
Long COVID (PASC) Is Maintained by a Self-Sustaining Pro-Inflammatory TLR4/RAGE-Loop of S100A8/A9 > TLR4/RAGE Signalling, Inducing Chronic Expression of IL-1b, IL-6 and TNFa: Anti-Inflammatory Ezrin Peptides as Potential Therapy

Abstract
:1. Introduction
2. Long COVID and Post-Acute Sequelae of COVID-19 (PASC)
2.1. Prevalence of Long COVID/PASC
2.2. The Frequency of Long COVID/PASC Due to Infection and Re-Infection
3. SARS-CoV-2 Infection, Acute COVID-19 and Long COVID
3.1. Markers of Immune Perturbation after Acute COVID-19
3.2. Complexity of Symptoms in long COVID/PASC Patients
3.3. Long COVID/PASC Compared and Contrasted with ME/CFS
4. Chronic Expression of Pro-Inflammatory Cytokines in Long COVID/PASC
4.1. IL-1b, IL-6 and TNFa Expression in Long COVID/PASC
4.2. IL-6 > IL-6R Driven S100A8/A9 Expression in Acute COVID-19
5. Other Pro-Inflammatory Factors in Acute COVID-19 (Metabolic Reprogramming, AGEs and HMGB1)
5.1. Virally Induced Metabolic Re-Programming
5.2. The Role of AGEs in Acute COVID-19
5.3. AGE > RAGE-Signalling in Acute COVID-19
5.4. SARS-CoV-2 Spike-Protein Binds mRAGE to Infect Lung Monocytes
5.5. Virus Induction of HMGB1 > RAGE Signalling in Acute COVID-19
5.6. S100A8/A9 > RAGE-Signalling in Acute COVID-19 and Long COVID
6. A Brief Review of S100A8/A9 Research
6.1. S100A8/A9 Ca2+-Binding Protein
6.2. Constitutive and Induced Expression of S100A8/A9
6.3. The Functions of S100A8/A9 > RAGE and TLR4 Signalling
6.4. Intra-Cellular Signalling Induced by S100A8/A9
6.5. S100A8/A9 over-Expression in Chronic Inflammatory Diseases
6.6. S100A8/A9 Inflammation in Rheumatoid Arthritis
6.7. S100A8/A9 Inflammation in Ulcerative Colitis
6.8. S100A8/A9 Inflammation in Cardiovascular Disease
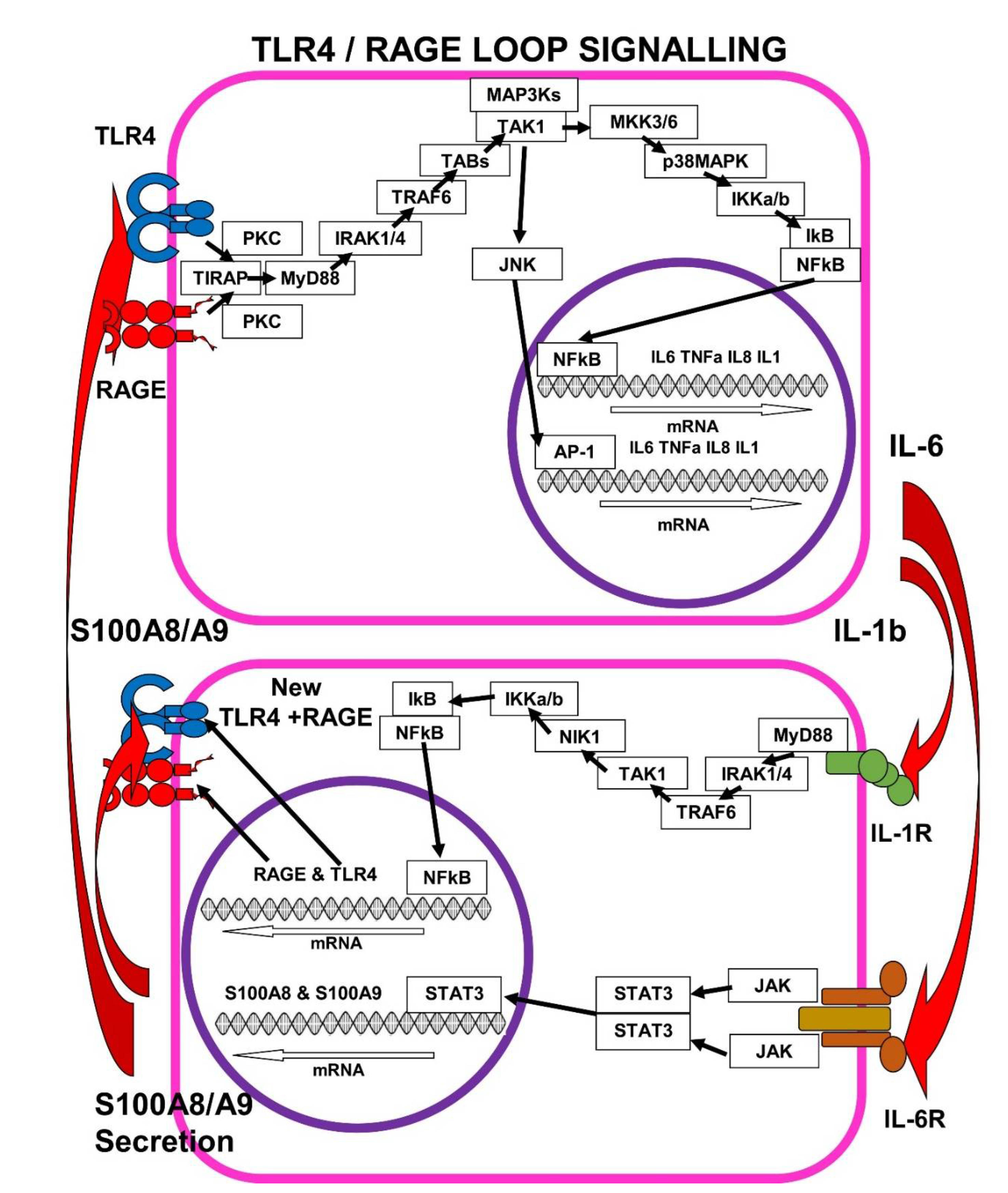
7. The Self-Sustaining TLR4/RAGE-Loop
7.1. IL-1b > IL-1R Signalling Enhances mRAGE and TLR4 Expression
7.2. IL-6 > IL-6R Signalling Enhances S100A8/A9 Expression
7.3. S100A8/A9 Pushes the TLR4/RAGE-Loop to Signal for More Cytokines
7.4. SARS-CoV-2 in hACE2 Mice and Inhibition of S100A8/A9 > RAGE-Signalling
7.5. SARS-CoV-2 in hACE2 Mice and Inhibition of S100A8/A9 >TLR4-Signalling
8. Ezrin Peptides as Potential Therapies for Long COVID/PASC
Ezrin Peptides HEP-1 and RepG3 Inhibit Pro-Inflammatory Cytokine Expression
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Tejerina, F.; Catalan, P.; Rodriguez-Grande, C.; Adan, J.; Rodriguez-Gonzalez, C.; Muñoz, P.; Aldamiz, T.; Diez, C.; Perez, L.; Fanciulli, C.; et al. Post-COVID-19 syndrome. SARS-CoV-2 RNA detection in plasma, stool, and urine in patients with persistent symptoms after COVID-19. BMC Infect. Dis. 2022, 22, 211. [Google Scholar] [CrossRef]
- Available online: https://www.ons.gov.uk/peoplepopulationandcommunity/healthandsocialcare/conditionsanddiseases/bulletins/prevalenceofongoingsymptomsfollowingcoronaviruscovid19infectionintheuk/7july2022 (accessed on 18 July 2022).
- Available online: https://coronavirus.data.gov.uk/details/cases (accessed on 18 July 2022).
- Al-Aly, Z.; Bowe, B.; Xie, Y. Outcomes of SARS-CoV-2 Reinfection. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Fernández-Lázaro, D.; Sánchez-Serrano, N.; Mielgo-Ayuso, J.; García-Hernández, J.L.; González-Bernal, J.J.; Seco-Calvo, J. Long COVID a New Derivative in the Chaos of SARS-CoV-2 Infection: The Emergent Pandemic? J. Clin. Med. 2021, 10, 5799. [Google Scholar] [CrossRef]
- Elicker, B. What Are the Long-term Pulmonary Sequelae of COVID-19 Infection? Radiology 2022, 304, 193–194. [Google Scholar] [CrossRef]
- Crook, H.; Raza, S.; Nowell, J.; Young, M.; Edison, P. Long covid—Mechanisms, risk factors, and Management. BMJ 2021, 374, n1648. [Google Scholar] [CrossRef] [PubMed]
- Tate, W.; Walker, M.; Sweetman, E.; Helliwell, A.; Peppercorn, K.; Edgar, C.; Blair, A.; Chatterjee, A. Molecular Mechanisms of Neuroinflammation in ME/CFS and Long COVID to Sustain Disease and Promote Relapses. Front. Neurol. 2022, 13, 877772. [Google Scholar] [CrossRef] [PubMed]
- Hornig, M.; Montoya, J.G.; Klimas, N.G.; Levine, S.; Felsenstein, D.; Bateman, L.; Peterson, D.L.; Gottschalk, C.G.; Schultz, A.F.; Che, X.; et al. Distinct plasma immune signatures in ME/CFS are present early in the course of illness. Sci. Adv. 2015, 1, e1400121. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.J.; Lu, S.; Tang, A.F.; Durstenfeld, M.S.; Ho, H.E.; Goldberg, S.A.; Forman, C.A.; Munter, S.E.; Hoh, R.; Tai, V.; et al. Markers of Immune Activation and Inflammation in Individuals With Postacute Sequelae of Severe Acute Respiratory Syndrome Coronavirus 2 Infection. J. Infect. Dis. 2021, 224, 1839–1848. [Google Scholar] [CrossRef]
- Schultheiβ, C.; Willscher, E.; Paschold, L.; Gottschick, C.; Klee, B.; Henkes, S.S.; Bosurgi, L.; Dutzmann, J.; Sedding, D.; Frese, T.; et al. The IL-1b, IL-6, and TNF cytokine triad is associated with post-acute sequelae of COVID-19. Cell Rep. Med. 2022, 3, 100663. [Google Scholar] [CrossRef]
- Unterman, A.; Sumida, T.S.; Nouri, N.; Yan, X.; Zhao, A.Y.; Gasque, V.; Schupp, J.C.; Asashima, H.; Liu, Y.; Cosme, C.; et al. Single-cell multi-omics reveals dyssynchrony of the innate and adaptive immune system in progressive COVID-19. Nat. Commun. 2022, 13, 440. [Google Scholar] [CrossRef]
- Allen, C.N.S.; Santerre, M.; Arjona, S.P.; Ghaleb, L.J.; Herzi, M.; Llewellyn, M.D.; Shcherbik, N.; Sawaya, B.E. SARS-CoV-2 Causes Lung Inflammation through Metabolic Reprogramming and RAGE. Viruses 2022, 14, 983. [Google Scholar] [CrossRef] [PubMed]
- Sellegounder, D.; Zafari, P.; Rajabinejad, M.; Taghadosi, M.; Kapahi, P. Advanced glycation end products (AGEs) and its receptor, RAGE, modulate age-dependent COVID-19 morbidity and mortality. Int. Immunopharmacol. 2021, 98, 107806. [Google Scholar] [CrossRef] [PubMed]
- Senatus, L.M.; Schmidt, A.M. The AGE-RAGE Axis: Implications form Age-Associated Arterial Diseases. Front. Genet. 2017, 8, 187. [Google Scholar] [CrossRef]
- Chiappalupi, S.; Salvadori, L.; Donato, R.; Riuzzi, F.; Sorci, G. Hyperactivated RAGE in Comorbidities as a Risk Factor for Severe COVID-19—The Role of RAGE-RAS Crosstalk. Biomolecules 2021, 11, 876. [Google Scholar] [CrossRef] [PubMed]
- Saputra, G.; Yudhawati, R.; Fitriah, M. Association of soluble receptor for advanced glycation end-products (sRAGE) serum on COVID-19 severity: A cross-sectional study. Ann. Med. Surg. 2022, 74, 103303. [Google Scholar] [CrossRef]
- Angioni, R.; Bonfanti, M.; Caporale, N.; Sánchez-Rodríguez, R.; Munari, F.; Savino, A.; Buratto, D.; Pagani, I.; Bertoldi, N.; Zanon, C.; et al. RAGE engagement by SARS-CoV-2 enables monocyte infection and underlies COVID-19 severity. bioRxiv 2022, preprint. [Google Scholar] [CrossRef]
- Al-kuraishy, H.; Al-Gareeb, A.; Alkazmi, L.; Habotta, O.; Batiha, G. High-mobility group box 1 (HMGB1) in COVID-19: Extrapolation of dangerous liaisons. Inflammopharmacology 2022, 30, 811–820. [Google Scholar] [CrossRef]
- Deguchi, A.; Yamamoto, T.; Shibata, N.; Maru, Y. S100A8 may govern hyper-inflammation in severe COVID-19. FASEB J. 2021, 35, e21798. [Google Scholar] [CrossRef]
- Chapuis, N.; Ibrahimi, N.; Belmondo, T.; Goulvestre, C.; Berger, A.E.; Mariaggi, A.A.; Andrieu, M.; Chenevier-Gobeaux, C.; Bayle, A.; Campos, L.; et al. Dynamics of circulating calprotectin accurately predict the outcome of moderate COVID-19 patients. eBioMedicine 2022, 80, 104077. [Google Scholar] [CrossRef]
- Mao, Q.; Wang, C.; Wen, W.; Zhou, M.; Tang, J.; Chen, C.; Cheng, Y.; Wu, Q.; Zhang, X.; Feng, Z.; et al. A meta-analysis of the association between calprotectin and the severity of COVID-19. J. Infect. 2022, 84, e31–e33. [Google Scholar] [CrossRef]
- Mellett, L.; Khader, S.A. S100A8/A9 in COVID-19 pathogenesis: Impact on clinical outcomes. Cytokine Growth Factor Rev. 2022, 63, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Silvin, A.; Chapuis, N.; Dunsmore, G.; Goubet, A.G.; Dubuisson, A.; Derosa, L.; Almire, C.; Hénon, C.; Kosmider, O.; Droin, N.; et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell 2020, 182, 1401–1418.e18. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Long, X.; Xu, Q.; Tan, J.; Wang, G.; Cao, Y.; Wei, J.; Luo, H.; Zhu, H.; Huang, L.; et al. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 992–994. [Google Scholar] [CrossRef]
- Ryan, F.J.; Hope, C.M.; Masavuli, M.G.; Lynn, M.A.; Mekonnen, Z.A.; Yeow, A.E.L.; Garcia-Valtanen, P.; Al-Delfi, Z.; Gummow, J.; Ferguson, C.; et al. Long-term perturbation of the peripheral immune system months after SARS-CoV-2 infection. BMC Med. 2022, 20, 26. [Google Scholar] [CrossRef] [PubMed]
- Cheon, I.S.; Li, C.; Son, Y.M.; Goplen, N.P.; Wu, Y.; Cassmann, T.; Wang, Z.; Wei, X.; Tang, J.; Li, Y.; et al. Immune signatures underlying post-acute COVID-19 lung sequelae. Sci. Immunol. 2021, 6, eabk1741. [Google Scholar] [CrossRef] [PubMed]
- Zervides, K.A.; Jern, A.; Nystedt, J.; Gullstrand, B.; Nilsson, P.C.; Sundgren, P.C.; Bengtsson, A.A.; Jönsen, A. Serum S100A8/A9 concentrations are associated with neuropsychiatric involvement in systemic lupus erythematosus: A cross-sectional study. BMC Rheumatol. 2022, 6, 38. [Google Scholar] [CrossRef]
- Gonzalez, L.; Karin Garrie, K.; Mark, D.; Turner, M. Role of S100 proteins in health and disease. BBA—Mol. Cell Res. 2020, 1867, 118677. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Braunstein, Z.; Toomey, A.C.; Zhong, J.; Rao, X. S100 Proteins As an Important Regulator of Macrophage Inflammation. Front. Immunol. 2018, 8, 1908. [Google Scholar] [CrossRef]
- Donato, R.; R cannon, B.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 Proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef]
- Hsu, K.; Champaiboon, C.; Guenther, B.D.; Sorenson, B.S.; Khammanivong, A.; Ross, K.F.; Geczy, C.L.; Herzberg, M.C. Anti-Infective Protective Properties of S100 Calgranulins. Antiinflamm Antiallergy Agents Med. Chem. 2009, 8, 290–305. [Google Scholar] [CrossRef] [Green Version]
- Henke, M.; Renner, A.; Rubin, B.; Gyves, J.; Eva Lorenz, E.J.; Koo, J. Up-Regulation of S100A8 And S100A9 Protein in Bronchial Epithelial Cells by Lipopolysaccharide. Exp. Lung. Res. 2006, 32, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Yen, T.; Harrison, C.A.; Devery, J.M.; Leong, S.; Iismaa, S.E.; Yoshimura, T.; Geczy, C.L. Induction of the S100 chemotactic protein, CP-10, in murine microvascular endothelial cells by proinflammatory stimuli. Blood 1997, 90, 4812. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Yen, T.; Carolyn, L. Geczy IL-10 Up-Regulates Macrophage Expression of the S100 Protein S100A8. J. Immunol. 2001, 166, 6358–6366. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE Mediates a Novel Proinflammatory Axis: A Central Cell Surface Receptor for S100/Calgranulin Polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef]
- Leclerc, E.; Fritz, G.; Vetter, S.; Heizmann, C. Binding of S100 proteins to RAGE: An update. Biochim. Biophys. Acta 2009, 1793, 993–1007. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Murata, H.; Yamamoto, K.I.; Ono, T.; Sakaguchi, Y.; Motoyama, A.; Hibino, T.; Kataoka, K.; Huh, N.H. TIRAP, an Adaptor Protein for TLR2/4, Transduces a Signal from RAGE Phosphorylated upon Ligand Binding. PLoS ONE 2011, 6, e23132. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef]
- Ma, L.; Sun, P.; Zhang, J.C.; Zhang, Q.; Yao, S.L. Proinflammatory effects of S100A8/A9 via TLR4 and RAGE signaling pathways in BV-2 microglial cells. Int. J. Mol. Med. 2017, 40, 31–38. [Google Scholar] [CrossRef]
- Sunahori, K.; Yamamura, M.; Yamana, J.; Takasugi, K.; Kawashima, M.; Yamamoto, H.; Chazin, W.J.; Nakatani, Y.; Yui, S.; Makino, H. The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and p38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, R69. [Google Scholar] [CrossRef]
- Lee, M.J.; Lee, J.K.; Choi, J.W.; Lee, C.S.; Sim, J.H.; Cho, C.H.; Lee, K.H.; Cho, I.H.; Chung, M.H.; Kim, H.R.; et al. Interleukin-6 Induces S100A9 Expression in Colonic Epithelial Cells through STAT3 Activation in Experimental Ulcerative Colitis. PLoS ONE 2012, 7, e38801. [Google Scholar] [CrossRef] [Green Version]
- Cotoi, O.; Dunér, P.; Ko, N.; Hedblad, B.; Nilsson, J.; Björkbacka, H.; Schiopu, A. Plasma S100A8/A9 Correlates With Blood Neutrophil Counts, Traditional Risk Factors, and Cardiovascular Disease in Middle-Aged Healthy Individuals. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 202. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.J.; Oh, H.J.; Jung, Y.O.; Cho, M.L.; Lee, S.Y.; Yu, J.G.; Park, M.K.; Kim, H.R.; Lee, S.H.; Park, S.H.; et al. The expression of the receptor for advanced glycation end-products (RAGE) in RA-FLS is induced by IL-17 via Act-1. Arthritis Res. Ther. 2011, 13, R113. Available online: http://arthritis-research.com/content/13/4/R113 (accessed on 22 July 2022). [CrossRef] [PubMed]
- Yan, Z.-Q. Regulation of TLR4 Expression Is a Tale About Tail. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2582–2584. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Qian, Y.; Li, Z.; Fan, E.K.; Li, Y.; Wu, L.; Billiar, T.R.; Wilson, M.A.; Shi, X.; Fan, J. TLR4-Upregulated IL-1β and IL-1RI Promote Alveolar Macrophag Pyroptosis and Lung Inflammation through an Autocrine Mechanism. Sci. Rep. 2016, 6, 31663. [Google Scholar] [CrossRef]
- Jauch-Speer, S.L.; Herrera-Rivero, M.; Ludwig, N.; De Carvalho, B.C.V.; Martens, L.; Wolf, J.; Chasan, A.I.; Witten, A.; Markus, B.; Schieffer, B.; et al. C/EBPδ-induced epigenetic changes control the dynamic gene transcription of S100a8 and S100a9. ELife 2022, 11, e75594. [Google Scholar] [CrossRef]
- Hsu, K.; Chung, Y.M.; Endoh, Y.; Geczy, C.L. TLR9 Ligands Induce S100A8 in Macrophages via a STAT3-Dependent Pathway which Requires IL-10 and PGE2. PLoS ONE 2014, 9, e103629. [Google Scholar] [CrossRef]
- Vogl, T.; Stratis, A.; Wixler, V.; Völler, T.; Thurainayagam, S.; Jorch, S.K.; Zenker, S.; Dreiling, A.; Chakraborty, D.; Fröhling, M.; et al. Autoinhibitory regulation of S100A8/S100A9 alarmin activity locally restricts sterile inflammation. J. Clin. Investig. 2018, 128, 1852–1866. [Google Scholar] [CrossRef]
- Guo, Q.; Zhao, Y.; Li, J.; Liu, J.; Yang, X.; Guo, X.; Kuang, M.; Xia, H.; Zhang, Z.; Cao, L.; et al. Induction of alarmin S100A8/A9 mediates activation of aberrant neutrophils in the pathogenesis of COVID-19. Cell Host Microbe 2021, 29, 222–235. [Google Scholar] [CrossRef]
- Jessop, F.; Schwarz, B.; Scott, D.; Lydia MRoberts, L.; Bohrnsen, E.; Hoidal, J.; Bosio, M. Impairing RAGE signaling promotes survival and limits disease pathogenesis following SARS-CoV-2 infection in mice. JCI Insight. 2022, 7, e155896. [Google Scholar] [CrossRef]
- Sohn, K.M.; Lee, S.G.; Kim, H.J.; Cheon, S.; Jeong, H.; Lee, J.; Kim, I.S.; Silwal, P.; Kim, Y.J.; Paik, S.; et al. COVID-19 patients upregulate toll-like receptor 4-mediated inflammatory signalling that mimics bacterial sepsis. bioRxiv 2020, preprint. [Google Scholar] [CrossRef]
- O’Kelly, B.; Vidal, L.; McHugh, T.; Woo, J.; Avramovic, G.; Lambert, J. Safety and efficacy of low dose naltrexone in a long covid cohort; an interventional pre-post study. Brain Behav. Immun.—Health 2022, 24, 100485. [Google Scholar] [CrossRef] [PubMed]
- Chulkina, M.; Pichugin, A.; Ataullakhanov, R. Pharmaceutical grade synthetic peptide Thr-Glu-Lys-Lys-Arg-Arg-Glu-Thr-Val-Glu-A,rg-Glu-Lys-Glu ameliorates DSS-induced murine colitis by reducing the number and pro-inflammatory activity of colon tissue-infiltrating Ly6G+ granulocytes and Ly6C+ monocytes. Peptides 2020, 132, 170364. [Google Scholar] [CrossRef] [PubMed]
- Holms, R.D.; Ataullakhanov, R.I. Ezrin Peptide Therapy from HIV to COVID: Inhibition of Inflammation and Amplification of Adaptive Anti-Viral Immunity. Int. J. Mol. Sci. 2021, 22, 11688. [Google Scholar] [CrossRef] [PubMed]
- Holms, R.D. The COVID-19 Cell Signalling Problem: Spike, RAGE, PKC, p38, NFkB & IL-6 Hyper-Expression and the Human Ezrin Peptide, VIP, PKA-CREB Solution. Immuno 2022, 2, 260–282. [Google Scholar] [CrossRef]
- Holms, R.D.; Ataullakhanov, R.I. Ezrin Peptide Therapy: A Potential Treatment for COVID. J. Bioprocess. Biotech. 2021, 12, 1–4. [Google Scholar]


{kind=link}
{kind=link}
| At least 1 sequela: | 1st infect, 357 | re-infect: 553 | per 1000 persons at 6 months |
| Mental problem: | 1st infect, 146 | re-infect: 227 | per 1000 persons at 6 months |
| Neurologic: | 1st infect, 100 | re-infect: 136 | per 1000 persons at 6 months |
| Hospitalization: | 1st infect, 52 | re-infect: 148 | per 1000 persons at 6 months |
| Musculo-skeletal: | 1st infect, 46 | re-infect: 59 | per 1000 persons at 6 months |
| Gastro-intestinal: | 1st infect, 42 | re-infect: 69 | per 1000 persons at 6 months |
| Cardio-vascular: | 1st infect, 38 | re-infect: 88 | per 1000 persons at 6 months |
| Diabetic: | 1st infect, 36 | re-infect: 58 | per 1000 persons at 6 months |
| Pulmonary: | 1st infect, 35 | re-infect: 86 | per 1000 persons at 6 months |
| Fatigue: | 1st infect, 25 | re-infect: 58 | per 1000 persons at 6 months |
| Coagulation-blood: | 1st infect, 22 | re-infect: 48 | per 1000 persons at 6 months |
| Kidneys: | 1st infect, 21 | re-infect: 35 | per 1000 persons at 6 months |
| All-cause mortality: | 1st infect, 21 | re-infect: 45 | per 1000 persons at 6 months |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holms, R.D. Long COVID (PASC) Is Maintained by a Self-Sustaining Pro-Inflammatory TLR4/RAGE-Loop of S100A8/A9 > TLR4/RAGE Signalling, Inducing Chronic Expression of IL-1b, IL-6 and TNFa: Anti-Inflammatory Ezrin Peptides as Potential Therapy. Immuno 2022, 2, 512-533. https://doi.org/10.3390/immuno2030033
Holms RD. Long COVID (PASC) Is Maintained by a Self-Sustaining Pro-Inflammatory TLR4/RAGE-Loop of S100A8/A9 > TLR4/RAGE Signalling, Inducing Chronic Expression of IL-1b, IL-6 and TNFa: Anti-Inflammatory Ezrin Peptides as Potential Therapy. Immuno. 2022; 2(3):512-533. https://doi.org/10.3390/immuno2030033
Chicago/Turabian StyleHolms, Rupert Donald. 2022. "Long COVID (PASC) Is Maintained by a Self-Sustaining Pro-Inflammatory TLR4/RAGE-Loop of S100A8/A9 > TLR4/RAGE Signalling, Inducing Chronic Expression of IL-1b, IL-6 and TNFa: Anti-Inflammatory Ezrin Peptides as Potential Therapy" Immuno 2, no. 3: 512-533. https://doi.org/10.3390/immuno2030033
APA StyleHolms, R. D. (2022). Long COVID (PASC) Is Maintained by a Self-Sustaining Pro-Inflammatory TLR4/RAGE-Loop of S100A8/A9 > TLR4/RAGE Signalling, Inducing Chronic Expression of IL-1b, IL-6 and TNFa: Anti-Inflammatory Ezrin Peptides as Potential Therapy. Immuno, 2(3), 512-533. https://doi.org/10.3390/immuno2030033