Pharmacological Inhibition of CCR2 Signaling Exacerbates Exercise-Induced Inflammation Independently of Neutrophil Infiltration and Oxidative Stress




Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Protocols
2.3. Real-Time PCR
2.4. Histological Analysis
2.5. ELISA and TBARS Assay
2.6. Statistical Analysis
3. Results
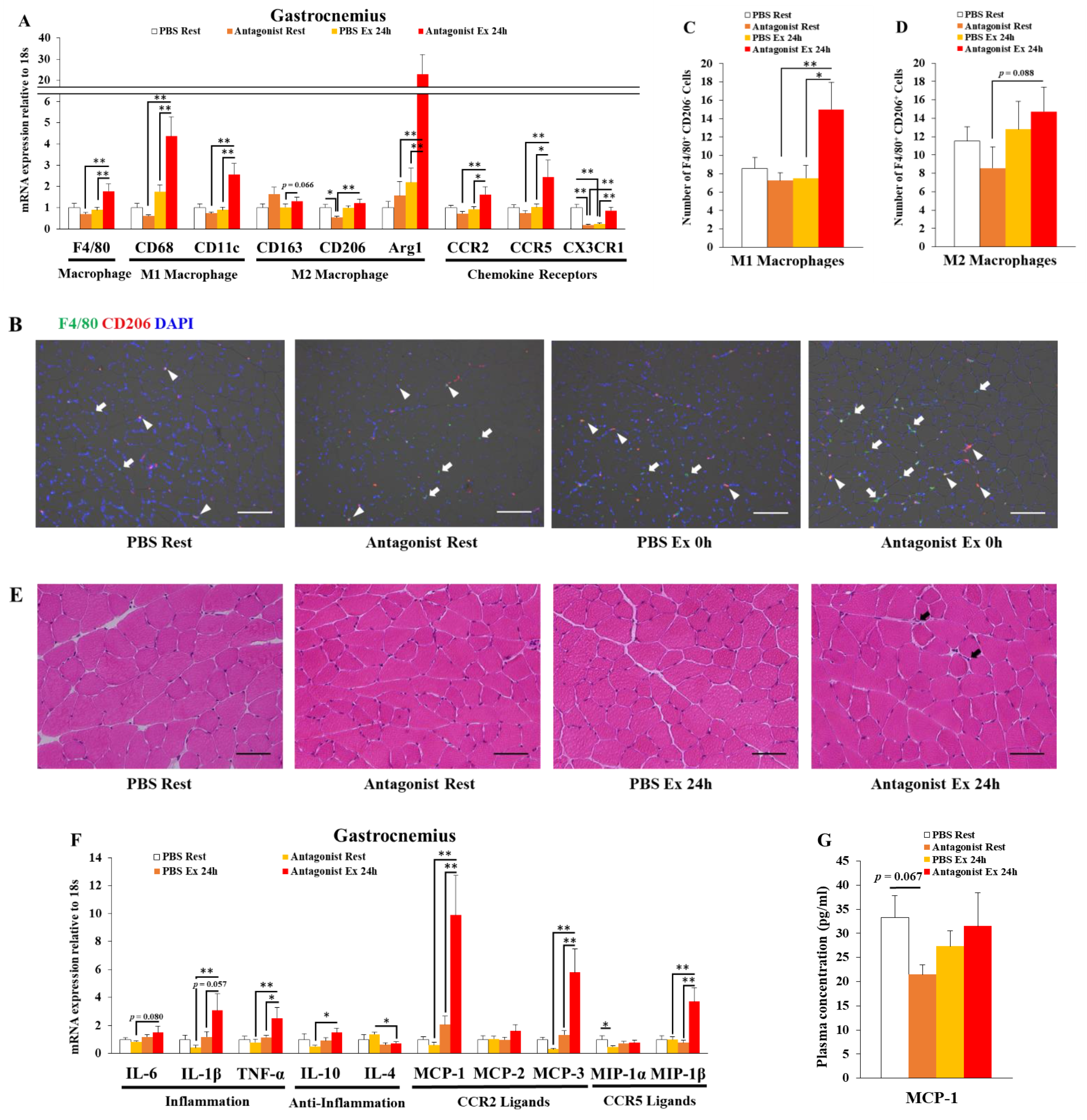
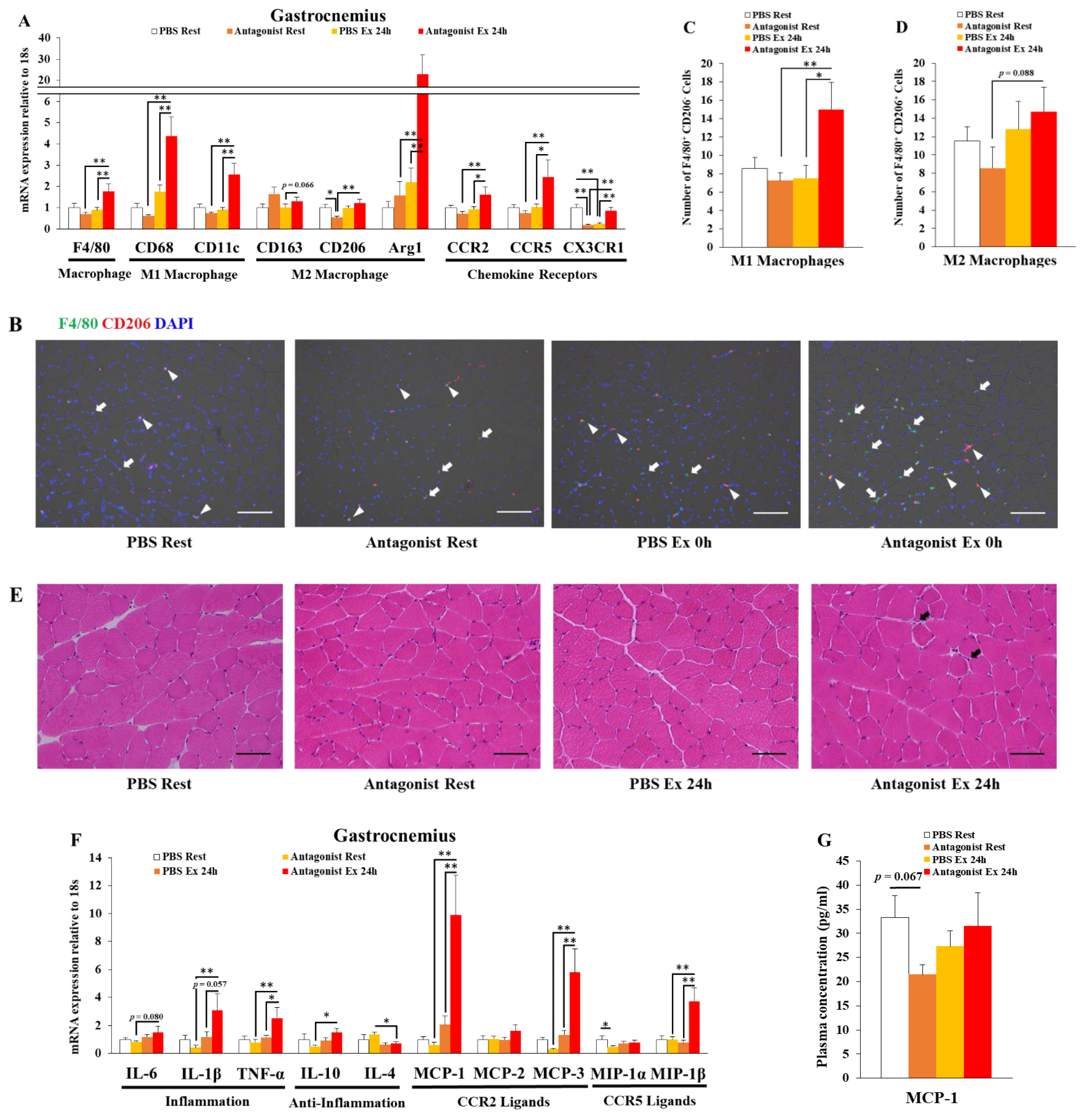
3.1. Inhibition of CCR2 Signaling Exacerbates Macrophage Infiltration and Inflammation 24 h after Exercise in Muscle
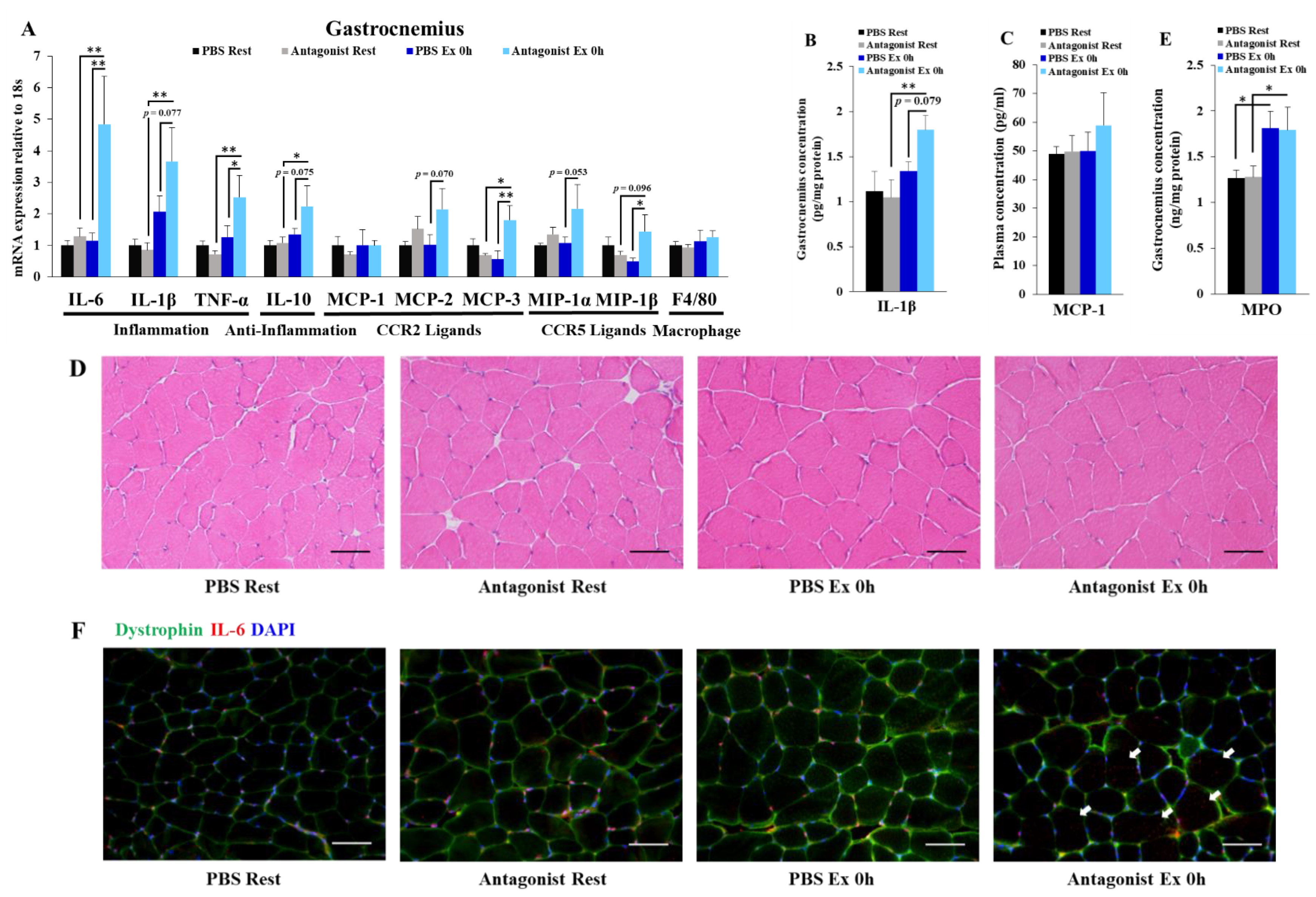
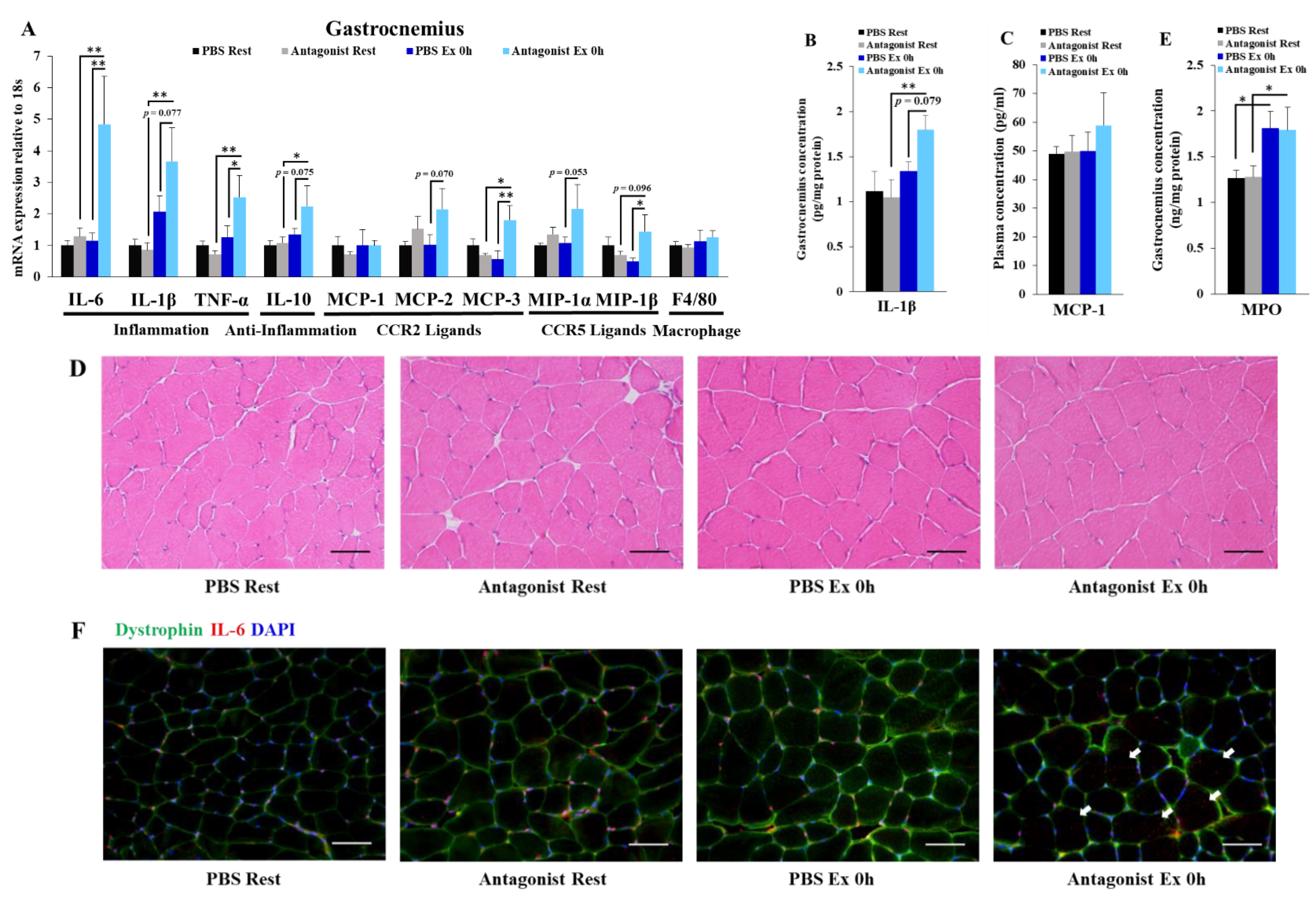
3.2. Inhibition of CCR2 Signaling Exacerbates Exercise-Induced Inflammation Immediately after Exercise Independently of Neutrophil Infiltration in Muscle
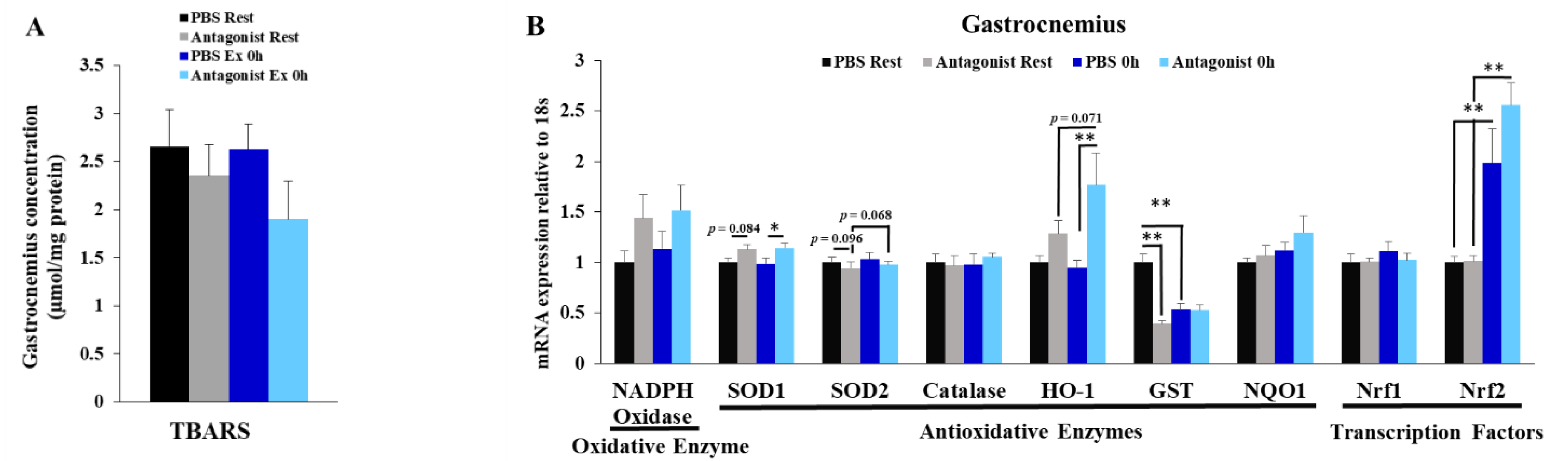
3.3. Inhibition of CCR2 Signaling Does Not Influence Muscle Oxidative Stress Immediately after Exercise
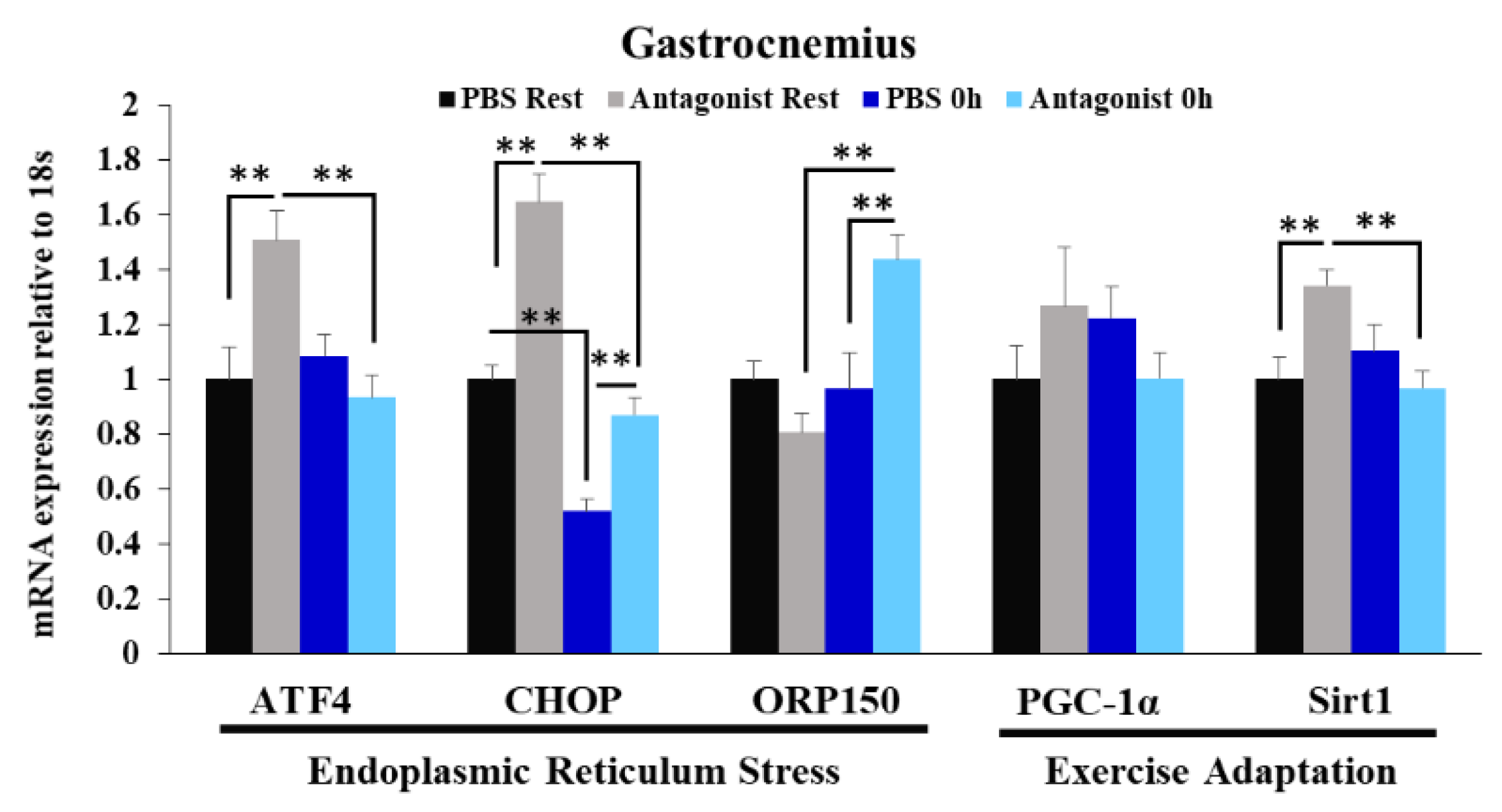
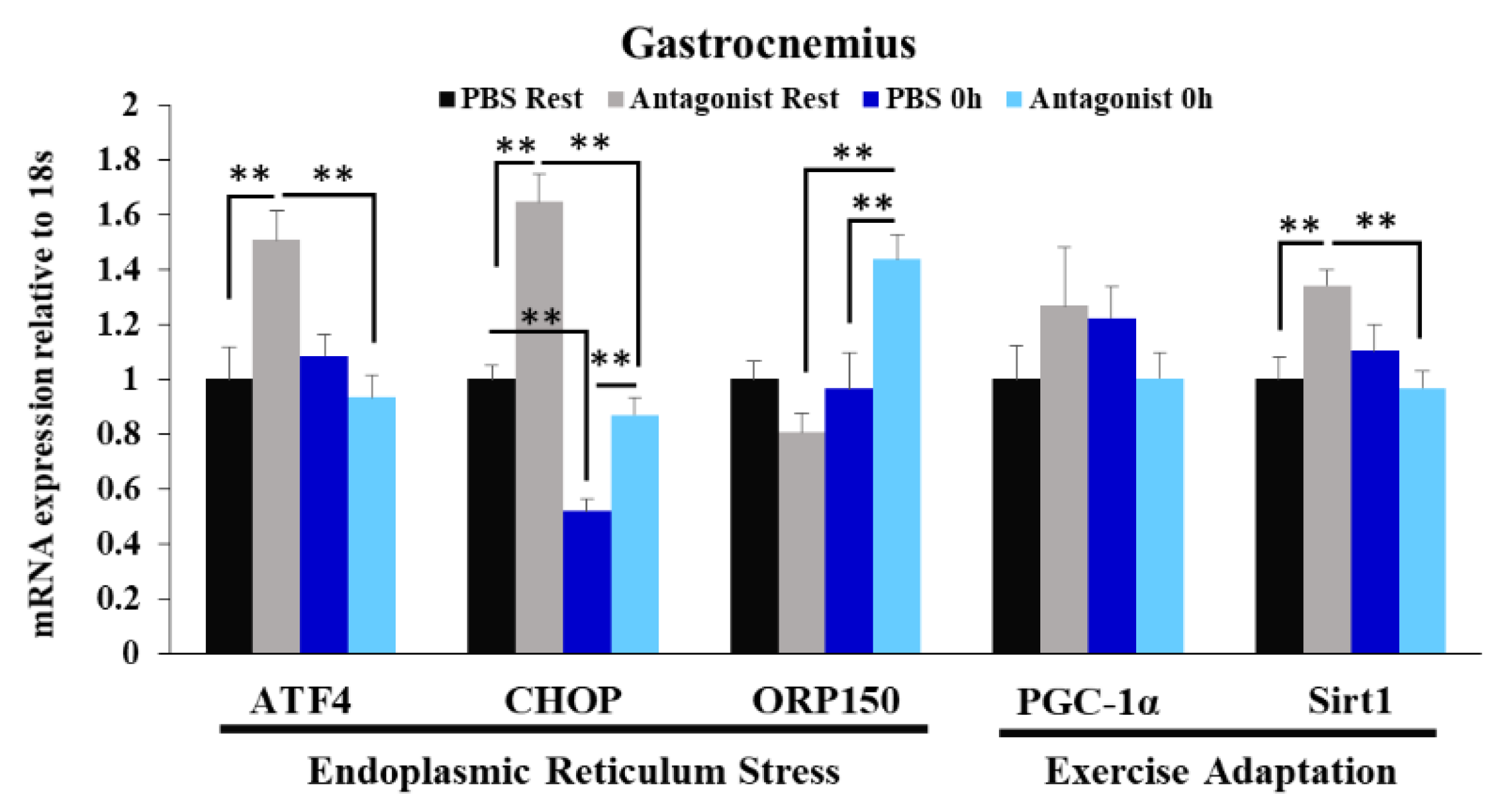
3.4. Effects of CCR2 Signaling Inhibition on Muscle Gene Expression of ER Stress Marker and Exercise Adaptation-Related Genes Immediately after Exercise
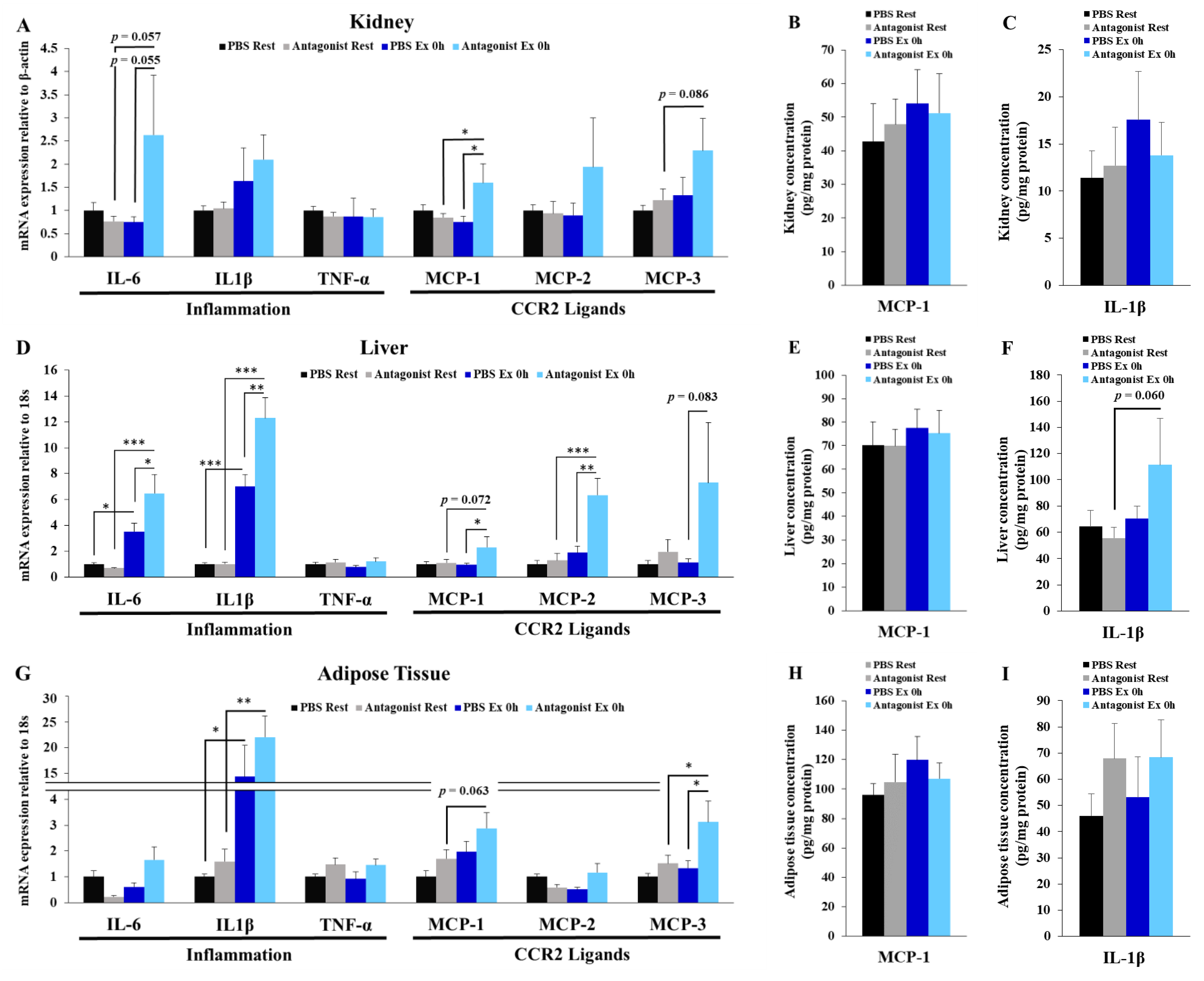
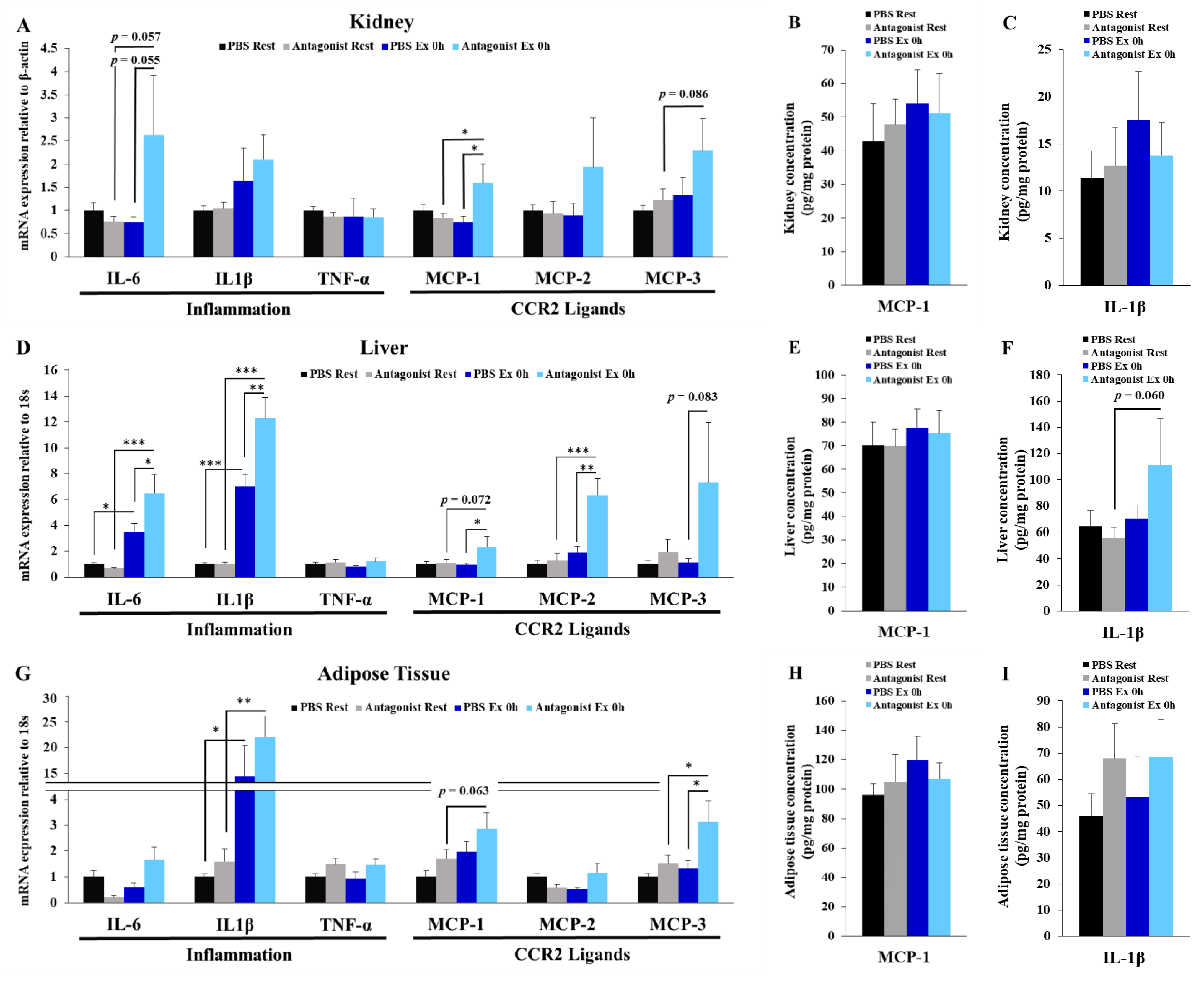
3.5. Inhibition of CCR2 Signaling Exacerbates Exercise-Induced Inflammation in Kidney, Liver, and Adipose Tissues
3.6. CCR2 Ligand-Producing Organs
4. Discussion
5. Limitation
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gleeson, M.; Bishop, N.C.; Stensel, D.J.; Lindley, M.R.; Mastana, S.S.; Nimmo, M.A. The Anti-Inflammatory Effects of Exercise: Mechanisms and Implications for the Prevention and Treatment of Disease. Nat. Rev. Immunol. 2011, 11, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Saleem, A.; Tarnopolsky, M.A. The Potential of Endurance Exercise-Derived Exosomes to Treat Metabolic Diseases. Nat. Rev. Endocrinol. 2016, 12, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Sylow, L.; Kleinert, M.; Richter, E.A.; Jensen, T.E. Exercise-Stimulated Glucose Uptake-Regulation and Implications for Glycaemic Control. Nat. Rev. Endocrinol. 2017, 13, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.K.; Labban, J.D.; Gapin, J.I.; Etnier, J.L. The Effects of Acute Exercise on Cognitive Performance: A Meta-Analysis. Brain Res. 2012, 1453, 87–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Nakaji, S.; Yamada, M.; Totsuka, M.; Sato, K.; Sugawara, K. Systemic Inflammatory Response to Exhaustive Exercise. Cytokine Kinetics. Exerc. Immunol. Rev. 2002, 8, 6–48. [Google Scholar] [PubMed]
- Suzuki, K.; Nakaji, S.; Yamada, M.; Liu, Q.; Kurakake, S.; Okamura, N.; Kumae, T.; Umeda, T.; Sugawara, K. Impact of a Competitive Marathon Race on Systemic Cytokine and Neutrophil Responses. Med. Sci. Sports Exerc. 2003, 35, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Tominaga, T.; Ruhee, R.T.; Ma, S. Characterization and Modulation of Systemic Inflammatory Response to Exhaustive Exercise in Relation to Oxidative Stress. Antioxidants 2020, 9, 401. [Google Scholar] [CrossRef]
- Tominaga, T.; Ma, S.; Sugama, K.; Kanda, K.; Omae, C.; Choi, W.; Hashimoto, S.; Aoyama, K.; Yoshikai, Y.; Suzuki, K. Changes in Urinary Biomarkers of Organ Damage, Inflammation, Oxidative Stress, and Bone Turnover Following a 3000-m Time Trial. Antioxidants 2021, 10, 79. [Google Scholar] [CrossRef]
- Tominaga, T.; Ikemura, T.; Yada, K.; Kanda, K.; Sugama, K.; Ma, S.; Choi, W.; Araya, M.; Huang, J.; Nakamura, N.; et al. The Effects of Beverage Intake after Exhaustive Exercise on Organ Damage, Inflammation and Oxidative Stress in Healthy Males. Antioxidants 2021, 10, 866. [Google Scholar] [CrossRef]
- Tominaga, T.; Ma, S.; Saitou, K.; Suzuki, K. Glucose Ingestion Inhibits Endurance Exercise-Induced IL-6 Producing Macrophage Infiltration in Mice Muscle. Nutrients 2019, 11, 1496. [Google Scholar] [CrossRef] [Green Version]
- Kawanishi, N.; Mizokami, T.; Niihara, H.; Yada, K.; Suzuki, K. Neutrophil Depletion Attenuates Muscle Injury after Exhaustive Exercise. Med. Sci. Sports Exerc. 2016, 48, 1917–1924. [Google Scholar] [CrossRef]
- Kawanishi, N.; Mizokami, T.; Niihara, H.; Yada, K.; Suzuki, K. Macrophage Depletion by Clodronate Liposome Attenuates Muscle Injury and Inflammation Following Exhaustive Exercise. Biochem. Biophys. Rep. 2016, 5, 146–151. [Google Scholar] [CrossRef] [Green Version]
- Miyatake, S.; Bilan, P.J.; Pillon, N.J.; Klip, A. Contracting C2C12 Myotubes Release CCL2 in an NF-ΚB-Dependent Manner to Induce Monocyte Chemoattraction. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E160–E170. [Google Scholar] [CrossRef] [Green Version]
- Evers-van Gogh, I.J.A.; Alex, S.; Stienstra, R.; Brenkman, A.B.; Kersten, S.; Kalkhoven, E. Electric Pulse Stimulation of Myotubes as an In Vitro Exercise Model: Cell-Mediated and Non-Cell-Mediated Effects. Sci. Rep. 2015, 5, 10944. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Nyasha, M.R.; Koide, M.; Tsuchiya, M.; Suzuki, N.; Hagiwara, Y.; Aoki, M.; Kanzaki, M. In Vitro Exercise Model Using Contractile Human and Mouse Hybrid Myotubes. Sci. Rep. 2019, 9, 11914. [Google Scholar] [CrossRef]
- Martinez, C.O.; McHale, M.J.; Wells, J.T.; Ochoa, O.; Michalek, J.E.; McManus, L.M.; Shireman, P.K. Regulation of Skeletal Muscle Regeneration by CCR2-Activating Chemokines Is Directly Related to Macrophage Recruitment. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R832–R842. [Google Scholar] [CrossRef] [Green Version]
- Saini, J.; McPhee, J.S.; Al-Dabbagh, S.; Stewart, C.E.; Al-Shanti, N. Regenerative Function of Immune System: Modulation of Muscle Stem Cells. Ageing Res. Rev. 2016, 27, 67–76. [Google Scholar] [CrossRef]
- Warren, G.L.; Hulderman, T.; Mishra, D.; Gao, X.; Millecchia, L.; O’Farrell, L.; Kuziel, W.A.; Simeonova, P.P. Chemokine Receptor CCR2 Involvement in Skeletal Muscle Regeneration. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 413–415. [Google Scholar] [CrossRef]
- Blanc, R.S.; Kallenbach, J.G.; Bachman, J.F.; Mitchell, A.; Paris, N.D.; Chakkalakal, J.V. Inhibition of Inflammatory CCR2 Signaling Promotes Aged Muscle Regeneration and Strength Recovery after Injury. Nat. Commun. 2020, 11, 4167. [Google Scholar] [CrossRef]
- Kamei, N.; Tobe, K.; Suzuki, R.; Ohsugi, M.; Watanabe, T.; Kubota, N.; Ohtsuka-Kowatari, N.; Kumagai, K.; Sakamoto, K.; Kobayashi, M.; et al. Overexpression of Monocyte Chemoattractant Protein-1 in Adipose Tissues Causes Macrophage Recruitment and Insulin Resistance. J. Biol. Chem. 2006, 281, 26602–26614. [Google Scholar] [CrossRef] [Green Version]
- Sell, H.; Dietze-Schroeder, D.; Kaiser, U.; Eckel, J. Monocyte Chemotactic Protein-1 Is a Potential Player in the Negative Cross-Talk between Adipose Tissue and Skeletal Muscle. Endocrinology 2006, 147, 2458–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawashima, M.; Kawanishi, N.; Tominaga, T.; Suzuki, K.; Miyazaki, A.; Nagata, I.; Miyoshi, M.; Miyakawa, M.; Sakuraya, T.; Sonomura, T.; et al. Icing after Eccentric Contraction-Induced Muscle Damage Perturbs the Disappearance of Necrotic Muscle Fibers and Phenotypic Dynamics of Macrophages in Mice. J. Appl. Physiol. Bethesda Md 1985 2021, 130, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Kitade, H.; Sawamoto, K.; Nagashimada, M.; Inoue, H.; Yamamoto, Y.; Sai, Y.; Takamura, T.; Yamamoto, H.; Miyamoto, K.; Ginsberg, H.N.; et al. CCR5 Plays a Critical Role in Obesity-Induced Adipose Tissue Inflammation and Insulin Resistance by Regulating Both Macrophage Recruitment and M1/M2 Status. Diabetes 2012, 61, 1680–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 Signaling Pathway: Pivotal Roles in Inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 Suppresses Macrophage Inflammatory Response by Blocking Proinflammatory Cytokine Transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [Green Version]
- Hasnain, S.Z.; Lourie, R.; Das, I.; Chen, A.C.-H.; McGuckin, M.A. The Interplay between Endoplasmic Reticulum Stress and Inflammation. Immunol. Cell Biol. 2012, 90, 260–270. [Google Scholar] [CrossRef]
- Egan, B.; Zierath, J.R. Exercise Metabolism and the Molecular Regulation of Skeletal Muscle Adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, J.; Sansbury, B.E.; Holden, C.R.; Tang, Y.; Wong, B.; Wysoczynski, M.; Rodriguez, J.; Bhatnagar, A.; Hill, B.G.; Spite, M. CCR7 Maintains Nonresolving Lymph Node and Adipose Inflammation in Obesity. Diabetes 2016, 65, 2268–2281. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Foster, S.R.; Shah, A.D.; Kleifeld, O.; Canals, M.; Schittenhelm, R.B.; Stone, M.J. Phosphoproteomic Characterization of the Signaling Network Resulting from Activation of the Chemokine Receptor CCR2. J. Biol. Chem. 2020, 295, 6518–6531. [Google Scholar] [CrossRef] [Green Version]
- Ogilvie, P.; Bardi, G.; Clark-Lewis, I.; Baggiolini, M.; Uguccioni, M. Eotaxin Is a Natural Antagonist for CCR2 and an Agonist for CCR5. Blood 2001, 97, 1920–1924. [Google Scholar] [CrossRef]
- Ogilvie, P.; Paoletti, S.; Clark-Lewis, I.; Uguccioni, M. Eotaxin-3 Is a Natural Antagonist for CCR2 and Exerts a Repulsive Effect on Human Monocytes. Blood 2003, 102, 789–794. [Google Scholar] [CrossRef] [Green Version]
- Schild, M.; Eichner, G.; Beiter, T.; Zügel, M.; Krumholz-Wagner, I.; Hudemann, J.; Pilat, C.; Krüger, K.; Niess, A.M.; Steinacker, J.M.; et al. Effects of Acute Endurance Exercise on Plasma Protein Profiles of Endurance-Trained and Untrained Individuals over Time. Mediat. Inflamm. 2016, 2016, 4851935. [Google Scholar] [CrossRef] [Green Version]
- Little, H.C.; Tan, S.Y.; Cali, F.M.; Rodriguez, S.; Lei, X.; Wolfe, A.; Hug, C.; Wong, G.W. Multiplex Quantification Identifies Novel Exercise-Regulated Myokines/Cytokines in Plasma and in Glycolytic and Oxidative Skeletal Muscle *. Mol. Cell. Proteom. 2018, 17, 1546–1563. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.; Lekstrom-Himes, J.; Donaldson, D.; Lee, Y.; Hu, M.; Xu, J.; Wyant, T.; Davidson, M. Effect of CC Chemokine Receptor 2 CCR2 Blockade on Serum C-Reactive Protein in Individuals at Atherosclerotic Risk and With a Single Nucleotide Polymorphism of the Monocyte Chemoattractant Protein-1 Promoter Region. Am. J. Cardiol. 2011, 107, 906–911. [Google Scholar] [CrossRef]
- White, G.E.; Iqbal, A.J.; Greaves, D.R. CC Chemokine Receptors and Chronic Inflammation—Therapeutic Opportunities and Pharmacological Challenges. Pharmacol. Rev. 2013, 65, 47–89. [Google Scholar] [CrossRef] [Green Version]
- Furuichi, K.; Wada, T.; Iwata, Y.; Kitagawa, K.; Kobayashi, K.-I.; Hashimoto, H.; Ishiwata, Y.; Asano, M.; Wang, H.; Matsushima, K.; et al. CCR2 Signaling Contributes to Ischemia-Reperfusion Injury in Kidney. J. Am. Soc. Nephrol. 2003, 14, 2503–2515. [Google Scholar] [CrossRef] [Green Version]
- Mirzadegan, T.; Diehl, F.; Ebi, B.; Bhakta, S.; Polsky, I.; McCarley, D.; Mulkins, M.; Weatherhead, G.S.; Lapierre, J.M.; Dankwardt, J.; et al. Identification of the Binding Site for a Novel Class of CCR2b Chemokine Receptor Antagonists: Binding to a Common Chemokine Receptor Motif within the Helical Bundle. J. Biol. Chem. 2000, 275, 25562–25571. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, A.J.; Enten, G.A.; Gao, X.; Majetschak, M. Chemokine Receptor Antagonists with A1-Adrenergic Receptor Blocker Activity. J. Basic Clin. Physiol. Pharmacol. 2021. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward | Reverse | |
|---|---|---|
| 18 s | TTCTGGCCAACGGTCTAGACAAC | CCAGTGGTCTTGGTGTGCTGA |
| Arginase1 | CTCCAAGCCAAAGTCCTTAGAG | AGGAGCTGTCATTAGGGACATC |
| ATF4 | AACCTCATGGGTTCTCCAGCGA | CTCCAACATCCAATCTGTCCCG |
| β-actin | GCGGACTGTTACTGAGCTGCGT | TGCTGTCGCCTTCACCGTTCC |
| CAT | ACATGGTCTGGGACTTCTGG | CAAGTTTTTGATGCCCTGGT |
| CCR2 | ACAGCTCAGGATTAACAGGGACTTG | ACCACTTGCATCCACACATGAC |
| CCR5 | CATCCGTTCCCCCTACAAGA | GGAACTGACCCTTGAAAATCCA |
| CD11c | CTGGATAGCCTTTCTTCTGCTG | GCACACTGTGTCCGAACTC |
| CD163 | GGGTCATTCAGAGGCACACTG | CTGGCTGTCCTGTCAAGGCT |
| CD206 | CAAGGAAGGTTGGCATTTGT | CCTTTCAGTCCTTTGCAAGC |
| CD68 | CTTCCCACAGGCAGCACAG | AATGATGAGAGGCAGCAAGAGG |
| CHOP | TATCTCATCCCCAGGAAACG | TATCTCATCCCCAGGAAACG |
| CX3CL1 | ACGAAATGCGAAATCATGTGC | CTGTGTCGTCTCCAGGACAA |
| F4/80 | CTTTGGCTATGGGCTTCCAGTC | GCAAGGAGGACAGAGTTTATCGTG |
| GSTm3 | GCTCTTACCACGTGCAGCTT | GGCTGGGAAGAGGAAATGGA |
| HO-1 | CACGCATATACCCGCTACCT | CCAGAGTGTTCATTCGAGCA |
| IL-10 | CGCAGCTCTAGGAGCATGTG | GCTCTTACTGACTGGCATGAG |
| IL-1β | GGGCCTCAAAGGAAAGAATC | TTGCTTGGGATCCACACTCT |
| IL-4 | GGTCTCAACCCCCAGCTAGT | GCCGATGATCTCTCTCAAGTGAT |
| IL-6 | AACGATGATGCACTTGCAGA | TGGTACTCCAGAAGACCAGAGG |
| MCP-1 | CTTCTGGGCCTGCTGTTCA | CCAGCCTACTCATTGGGATCA |
| MCP-2 | AGAGACAGCCAAAGCTGGAA | CAGGCACCATCTGCTTGTAA |
| MCP-3 | CACATTCCTACAGACAGCTC | AGCTACAGAAGGATCACCAG |
| MIP-1α | ACTGCCTGCTGCTTCTCCTACA | ATGACACCTGGCTGGGAGCAAA |
| MIP-1β | ACCCTCCCACTTCCTGCTGTTT | CTGTCTGCCTCTTTTGGTCAGG |
| NADPH Oxidase | TTGGGTCAGCACTGGCTCTG | TGGCGGTGTGCAGTGCTATC |
| NQO1 | GGTATTACGATCCTCCCTCAACATC | GAGTACCTCCCATCCTCTCTTCTTC |
| Nrf1 | GTGGGACAGCAAGCGATTGTAC | CGCACCACATTCTCCAAAGG |
| Nrf2 | CTCGCTGGAAAAAGAAGTGG | CCGTCCAGGAGTTCAGAGAG |
| ORP150 | CAGACTGAAGAGGCGAAACC | TTCCTGTTCAGGTCCAGCTC |
| PGC-1α | AGCCGTGACCACTGACAACGAG | GCTGCATGGTTCTGAGTGCTAAG |
| Sirt1 | GCAACAGCATCTTGCCTGAT | GTGCTACTGGTCTCACTT |
| SOD1 | GAGACCTGGGCAATGTGACT | GTTTACTGCGCAATCCCAAT |
| SOD2 | TCAAGCGTGACTTTGGGTCT | AGCGGAATAAGGCCTGTTGT |
| TNF-α | CCTCCCTCTCATCAGTTCTA | ACTTGGTGGTTTGCTACGAC |
| Antibodies | Source | Identifier | Dilution (Concentration) |
|---|---|---|---|
| Goat anti-IL-6 antibody | R&D Systems, Minneapolis, MN, USA | Cat# AF406 | 10 µg/mL |
| Mouse anti-dystrophin antibody (clone 1808) | Abcam, Cambridge, UK | Cat# ab3149 | 1:400 |
| Rat anti-F4/80 antibody (clone BM8) | Biolegend, San Diego, CA, USA | Cat# 123101 | 1:100 |
| Goat anti-CD206 antibody | R&D Systems, Minneapolis, MN, USA | Cat# AF2535 | 2 µg/mL |
| Alexa Fluor 555-conjugated rabbit anti-goat IgG antibody | Thermo Fisher Scientific, Waltham, MA, USA | Cat# A21431 | 1:200 |
| Fluorescein-conjugate horse anti-mouse IgG antibody | Vector Laboratories, Burlington, ON, Canada | Cat# FI-2000 | 1:200 |
| Alexa Fluor 488-conjugated donkey anti-rat IgG antibody | Thermo Fisher Scientific, Waltham, MA, USA | Cat# A21208 | 1:200 |
| Alexa Fluor 555-conjugated donkey anti-goat IgG antibody | Abcam, Cambridge, UK | Cat# ab150130 | 1:200 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tominaga, T.; Huang, J.; Suzuki, K. Pharmacological Inhibition of CCR2 Signaling Exacerbates Exercise-Induced Inflammation Independently of Neutrophil Infiltration and Oxidative Stress. Immuno 2022, 2, 26-39. https://doi.org/10.3390/immuno2010003
Tominaga T, Huang J, Suzuki K. Pharmacological Inhibition of CCR2 Signaling Exacerbates Exercise-Induced Inflammation Independently of Neutrophil Infiltration and Oxidative Stress. Immuno. 2022; 2(1):26-39. https://doi.org/10.3390/immuno2010003
Chicago/Turabian StyleTominaga, Takaki, Jiapeng Huang, and Katsuhiko Suzuki. 2022. "Pharmacological Inhibition of CCR2 Signaling Exacerbates Exercise-Induced Inflammation Independently of Neutrophil Infiltration and Oxidative Stress" Immuno 2, no. 1: 26-39. https://doi.org/10.3390/immuno2010003
APA StyleTominaga, T., Huang, J., & Suzuki, K. (2022). Pharmacological Inhibition of CCR2 Signaling Exacerbates Exercise-Induced Inflammation Independently of Neutrophil Infiltration and Oxidative Stress. Immuno, 2(1), 26-39. https://doi.org/10.3390/immuno2010003