
Proteogenomics Analysis Reveals Novel Micropeptides in Primary Human Immune Cells

 ,
, 
Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Bioinformatics Analysis
4.2. ORF Cloning and Confocal Microscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Orom, U.A.; Shiekhattar, R. Long non-coding RNAs usher in a new era in the biology of enhancers. Cell 2013, 154, 1190–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Cao, X. Long non-coding RNAs in innate immunity. Cell. Mol. Immunol. 2016, 13, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Zhang, J.; Wei, M.; Liu, B.; Dong, K. Emerging role of long non-coding RNA-encoded micropeptides in cancer. Cancer Cell Int. 2020, 20, 506. [Google Scholar] [CrossRef]
- Niu, L.; Lou, F.; Sun, Y.; Sun, L.; Cai, X.; Liu, Z.; Zhou, H.; Wang, H.; Wang, Z.; Bai, J.; et al. A micropeptide encoded by lncRNA MIR155HG suppresses autoimmune inflammation via modulating antigen presentation. Sci. Adv. 2020, 6, eaaz2059. [Google Scholar] [CrossRef]
- Bhatta, A.; Atianand, M.; Jiang, Z.; Crabtree, J.; Blin, J.; Fitzgerald, K.A. A Mitochondrial Micropeptide Is Required for Activation of the Nlrp3 Inflammasome. J. Immunol. 2020, 204, 428–437. [Google Scholar] [CrossRef]
- Jackson, R.; Kroehling, L.; Khitun, A.; Bailis, W.; Jarret, A.; York, A.G.; Khan, O.M.; Brewer, J.R.; Skadow, M.H.; Duizer, C.; et al. The translation of non-canonical open reading frames controls mucosal immunity. Nature 2018, 564, 434–438. [Google Scholar] [CrossRef]
- Makarewich, C.A.; Olson, E.N. Mining for Micropeptides. Trends Cell Biol. 2017, 27, 685–696. [Google Scholar] [CrossRef]
- Renuse, S.; Chaerkady, R.; Pandey, A. Proteogenomics. Proteomics 2011, 11, 620–630. [Google Scholar] [CrossRef]
- Subbannayya, Y.; Pinto, S.M.; Gowda, H.; Prasad, T.S. Proteogenomics for understanding oncology: Recent advances and future prospects. Expert Rev. Proteom. 2016, 13, 297–308. [Google Scholar] [CrossRef]
- Othoum, G.; Coonrod, E.; Zhao, S.; Dang, H.X.; Maher, C.A. Pan-cancer proteogenomic analysis reveals long and circular non-coding RNAs encoding peptides. NAR Cancer 2020, 2, zcaa015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.X.; Xiao, Q.X.; Liu, Q.; Deng, R.; Bian, J.; Deng, I.B.; Al-Hawwas, M.; Yu, F.X. Microarray Expression Profiles of lncRNAs and mRNAs in Postoperative Cognitive Dysfunction. Front. Neurosci. 2018, 12, 694. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotti, C.; De Mattei, M.; Mazziotta, C.; Taraballi, F.; Rotondo, J.C.; Tognon, M.; Martini, F. Long Non-coding RNAs and MicroRNAs Interplay in Osteogenic Differentiation of Mesenchymal Stem Cells. Front. Cell Dev. Biol. 2021, 9, 646032. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [Green Version]
- Nomura, Y.; Dohmae, N. Discovery of a small protein-encoding cis-regulatory overlapping gene of the tumor suppressor gene Scribble in humans. Commun. Biol. 2021, 4, 1098. [Google Scholar] [CrossRef]
- Liu, C.; Peng, Z.; Li, P.; Fu, H.; Feng, J.; Zhang, Y.; Liu, T.; Liu, Y.; Liu, Q.; Liu, Q.; et al. lncRNA RMST Suppressed GBM Cell Mitophagy through Enhancing FUS SUMOylation. Mol. Ther. Nucleic Acids 2020, 19, 1198–1208. [Google Scholar] [CrossRef]
- Ng, S.Y.; Bogu, G.K.; Soh, B.S.; Stanton, L.W. The long non-coding RNA RMST interacts with SOX2 to regulate neurogenesis. Mol. Cell 2013, 51, 349–359. [Google Scholar] [CrossRef] [Green Version]
- van Alphen, C.; Cucchi, D.G.J.; Cloos, J.; Schelfhorst, T.; Henneman, A.A.; Piersma, S.R.; Pham, T.V.; Knol, J.C.; Jimenez, C.R.; Janssen, J. The influence of delay in mononuclear cell isolation on acute myeloid leukemia phosphorylation profiles. J. Proteom. 2021, 238, 104134. [Google Scholar] [CrossRef]
- Mathieson, T.; Franken, H.; Kosinski, J.; Kurzawa, N.; Zinn, N.; Sweetman, G.; Poeckel, D.; Ratnu, V.S.; Schramm, M.; Becher, I.; et al. Systematic analysis of protein turnover in primary cells. Nat. Commun. 2018, 9, 689. [Google Scholar] [CrossRef] [Green Version]
- de Azambuja Rodrigues, P.M.; Valente, R.H.; Brunoro, G.V.F.; Nakaya, H.T.I.; Araujo-Pereira, M.; Bozza, P.T.; Bozza, F.A.; Trugilho, M.R.O. Proteomics reveals disturbances in the immune response and energy metabolism of monocytes from patients with septic shock. Sci. Rep. 2021, 11, 15149. [Google Scholar] [CrossRef]
- Muller, M.M.; Lehmann, R.; Klassert, T.E.; Reifenstein, S.; Conrad, T.; Moore, C.; Kuhn, A.; Behnert, A.; Guthke, R.; Driesch, D.; et al. Global analysis of glycoproteins identifies markers of endotoxin tolerant monocytes and GPR84 as a modulator of TNFalpha expression. Sci. Rep. 2017, 7, 838. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, Y.; Jia, L.; Zhang, C.; Cao, W.; Alam, N.; Wang, R.; Wang, W.; Bai, L.; Zhao, S.; et al. TMT-based quantitative proteomic profiling of human monocyte-derived macrophages and foam cells. Proteome Sci. 2022, 20, 1. [Google Scholar] [CrossRef]
- Dietze, R.; Hammoud, M.K.; Gomez-Serrano, M.; Unger, A.; Bieringer, T.; Finkernagel, F.; Sokol, A.M.; Nist, A.; Stiewe, T.; Reinartz, S.; et al. Phosphoproteomics identify arachidonic-acid-regulated signal transduction pathways modulating macrophage functions with implications for ovarian cancer. Theranostics 2021, 11, 1377–1395. [Google Scholar] [CrossRef] [PubMed]
- Dyring-Andersen, B.; Lovendorf, M.B.; Coscia, F.; Santos, A.; Moller, L.B.P.; Colaco, A.R.; Niu, L.; Bzorek, M.; Doll, S.; Andersen, J.L.; et al. Spatially and cell-type resolved quantitative proteomic atlas of healthy human skin. Nat. Commun. 2020, 11, 5587. [Google Scholar] [CrossRef] [PubMed]
- Soderholm, S.; Kainov, D.E.; Ohman, T.; Denisova, O.V.; Schepens, B.; Kulesskiy, E.; Imanishi, S.Y.; Corthals, G.; Hintsanen, P.; Aittokallio, T.; et al. Phosphoproteomics to Characterize Host Response During Influenza A Virus Infection of Human Macrophages. Mol. Cell. Proteom. 2016, 15, 3203–3219. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Gaffrey, M.J.; Thomas, D.G.; Weber, T.J.; Hess, B.M.; Weitz, K.K.; Piehowski, P.D.; Petyuk, V.A.; Moore, R.J.; Qian, W.J.; et al. A proteome-wide assessment of the oxidative stress paradigm for metal and metal-oxide nanomaterials in human macrophages. NanoImpact 2020, 17, 100194. [Google Scholar] [CrossRef] [PubMed]
- Gallud, A.; Kloditz, K.; Ytterberg, J.; Ostberg, N.; Katayama, S.; Skoog, T.; Gogvadze, V.; Chen, Y.Z.; Xue, D.; Moya, S.; et al. Cationic gold nanoparticles elicit mitochondrial dysfunction: A multi-omics study. Sci. Rep. 2019, 9, 4366. [Google Scholar] [CrossRef] [Green Version]
- Nilsen, B.W.; Simon-Santamaria, J.; Ortengren, U.; Jensen, E.; Bruun, J.A.; Michelsen, V.B.; Sorensen, K.K. Dose- and time-dependent effects of triethylene glycol dimethacrylate on the proteome of human THP-1 monocytes. Eur. J. Oral Sci. 2018, 126, 345–358. [Google Scholar] [CrossRef]
- Xia, L.; Wang, T.D.; Shen, S.M.; Zhao, M.; Sun, H.; He, Y.; Xie, L.; Wu, Z.X.; Han, S.F.; Wang, L.S.; et al. Phosphoproteomics study on the activated PKCdelta-induced cell death. J. Proteome Res. 2013, 12, 4280–4301. [Google Scholar] [CrossRef]
- Hijazi, M.; Smith, R.; Rajeeve, V.; Bessant, C.; Cutillas, P.R. Reconstructing kinase network topologies from phosphoproteomics data reveals cancer-associated rewiring. Nat. Biotechnol. 2020, 38, 493–502. [Google Scholar] [CrossRef]
- Chen, Y.; Ho, L.; Tergaonkar, V. sORF-Encoded MicroPeptides: New players in inflammation, metabolism, and precision medicine. Cancer Lett. 2021, 500, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Olexiouk, V.; Van Criekinge, W.; Menschaert, G. An update on sORFs.org: A repository of small ORFs identified by ribosome profiling. Nucleic Acids Res. 2018, 46, D497–D502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, C.; Muller, M.; Pak, H.; Harnett, D.; Huber, F.; Grun, D.; Leleu, M.; Auger, A.; Arnaud, M.; Stevenson, B.J.; et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat. Commun. 2020, 11, 1293. [Google Scholar] [CrossRef] [Green Version]
- Volders, P.J.; Anckaert, J.; Verheggen, K.; Nuytens, J.; Martens, L.; Mestdagh, P.; Vandesompele, J. LNCipedia 5: Towards a reference set of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, D135–D139. [Google Scholar] [CrossRef] [Green Version]
- Lorey, M.B.; Rossi, K.; Eklund, K.K.; Nyman, T.A.; Matikainen, S. Global Characterization of Protein Secretion from Human Macrophages Following Non-canonical Caspase-4/5 Inflammasome Activation. Mol. Cell Proteom. 2017, 16 (Suppl. S1), S187–S199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieckmann, J.C.; Geiger, R.; Hornburg, D.; Wolf, T.; Kveler, K.; Jarrossay, D.; Sallusto, F.; Shen-Orr, S.S.; Lanzavecchia, A.; Mann, M.; et al. Social network architecture of human immune cells unveiled by quantitative proteomics. Nat. Immunol. 2017, 18, 583–593. [Google Scholar] [CrossRef]
- Peng, J.; Elias, J.E.; Thoreen, C.C.; Licklider, L.J.; Gygi, S.P. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: The yeast proteome. J. Proteome Res. 2003, 2, 43–50. [Google Scholar] [CrossRef]
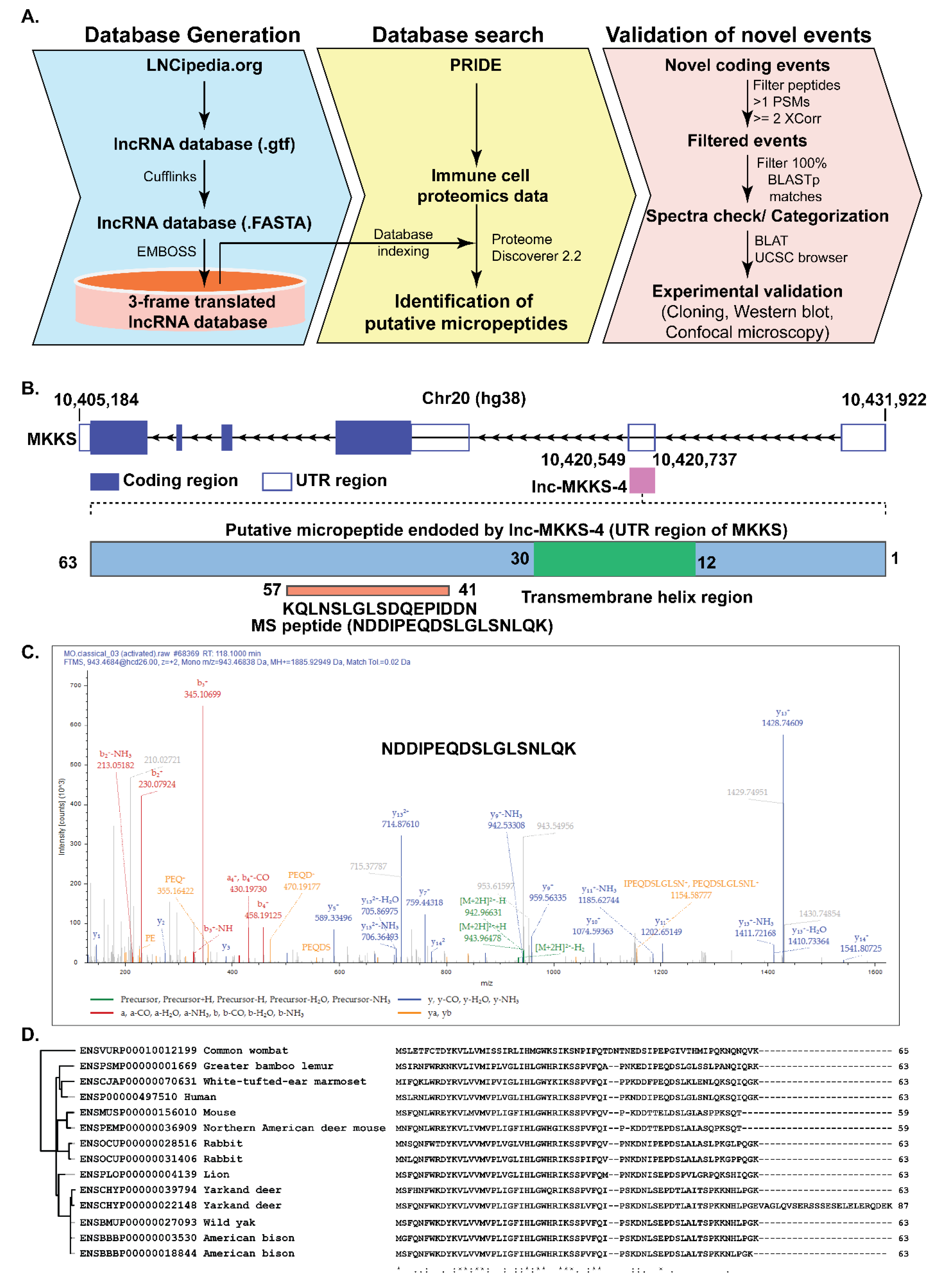
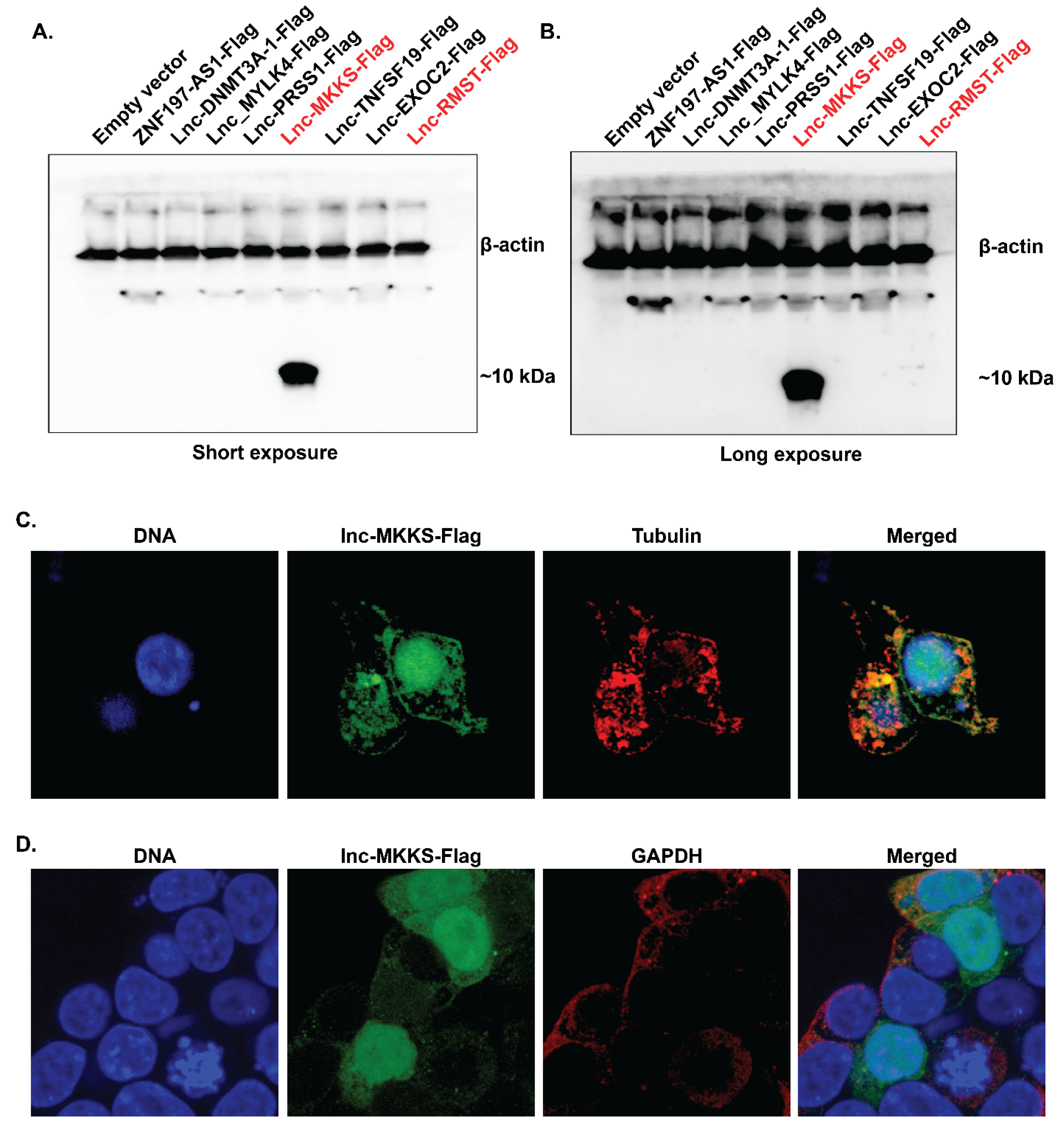

{kind=link}
{kind=link}
| Number of Putative Micropeptide Encoding Events | |
|---|---|
| Myeloid (CD1c+CD304−CD19−CD14−CD3−) Dendritic cells | 352 |
| Plasmacytoid (CD1c−CD304+CD19−CD14−CD3−) Dendritic cells | 384 |
| Classical monocyte (CD14+CD16−) cell proteome | 2513 |
| Classical monocyte (CD14+CD16−) cell secretome | 727 |
| Intermediate monocyte (CD14+CD16+) cell secretome | 291 |
| Non-classical monocyte (CD14dimCD16+) cell secretome | 665 |
| Primary macrophage extracellular vesicle (EV) | 390 |
| Primary macrophage rest-secretome (RS) | 608 |
| Total putative micropeptide encoding events | 5930 |
| Non-redundant putative micropeptide encoding events | 2815 |
| Very high confidence putative micropeptide encoding events | 185 |
| Peptide | Findings from Kim et al. | Findings from the Subbannayya et al. |
|---|---|---|
| AGNIELFSK | Described as a novel coding region exon of C15orf52, expressed in Adult adrenal gland | Originated from lnc-CCDC9B-3:3, maps to 3′ UTR of CCDC9B, Expressed in classical monocyte secretome |
| EAGAGAEAAAGSARPLGR | Described as a gene extension of NPLOC4, Expressed in B cells, Adult testis, Adult ovary, Fetal ovary | Originated from lnc-PDE6G-2:1, matches 5′ UTR of NPLOC4 gene, Expressed in classical monocyte proteome |
| NDDIPEQDSLGLSNLQK | Described as a novel coding region of MKKS, Ubiquitous expression in adult tissues including immune cells (Monocytes, CD4+ T cells, B cells, CD8+ T cells | Originated from lnc-MKKS-4:1, Matches UTR region of MKKS gene, Expressed in classical monocyte proteome |
| NSTLSEPGSGR | Described as a pseudogene (HDGFP1), Expressed in CD8+ T cells | Originated from lnc-OR13H1-3:1, Matches HDGFP pseudogene NST00000405943.3, Expressed in Primary macrophages resting secretome |
| TQNNLESDYLAR | Described as a C-terminal protein extension of AAK1, Expressed in several regions, including Monocytes, CD4+ T cells, CD8+ T cells and B cells | Originated from lnc-AAK1-1:2, Matches intronic region of AAK1, Expressed in Classical monocyte proteome |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subbannayya, Y.; Bhatta, A.; Pinto, S.M.; Fitzgerald, K.A.; Kandasamy, R.K. Proteogenomics Analysis Reveals Novel Micropeptides in Primary Human Immune Cells. Immuno 2022, 2, 283-292. https://doi.org/10.3390/immuno2020018
Subbannayya Y, Bhatta A, Pinto SM, Fitzgerald KA, Kandasamy RK. Proteogenomics Analysis Reveals Novel Micropeptides in Primary Human Immune Cells. Immuno. 2022; 2(2):283-292. https://doi.org/10.3390/immuno2020018
Chicago/Turabian StyleSubbannayya, Yashwanth, Ankit Bhatta, Sneha M. Pinto, Katherine A. Fitzgerald, and Richard K. Kandasamy. 2022. "Proteogenomics Analysis Reveals Novel Micropeptides in Primary Human Immune Cells" Immuno 2, no. 2: 283-292. https://doi.org/10.3390/immuno2020018
APA StyleSubbannayya, Y., Bhatta, A., Pinto, S. M., Fitzgerald, K. A., & Kandasamy, R. K. (2022). Proteogenomics Analysis Reveals Novel Micropeptides in Primary Human Immune Cells. Immuno, 2(2), 283-292. https://doi.org/10.3390/immuno2020018