
Recent Progress on the Roles of Regulatory T Cells in IgG4-Related Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. History of Regulatory T cells
3. Regulatory T Cells in Animal Models of IgG4-Related Disease
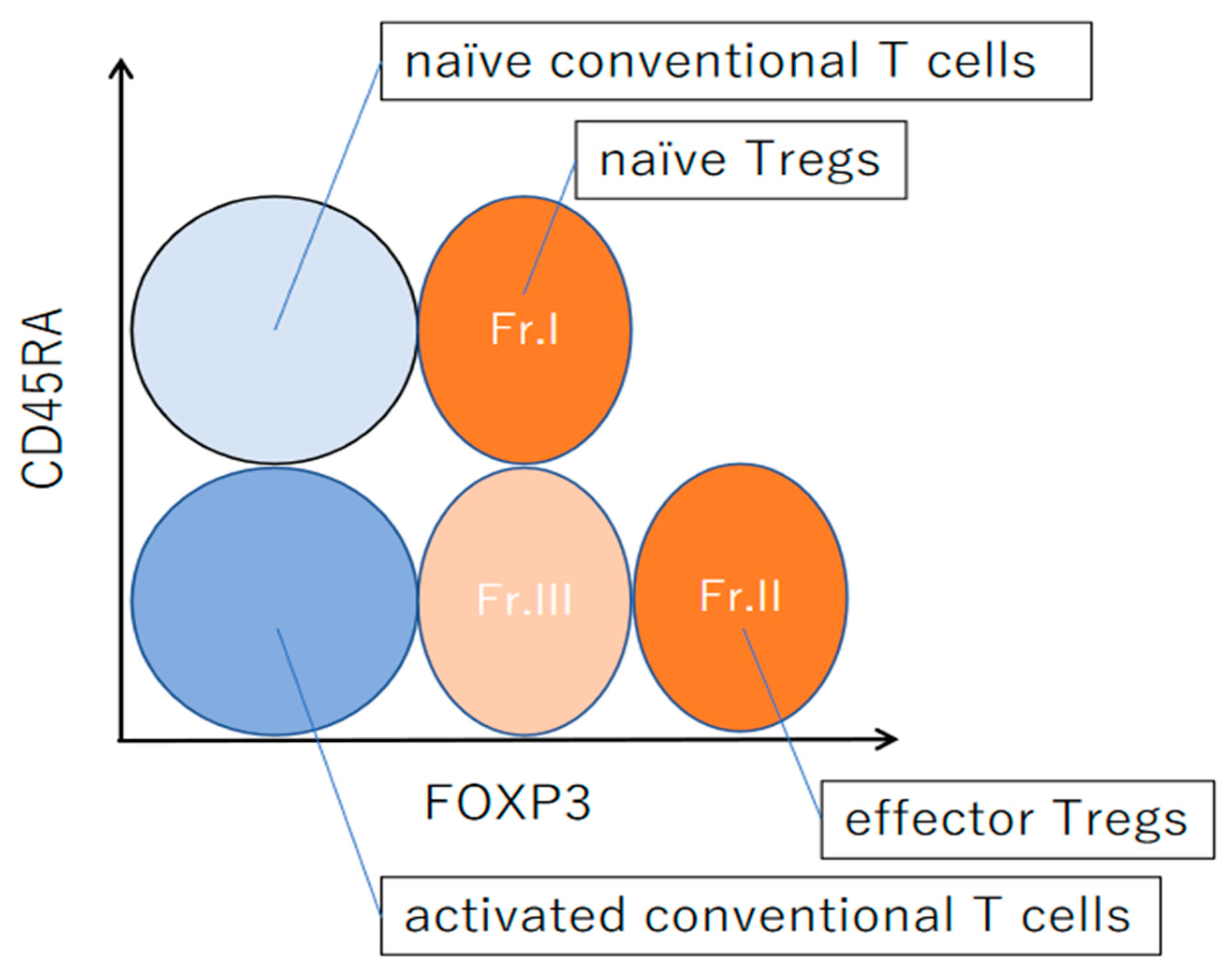
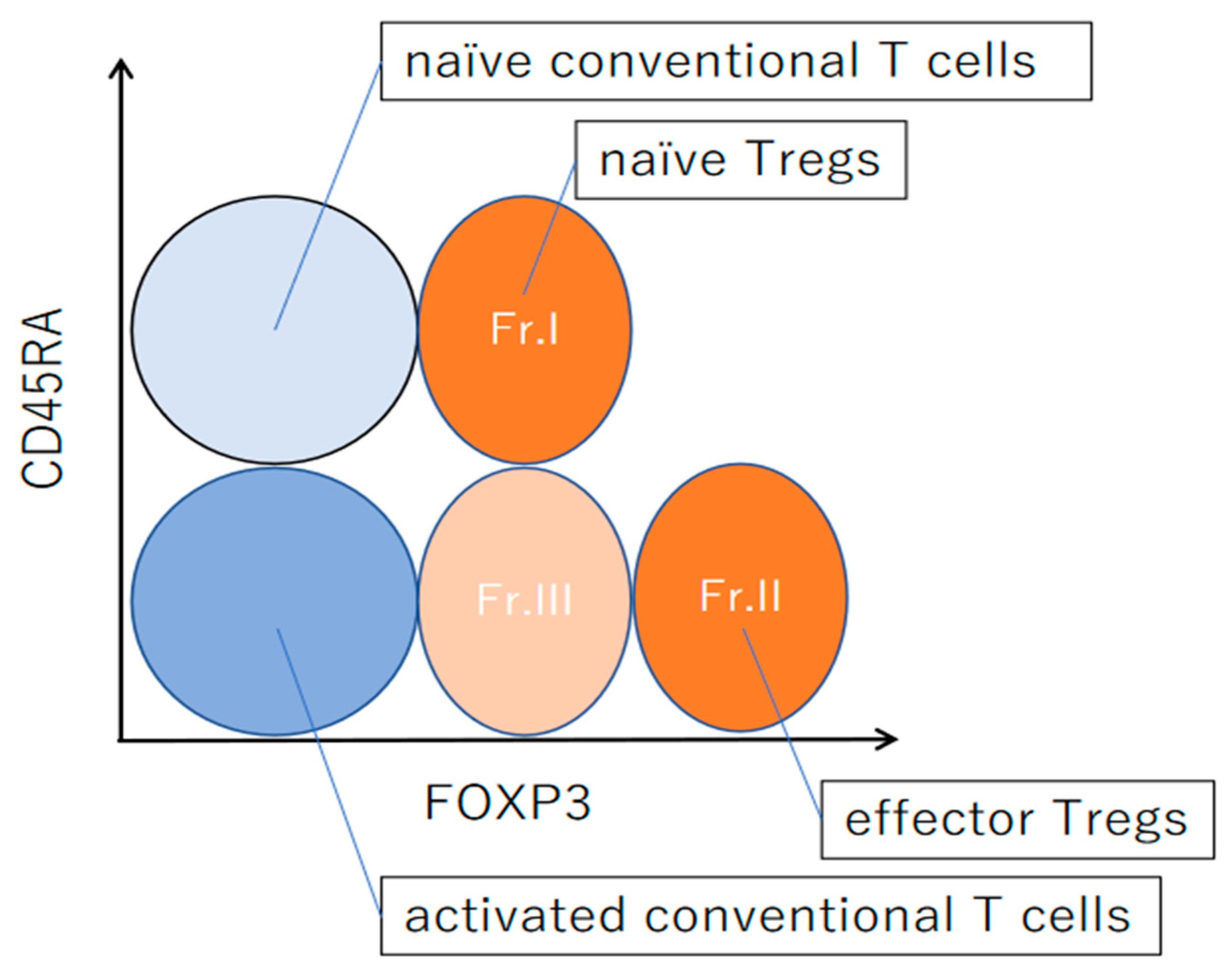
4. Differentiation of Regulatory T Cells in Humans
5. Regulatory T Cells in IgG4-Related Disease
5.1. Circulating Regulatory T Cells in Patients with IgG4-Related Disease
5.2. Regulatory T Cells in the Involved Organ in IgG4-Related Disease
6. Functional Disorders of Regulatory T Cells in IgG4-Related Disease
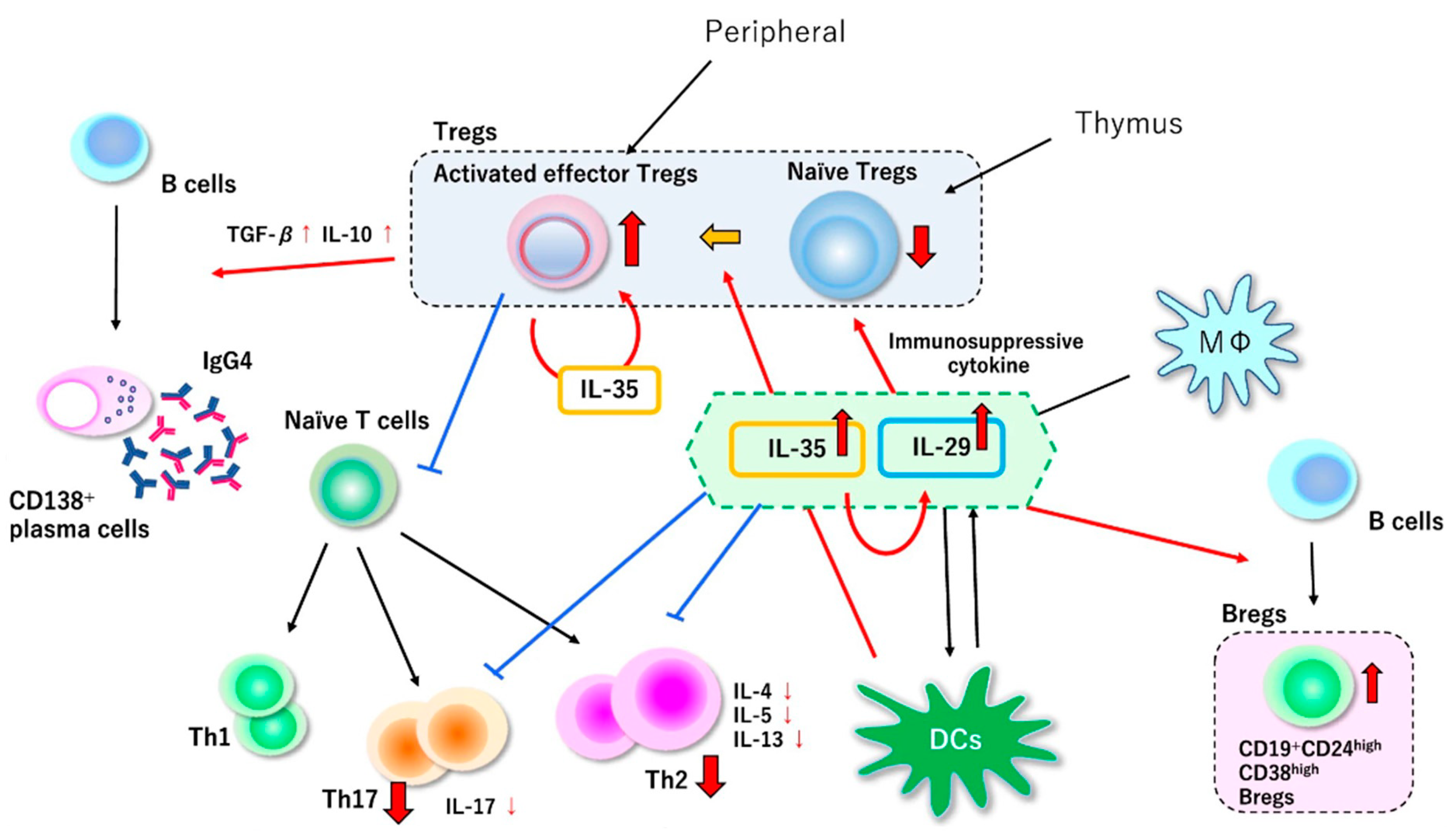
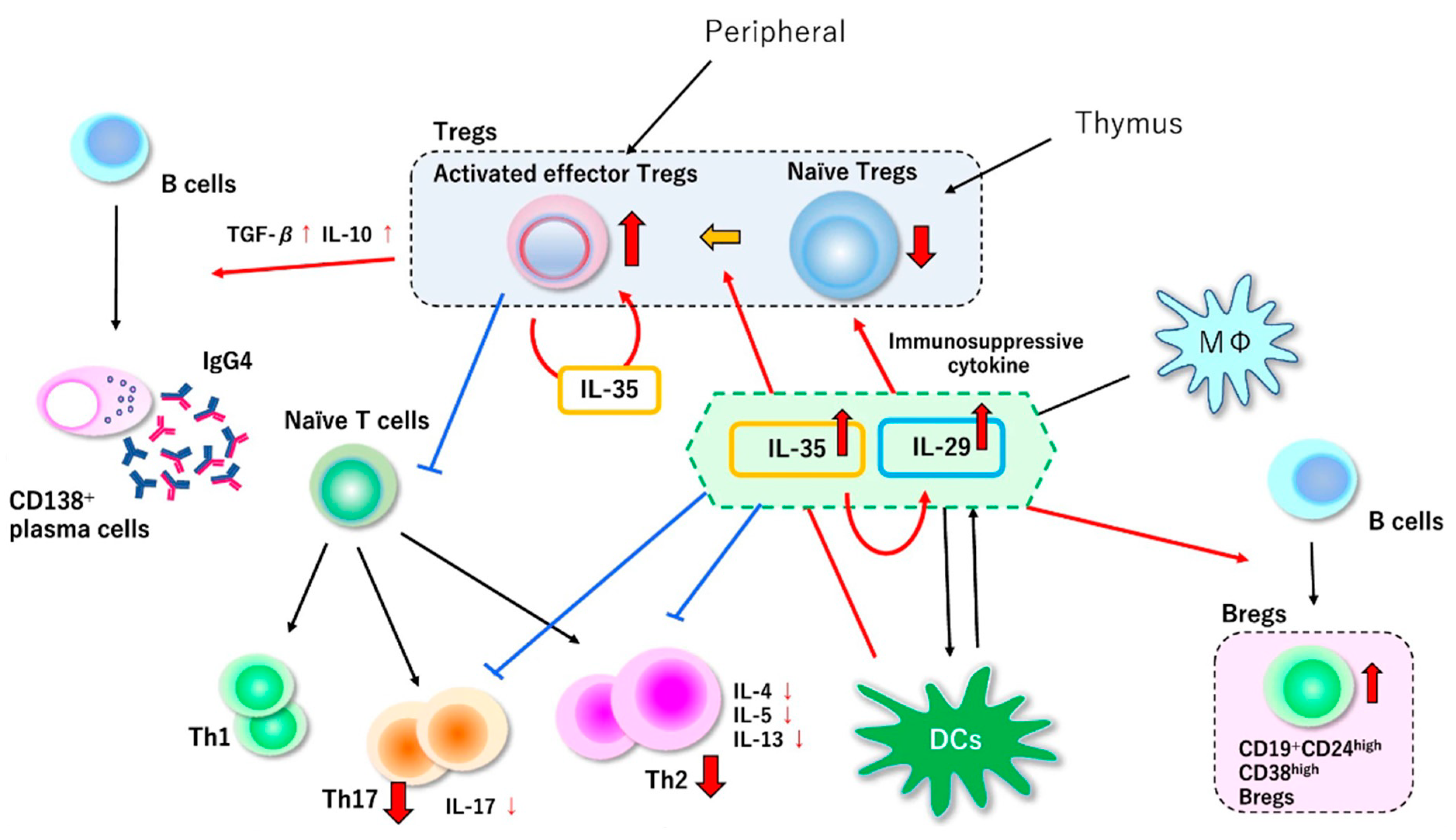
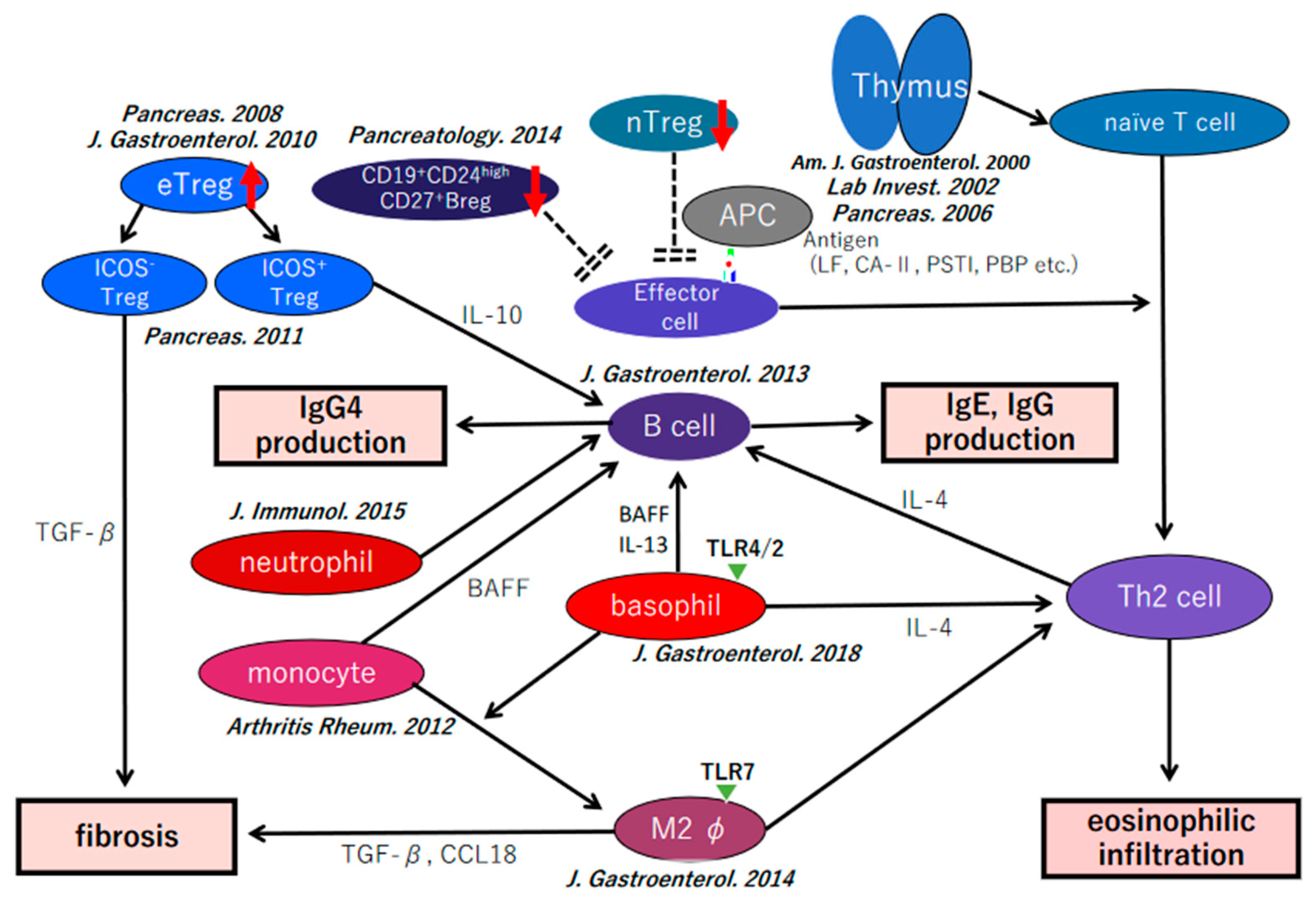
7. Our Hypothesis of the Pathophysiology of IgG4-Related Disease
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kawaguchi, K.; Koike, M.; Tsuruta, K.; Okamoto, A.; Tabata, I.; Fujita, N. Lymphoplasmacytic sclerosing pancreatitis with cholangitis: A variant of primary sclerosing cholangitis extensively involving pancreas. Hum. Pathol. 1991, 22, 387–395. [Google Scholar] [CrossRef]
- Umehara, H.; Okazaki, K.; Kawa, S.; Takahashi, H.; Goto, H.; Matsui, S.; Ishizaka, N.; Akamizu, T.; Sato, Y.; Kawano, M.; et al. The 2020 revised comprehensive diagnostic (RCD) criteria for IgG4-RD. Mod. Rheumatol. 2021, 31, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Hamano, H.; Kawa, S.; Horiuchi, A.; Unno, H.; Furuya, N.; Akamatsu, T.; Fukushima, M.; Nikaido, T.; Nakayama, K.; Usuda, N.; et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N. Engl. J. Med. 2001, 344, 732–738. [Google Scholar] [CrossRef]
- Yoshida, K.; Toki, F.; Takeuchi, T.; Watanabe, S.; Shiratori, K.; Hayashi, N. Chronic pancreatitis caused by an autoimmune abnormality. Dig. Dis. Sci. 1995, 40, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Nakajima, H.; Egawa, N.; Funata, N.; Tsuruta, K.; Okamoto, A. IgG4-related sclerosing disease incorporating sclerosing pancreatitis, cholangitis, sialadenitis and retroperitoneal fibrosis with lymphadenopathy. Pancreatology 2006, 6, 132–137. [Google Scholar] [CrossRef]
- Yamamoto, M.; Takahashi, H.; Ohara, M.; Suzuki, C.; Naishiro, Y.; Yamamoto, H.; Shinomura, Y.; Imai, K. A new conceptualization for Mikulicz’s disease as an IgG4-related plasmacytic disease. Mod. Rheumatol. 2006, 16, 335–340. [Google Scholar] [CrossRef]
- Masaki, Y.; Dong, L.; Kurose, N.; Kitagawa, K.; Morikawa, Y.; Yamamoto, M.; Takahashi, H.; Shinomura, Y.; Imai, K.; Saeki, T.; et al. Proposal for a new clinical entity, IgG4-positive multiorgan lymphoproliferative syndrome: Analysis of 64 cases of IgG4-related disorders. Ann. Rheum. Dis. 2009, 68, 1310–1315. [Google Scholar] [CrossRef]
- Umehara, H.; Okazaki, K.; Masaki, Y.; Kawano, M.; Yamamoto, M.; Saeki, T.; Matsui, S.; Sumida, T.; Mimori, T.; Tanaka, Y.; et al. A novel clinical entity, IgG4-related disease (IgG4RD): General concept and details. Mod. Rheumatol. 2012, 22, 1–14. [Google Scholar] [CrossRef]
- Shimosegawa, T.; Chari, S.T.; Frulloni, L.; Kamisawa, T.; Kawa, S.; Mino-Kenudson, M.; Kim, M.H.; Klöppel, G.; Lerch, M.M.; Löhr, M.; et al. International consensus diagnostic criteria for autoimmune pancreatitis: Guidelines of the International Association of Pancreatology. Pancreas 2011, 40, 352–358. [Google Scholar] [CrossRef]
- Notohara, K.; Burgart, L.J.; Yadav, D.; Chari, S.; Smyrk, T.C. Idiopathic chronic pancreatitis with periductal lymphoplasmacytic infiltration: Clinicopathologic features of 35 cases. Am. J. Surg. Pathol. 2003, 27, 1119–1127. [Google Scholar] [CrossRef]
- Zamboni, G.; Lüttges, J.; Capelli, P.; Frulloni, L.; Cavallini, G.; Pederzoli, P.; Leins, A.; Longnecker, D.; Klöppel, G. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: A study on 53 resection specimens and 9 biopsy specimens. Virchows Arch. 2004, 445, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; Vieira, P.; Fiorentino, D.F.; Trounstine, M.L.; Khan, T.A.; Mosmann, T.R. Homology of cytokine synthesis inhibitory factor (IL-10) to the Epstein–Barr virus gene BCRFI. Science 1990, 248, 1230–1234. [Google Scholar] [CrossRef] [PubMed]
- Zen, Y.; Fujii, T.; Harada, K.; Kawano, M.; Yamada, K.; Takahira, M.; Nakanuma, Y. Th2 and regulatory immune reactions are increased in immunoglobin G4-related sclerosing pancreatitis and cholangitis. Hepatology 2007, 45, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Groux, H.; O’Garra, A.; Bigler, M.; Rouleau, M.; Antonenko, S.; de Vries, J.E.; Roncarolo, M.G. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 1997, 389, 737–742. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Mizoguchi, A.; Mizoguchi, E.; Takedatsu, H.; Blumberg, R.S.; Bhan, A.K. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity 2002, 16, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Fujita, S.; Seino, K.; Sato, K.; Sato, Y.; Eizumi, K.; Yamashita, N.; Taniguchi, M.; Sato, K. Regulatory dendritic cells act as regulators of acute lethal systemic inflammatory response. Blood 2006, 107, 3656–3664. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, S.; Kurishima, A.; Inaba, M.; Ando, Y.; Fukui, T.; Uchida, K.; Nishio, A.; Iwai, H.; Yokoi, T.; Ito, T.; et al. Amelioration of 2,4,6-trinitrobenzene sulfonic acid-induced colitis in mice by immunoregulatory dendritic cells. J. Gastroenterol. 2011, 46, 1368–1381. [Google Scholar] [CrossRef]
- Lechner, A.; Bohnacker, S.; Esser-von Bieren, J. Macrophage regulation & function in helminth infection. Semin Immunol. 2021, 53, 101526. [Google Scholar]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T cells and Human Disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar]
- Baecher-Allan, C.; Brown, J.A.; Freeman, G.J.; Hafler, D.A. CD4+CD25 high regulatory cells in human peripheral blood. J. Immunol. 2001, 167, 1245–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieckmann, D.; Plottner, H.; Berchtold, S.; Berger, T.; Schuler, G. Ex vivo isolation and characterization of CD4(+) CD25(+) T cells with regulatory properties from human blood. J. Exp. Med. 2001, 193, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Jonuleit, H.; Schmitt, E.; Stassen, M.; Tuettenberg, A.; Knop, J.; Enk, A.H. Identification and functional characterization of human CD4(+) CD25(+) T cells with regulatory properties isolated from peripheral blood. J. Exp. Med. 2001, 193, 1285–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levings, M.K.; Sangregorio, R.; Roncarolo, M.G. Human CD25(+) CD4(+) T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J. Exp. Med. 2001, 193, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [Green Version]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Khattri, R.; Cox, T.; Yasayko, S.A.; Ramsdell, F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 2003, 4, 337–342. [Google Scholar] [CrossRef]
- Morikawa, H.; Sakaguchi, S. Genetic and epigenetic basis of Treg cell development and function: From a FoxP3-centered view to an epigenome-defined view of natural Treg cells. Immunol. Rev. 2014, 259, 192–205. [Google Scholar] [CrossRef]
- Katoh, H.; Zheng, P.; Liu, Y. FOXP3: Genetic and epigenetic implications for autoimmunity. J. Autoimmun. 2013, 41, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular mechanisms of Treg-mediated T cell suppression. Front. Immunol. 2012, 3, 51. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T. Regulatory T cells: How do they suppress immune responses? Int. Immunol. 2009, 21, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Roncarolo, M.G.; Gregori, S.; Battaglia, M.; Bacchetta, R.; Fleischhauer, K.; Levings, M.K. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol. Rev. 2006, 212, 28–50. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Miyara, M.; Costantino, C.M.; Hafler, D.A. FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010, 10, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Crispin, J.C.; Martínez, A.; Alcocer-Varela, J. Quantification of regulatory T cells in patients with systemic lupus erythematosus. J. Autoimmun. 2003, 21, 273–276. [Google Scholar] [CrossRef]
- Cao, D.; van Vollenhoven, R.; Klareskog, L.; Trollmo, C.; Malmström, V. CD25bright CD4+ regulatory T cells are enriched in inflamed joints of patients with chronic rheumatic disease. Arthritis Res. Ther. 2004, 6, 335–346. [Google Scholar] [CrossRef] [Green Version]
- De Kleer, I.M.; Wedderburn, L.R.; Taams, L.S.; Patel, A.; Varsani, H.; Klein, M.; de Jager, W.; Pugayung, G.; Giannoni, F.; Rijkers, G.; et al. CD4CD25bright regulatory T cells actively regulate inflammation in the joints of patients with the remitting from of juvenile idiopathic arthritis. J. Immunol. 2004, 172, 6435–6443. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Malmström, V.; Baecher-Allan, C.; Hafler, D.; Klareskog, L.; Trollmo, C. Isolation and functional characterization of regulatory CD25bright CD4+ T cells from the target organ of patients with rheumatoid arthritis. Eur. J. Immunol. 2003, 33, 215–223. [Google Scholar] [CrossRef]
- Longhi, M.S.; Ma, Y.; Bogdanos, D.P.; Cheeseman, P.; Mieli-Vergani, G.; Vergani, D. Impairment of CD4+CD25+ regulatory T cells in autoimmune liver disease. J. Hepatol. 2004, 41, 31–37. [Google Scholar] [CrossRef]
- Boyer, O.; Saadoun, D.; Abriol, J.; Dodille, M.; Piette, J.C.; Cacoub, P.; Klatzmann, D. CD4+CD25+ regulatory T-cell deficiency in patients with hepatitis C-mixed cryoglobulinemia vasculitis. Blood 2004, 103, 3428–3430. [Google Scholar] [CrossRef]
- Furuno, K.; Yuge, T.; Kusuhara, K.; Takada, H.; Nishio, H.; Khajoee, V.; Ohno, T.; Hara, T. CD25+CD4+ regulatory T cells in patients with Kawasaki disease. J. Pediatr. 2004, 145, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Lindley, S.; Dayan, C.M.; Bishop, A.; Roep, B.O.; Peakman, M.; Tree, T.I. Defective suppressor function in CD4(+) CD25(+) T-cells from patients with type I diabetes. Diabetes 2005, 193, 1303–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putnam, A.L.; Vendrame, F.; Dotta, F.; Gottlieb, P.A. CD4+CD25high regulatory T cells in human autoimmune diabetes. J. Autoimmun. 2005, 24, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Putheti, P.; Pettersson, A.; Soderstrom, M.; Link, H.; Huang, Y.M. Circulating CD4+CD25+ T regulatory cells are not altered in multiple sclerosis and unaffected by disease-modulating drugs. J. Clin. Immunol. 2004, 24, 155–161. [Google Scholar] [CrossRef]
- Viglietta, V.; Baecher-Allan, C.; Weiner, H.L.; Hafler, D.A. Loss of functional suppression by CD4+CD25+ regulatory T patients with multiple sclerosis. J. Exp. Med. 2004, 199, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Gottenberg, J.E.; Lavie, F.; Abbed, K.; Gasnault, J.; Le Nevot, E.; Delfraissy, J.F.; Taoufik, Y.; Mariette, X. CD4 CD25high regulatory T cells are not impaired in patients with primary Sjögren’s syndrome. J. Autoimmun. 2005, 24, 235–242. [Google Scholar] [CrossRef]
- Balandina, A.; Lecart, S.; Dartevelle, P.; Saoudi, A.; Berrih-Aknin, S. Functional defect of regulatory CD4+CD25+ T cells in the thymus of patients with autoimmune Myasthenia Gravis. Blood 2005, 105, 734–741. [Google Scholar] [CrossRef] [Green Version]
- Fattorossi, A.; Battaglia, A.; Buzzonetti, A.; Ciaraffa, F.; Scambia, G.; Evoli, A. Circulating and thymic CD4+CD25+ T regulatory cells in myasthenia gravis: Effect of immunosuppressive treatment. Immunology 2005, 116, 134–141. [Google Scholar] [CrossRef]
- Van Amelsfort, J.M.; Jacobs, K.M.; Bijlsma, J.W.; Bijlsma, J.W.; Lafeber, F.P.; Taams, L.S. CD4+CD25+ regulatory T cells in rheumatoid arthritis: Differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum. 2004, 50, 2775–2785. [Google Scholar] [CrossRef]
- Ehrenstein, M.R.; Evans, J.G.; Singh, A.; Moore, S.; Warnes, G.; Isenberg, D.A.; Mauri, C. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNF alpha therapy. J. Exp. Med. 2004, 200, 277–285. [Google Scholar] [CrossRef]
- Möttönen, M.; Heikkinen, J.; Mustonen, I.; Isomäki, P.; Luukkainen, R.; Lassila, O. CD4+ CD25+ T cells with the phenotypic and functional characteristics of regulatory T cells are enriched in the synovial fluid of patients with rheumatoid arthritis. Clin. Exp. Immunol. 2004, 140, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Vigna-Pérez, M.; Abud-Mendoza, C.; Portillo-Salazar, H.; Alvarado-Sánchez, B.; Cuevas-Orta, E.; Moreno-Valdés, R.; Baranda, L.; Paredes-Saharopulos, O.; González-Amaro, R. Immune effects of therapy with adalimumab in patients with rheumatoid arthritis. Clin. Exp. Immunol. 2005, 141, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Makita, S.; Kanai, T.; Oshima, S.; Uraushihara, K.; Totsuka, T.; Sawada, T.; Nakamura, T.; Koganei, K.; Fukushima, T.; Watanabe, M. CD4+CD25bright T cells in human intestinal lamina propria as regulatory cells. J. Immunol. 2004, 173, 3119–3130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maul, J.; Loddenkemper, C.; Mundt, P.; Berg, E.; Giese, T.; Stallmach, A.; Zeitz, M.; Duchmann, R. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology 2005, 128, 1868–1878. [Google Scholar] [CrossRef]
- Nakajima, T.; Ueki-Maruyama, K.; Oda, T.; Ohsawa, Y.; Ito, H.; Seymour, G.J.; Yamazaki, K. Regulatory T-cells infiltrate periodontal disease tissues. J. Dent. Res. 2005, 84, 639–643. [Google Scholar] [CrossRef] [Green Version]
- Okui, T.; Ito, H.; Honda, T.; Amanuma, R.; Yoshie, H.; Yamazaki, K. Characterization of CD4+ FOXP3+ T-cell clones established from chronic inflammatory lesions. Oral Microbiol. Immunol. 2008, 23, 49–54. [Google Scholar] [CrossRef]
- Asada, M.; Nishio, A.; Uchida, K.; Kido, M.; Ueno, S.; Uza, N.; Kiriya, K.; Inoue, S.; Kitamura, H.; Ohashi, S.; et al. Identification of a novel autoantibody against pancreatic secretory trypsin inhibitor in patients with autoimmune pancreatitis. Pancreas 2006, 33, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Uchida, K.; Okazaki, K.; Nishi, T.; Uose, S.; Nakase, H.; Ohana, M.; Matsushima, Y.; Omori, K.; Chiba, T. Experimental immune-mediated pancreatitis in neonatally thymectomized mice immunized with carbonic anhydrase II and lactoferrin. Lab. Investig. 2002, 82, 411–424. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, Y.; Inaba, M.; Tsuda, M.; Quan, G.K.; Omae, M.; Ando, Y.; Uchida, K.; Okazaki, K.; Ikehara, S. The Wistar Bonn Kobori rat, a unique animal model for autoimmune pancreatitis with extrapancreatic exocrinopathy. Clin. Exp. Immunol. 2008, 152, 1–12. [Google Scholar] [CrossRef]
- Bowman, M.; Holt, G. Selective enhancement of systemic Th1 immunity in immunologically immature rats with an orally administered bacterial extract. Infect Immun. 2001, 69, 3719–3727. [Google Scholar] [CrossRef] [Green Version]
- Ueda, Y.; Katagiri, K.; Tomiyama, T.; Yasuda, K.; Habiro, K.; Katakai, T.; Ikehara, S.; Matsumoto, M.; Kinashi, T. Mst1 regulates integrin-dependent thymocyte trafficking and antigen recognition in the thymus. Nat. Commun. 2012, 3, 1098. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Ueda, Y.; Katakai, T.; Kondo, N.; Okazaki, K.; Kinashi, T. Antigen-specific suppression and immunological synapse formation by regulatory T cells require the Mst1 kinase. PLoS ONE 2013, 8, e73874. [Google Scholar] [CrossRef] [PubMed]
- Yamashina, M.; Nishio, A.; Nakayama, S.; Okazaki, T.; Uchida, K.; Fukui, T.; Okazaki, K. Comparative study on experimental autoimmune pancreatitis and its extrapancreatic involvement in mice. Pancreas 2012, 41, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Koyabu, M.; Uchida, K.; Sakaguchi, Y.; Fukata, N.; Kusuda, T.; Miyoshi, H.; Yoshida, K.; Sumimoto, K.; Mitsuyama, T.; Fukui, T.; et al. Possible involvement of Foxp3(+) regulatory T cells in the development of immune-mediated pancreatitis in MRL/Mp mice treated with polyinosinic:polycytidylic acid. Int. J. Rheumatol. 2013, 2013, 367325. [Google Scholar] [CrossRef]
- Seddiki, N.; Santner-Nanan, B.; Tangye, S.G.; Alexander, S.I.; Solomon, M.; Lee, S.; Nanan, R.; Fazekas de Saint Groth, B. Persistence of naive CD45RA+ regulatory T cells in adult life. Blood 2006, 107, 2830–2838. [Google Scholar] [CrossRef] [Green Version]
- Fritzsching, B.; Oberle, N.; Pauly, E.; Geffers, R.; Buer, J.; Poschl, J.; Krammer, P.; Linderkamp, O.; Suri-Payer, E. Naive regulatory T cells: A novel subpopulation defined by resistance toward CD95L-mediated cell death. Blood 2006, 108, 3371–3378. [Google Scholar] [CrossRef]
- Miyara, M.; Yoshioka, Y.; Kitoh, A.; Shima, T.; Wing, K.; Niwa, A.; Parizot, C.; Taflin, C.; Heike, T.; Valeyre, D.; et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009, 30, 899–911. [Google Scholar] [CrossRef] [Green Version]
- Wing, J.B.; Kitagawa, Y.; Locci, M.; Hume, H.; Tay, C.; Morita, T.; Kidani, Y.; Matsuda, K.; Inoue, T.; Kurosaki, T.; et al. A distinct subpopulation of CD25− T- follicular regulatory cells localizes in the germinal centers. Proc. Natl. Acad. Sci. USA 2017, 114, E6400–E6409. [Google Scholar] [CrossRef] [Green Version]
- Abbas, A.K.; Benoist, C.; Bluestone, J.A.; Campbell, D.J.; Ghosh, S.; Hori, S.; Jiang, S.; Kuchroo, V.K.; Mathis, D.; Roncarolo, M.G.; et al. Regulatory T cells: Recommendations to simplify the nomenclature. Nat. Immunol. 2013, 14, 307–308. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Miyoshi, H.; Uchida, K.; Taniguchi, T.; Yazumi, S.; Matsushita, M.; Takaoka, M.; Okazaki, K. Circulating naïve and CD4+CD25high regulatory T cells in patients with autoimmune pancreatitis. Pancreas 2008, 36, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ito, F.; Kamekura, R.; Yamamoto, M.; Takano, K.; Takaki, H.; Yabe, H.; Ikegami, I.; Shigehara, K.; Himi, T.; Takahashi, H.; et al. IL-10 + T follicular regulatory cells are associated with the pathogenesis of IgG4-related disease. Immunol Lett. 2019, 207, 56–563. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Hanabuchi, S.; Wang, Y.H.; Park, W.R.; Arima, K.; Bover, L.; Qin, F.X.; Gilliet, M.; Liu, Y.J. Two functional subsets of FOXP3+ regulatory T cells in human thymus and periphery. Immunity 2008, 28, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Kusuda, T.; Uchida, K.; Miyoshi, H.; Koyabu, M.; Satoi, S.; Takaoka, M.; Shikata, N.; Uemura, Y.; Okazaki, K. Involvement of inducible costimulator- and interleukin 10-positive regulatory T cells in the development of IgG4-related autoimmune pancreatitis. Pancreas 2011, 40, 1120–1130. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Delgoffe, G.M.; Guy, C.S.; Vignali, K.M.; Chaturvedi, V.; Fairweather, D.; Satoskar, A.R.; Garcia, K.C.; Hunter, C.A.; Drake, C.G.; et al. The composition and signaling of the IL-35 receptor are unconventional. Nat. Immunol. 2012, 13, 290–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Tanaka, T.; Nakamaru, K.; Tomiyama, T.; Yamaguchi, T.; Ando, Y.; Ikeura, T.; Fukui, T.; Uchida, K.; Nishio, A.; et al. Interleukin-35 promotes the differentiation of regulatory T cells and suppresses Th2 response in IgG4-related type 1 autoimmune pancreatitis. J. Gastroenterol. 2020, 55, 789–799. [Google Scholar] [CrossRef]
- Sumimoto, K.; Uchida, K.; Kusuda, T.; Mitsuyama, T.; Sakaguchi, Y.; Fukui, T.; Matsushita, M.; Takaoka, M.; Nishio, A.; Okazaki, K. The role of CD19+ CD24high CD38high and CD19+ CD24high CD27+ regulatory B cells in patients with type 1 autoimmune pancreatitis. Pancreatology 2014, 14, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Khosroshahi, A.; Carruthers, M.N.; Deshpande, V.; Unizony, S.; Bloch, D.B.; Stone, J.H. Rituximab for the treatment of IgG4-related disease: Lessons from 10 consecutive patients. Medicine 2012, 91, 57–66. [Google Scholar] [CrossRef]
- Mattoo, H.; Mahajan, V.S.; Della-Torre, E.; Sekigami, Y.; Carruthers, M.; Wallace, Z.S.; Deshpande, V.; Stone, J.H.; Pillai, S. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J. Allergy Clin. Immunol. 2014, 134, 679–687. [Google Scholar] [CrossRef] [Green Version]
- Sage, P.T.; Sharpe, A.H. T follicular regulatory cells in the regulation of B cell responses. Trends Immunol. 2015, 36, 410–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyabu, M.; Uchida, K.; Miyoshi, H.; Sakaguchi, Y.; Fukui, T.; Ikeda, H.; Takaoka, M.; Hirohara, J.; Nishio, A.; Uemura, Y.; et al. Analysis of regulatory T cells and IgG4-positive plasma cells among patients of IgG4-related sclerosing cholangitis and autoimmune liver diseases. J. Gastroenterol. 2010, 45, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Moriyama, M.; Nakashima, H.; Miyake, K.; Hayashida, J.N.; Maehara, T.; Shinozaki, S.; Kubo, Y.; Nakamura, S. Th2 and regulatory immune reactions contribute to IgG4 production and the initiation of Mikulicz disease. Arthritis Rheum. 2012, 64, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, I.; Yamada, K.; Fujii, H.; Inoue, D.; Umehara, H.; Yamagishi, M.; Yamaguchi, Y.; Nagata, M.; Matsumura, M.; Kawano, M. Clinical and histological changes associated with corticosteroid therapy in IgG4-related tubulointerstitial nephritis. Mod. Rheumatol. 2012, 22, 859–870. [Google Scholar] [CrossRef]
- Kawamura, E.; Hisano, S.; Nakashima, H.; Takeshita, M.; Saito, T. Immunohistological analysis for immunological response and mechanism of interstitial fibrosis in IgG4-related kidney disease. Mod. Rheumatol. 2015, 25, 571–578. [Google Scholar] [CrossRef]
- Fukuhara, T.; Tomiyama, T.; Yasuda, K.; Ueda, Y.; Ozaki, Y.; Son, Y.; Nomura, S.; Uchida, K.; Okazaki, K.; Kinashi, T. Hypermethylation of MST1 in IgG4-related autoimmune pancreatitis and rheumatoid arthritis. Biochem. Biophys. Res. Commun. 2015, 463, 968–974. [Google Scholar] [CrossRef]
- Uchida, K.; Okazaki, K. Clinical and pathophysiological aspects of type 1 autoimmune pancreatitis. J. Gastroenterol. 2018, 53, 475–483. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uchida, K. Recent Progress on the Roles of Regulatory T Cells in IgG4-Related Disease. Immuno 2022, 2, 430-442. https://doi.org/10.3390/immuno2020026
Uchida K. Recent Progress on the Roles of Regulatory T Cells in IgG4-Related Disease. Immuno. 2022; 2(2):430-442. https://doi.org/10.3390/immuno2020026
Chicago/Turabian StyleUchida, Kazushige. 2022. "Recent Progress on the Roles of Regulatory T Cells in IgG4-Related Disease" Immuno 2, no. 2: 430-442. https://doi.org/10.3390/immuno2020026
APA StyleUchida, K. (2022). Recent Progress on the Roles of Regulatory T Cells in IgG4-Related Disease. Immuno, 2(2), 430-442. https://doi.org/10.3390/immuno2020026