59, 253–281. [Google Scholar] [CrossRef]
- MacEdo, C.; Orkis, E.A.; Popescu, I.; Elinoff, B.D.; Zeevi, A.; Shapiro, R.; Lakkis, F.G.; Metes, D. Contribution of naïve and memory t-cell populations to the human alloimmune response. Am. J. Transplant. 2009, 9, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.I.; Morris, A.G.; Booth, L.J. Clinical significance of in vitro donor-specific hyporesponsiveness in renal allograft recipients as demonstrated by the MLR. Transpl. Int. 1994, 7, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Crespo, E.; Bestard, O. Biomarkers to assess donor-reactive T-cell responses in kidney transplant patients. Clin. Biochem. 2016, 49, 329–337. [Google Scholar] [CrossRef]
- Emerson, R.O.; Mathew, J.M.; Konieczna, I.M.; Robins, H.S.; Leventhal, J.R. Defining the alloreactive T cell repertoire using high-throughput sequencing of mixed lymphocyte reaction culture. PLoS ONE 2014, 9, e111934. [Google Scholar] [CrossRef]
- Crespo, E.; Lucia, M.; Cruzado, J.M.; Luque, S.; Melilli, E.; Manonelles, A.; Lloberas, N.; Torras, J.; Grinyó, J.M.; Bestard, O. Pre-transplant donor-specific T-cell alloreactivity is strongly associated with early acute cellular rejection in kidney transplant recipients not receiving T-cell depleting induction therapy. PLoS ONE 2015, 10, e117618. [Google Scholar] [CrossRef]
- Nickel, P.; Presber, F.; Bold, G.; Biti, D.; Schönemann, C.; Tullius, S.G.; Volk, H.D.; Reinke, P. Enzyme-linked immunosorbent spot assay for donor-reactive interferon-gamma-producing cells identifies T-cell presensitization and correlates with graft function at 6 and 12 months in renal-transplant recipients. Transplantation 2004, 78, 1640–1646. [Google Scholar] [CrossRef]
- Hricik, D.E.; Augustine, J.; Nickerson, P.; Formica, R.N.; Emilio, D.; Rush, D.; Newell, K.A.; Goebel, J.; Gibson, I.W.; Fairchild, R.L.; et al. Interferon Gamma ELISPOT Testing as a Risk-stratifying Biomarker for Kidney Transplant Injury: Results from the CTOT-01 Multecenter Study. Am. J. Transplant. 2015, 15, 3166–3173. [Google Scholar] [CrossRef]
- Girmanova, E.; Hruba, P.; Viklicky, O.; Slavcev, A. ELISpot assay and prediction of organ transplant rejection. Int. J. Immunogenet. 2022, 49, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Waanders, M.M.; Heidt, S.; Koekkoek, K.M.; Zoet, Y.M.; Doxiadis, I.I.N.; Amir, A.; Heemskerk, M.H.M.; Mulder, A.; Brand, A.; Roelen, D.L.; et al. Monitoring of indirect allorecognition: Wishful thinking or solid data? Tissue Antigens 2008, 71, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zachary, A.A.; Kopchaliiska, D.; Montgomery, R.A.; Leffell, M.S. HLA-specific B cells: I. A method for their detection, quantification, and isolation using HLA tetramers. Transplantation 2007, 83, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Wehmeier, C.; Karahan, G.E.; Heidt, S. HLA-specific memory B-cell detection in kidney transplantation: Insights and future challenges. Int. J. Immunogenet. 2020, 47, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Karahan, G.E.; Claas, F.H.J.; Heidt, S. Detecting the humoral alloimmune response: We need more than serum antibody screening. Transplantation 2015, 99, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Karahan, G.E.; de Vaal, Y.J.H.; Roelen, D.L.; Buchli, R.; Claas, F.H.J.; Heidt, S. Quantification of HLA class II-specific memory B cells in HLA-sensitized individuals. Hum. Immunol. 2015, 76, 129–136. [Google Scholar] [CrossRef]
- Quaglia, M.; Merlotti, G.; Guglielmetti, G.; Castellano, G.; Cantaluppi, V. Recent advances on biomarkers of early and late kidney graft dysfunction. Int. J. Mol. Sci. 2020, 21, 5404. [Google Scholar] [CrossRef]
- Wagner, J.C.; Tang, Q. CAR-Tregs as a Strategy for Inducing Graft Tolerance. Curr. Transpl. Rep. 2020, 7, 205–214. [Google Scholar] [CrossRef]
- Roemhild, A.; Otto, N.M.; Moll, G.; Abou-El-Enein, M.; Kaiser, D.; Bold, G.; Schachtner, T.; Choi, M.; Oellinger, R.; Landwehr-Kenzel, S.; et al. Regulatory T cells for minimising immune suppression in kidney transplantation: Phase I/IIa clinical trial. BMJ 2020, 371, m3734. [Google Scholar] [CrossRef]
- Cravedi, P.; Heeger, P.S. Immunologic monitoring in transplantation revisited. Curr. Opin. Organ Transplant. 2012, 17, 26–32. [Google Scholar] [CrossRef]
- Chopra, B.; Sureshkumar, K.K. Emerging role of cell-free DNA in kidney transplantation. World J. Exp. Med. 2021, 11, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Bloom, R.D.; Bromberg, J.S.; Poggio, E.D.; Bunnapradist, S.; Langone, A.J.; Sood, P.; Matas, A.J.; Mehta, S.; Mannon, R.B.; Sharfuddin, A.; et al. Cell-Free DNA and active rejection in kidney allografts. J. Am. Soc. Nephrol. 2017, 28, 2221–2232. [Google Scholar] [CrossRef] [PubMed]
- Carbone, D.P.; Gandara, D.R.; Antonia, S.J.; Zielinski, C.; Paz-Ares, L. Non-small-cell lung cancer: Role of the immune system and potential for immunotherapy. J. Thorac. Oncol. 2015, 10, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Magraner, L.; Miles, J.; Baker, C.L.; Applebee, C.J.; Lee, D.J.; Elsheikh, S.; Lashin, S.; Withers, K.; Watts, A.G.; Parry, R.; et al. High PD-1/PD-L1 checkpoint interaction infers tumor selection and therapeutic sensitivity to anti-PD-1/PD-L1 treatment. Cancer Res. 2020, 80, 4244–4257. [Google Scholar] [CrossRef]
- Rienda, B.; Elexpe, A.; Tolentino-Cortez, T.; Gulak, M.; Bruzos-Cidón, C.; Torrecilla, M.; Astigarraga, E.; Barreda-Gómez, G. Analysis of Acetylcholinesterase Activity in Cell Membrane Microarrays of Brain Areas as a Screening Tool to Identify Tissue Specific Inhibitors. Analytica 2021, 2, 25–36. [Google Scholar] [CrossRef]
- Fernández, R.; Garate, J.; Tolentino-Cortez, T.; Herraiz, A.; Lombardero, L.; Ducrocq, F.; Rodríguez-Puertas, R.; Trifilieff, P.; Astigarraga, E.; Barreda-Gómez, G.; et al. Microarray and Mass Spectrometry-Based Methodology for Lipid Profiling of Tissues and Cell Cultures. Anal. Chem. 2019, 91, 15967–15973. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).