Observations of, and Insights into, Cystic Fibrosis Mucus Heterogeneity in the Pre-Modulator Era: Sputum Characteristics, DNA and Glycoprotein Content, and Solubilization Time



Abstract
1. Introduction
2. Materials and Methods
2.1. Study Declarations
2.2. Collection and Chemical Analysis of Sputum Samples
2.3. Assessment of Variation in DNA Content within Individual Sputum Samples
2.4. Sputum Solubilization Studies
2.5. Collection, Chemical Analysis, and Solubilization of Bronchiolar Plugs
2.6. Electron Microscopy
3. Results
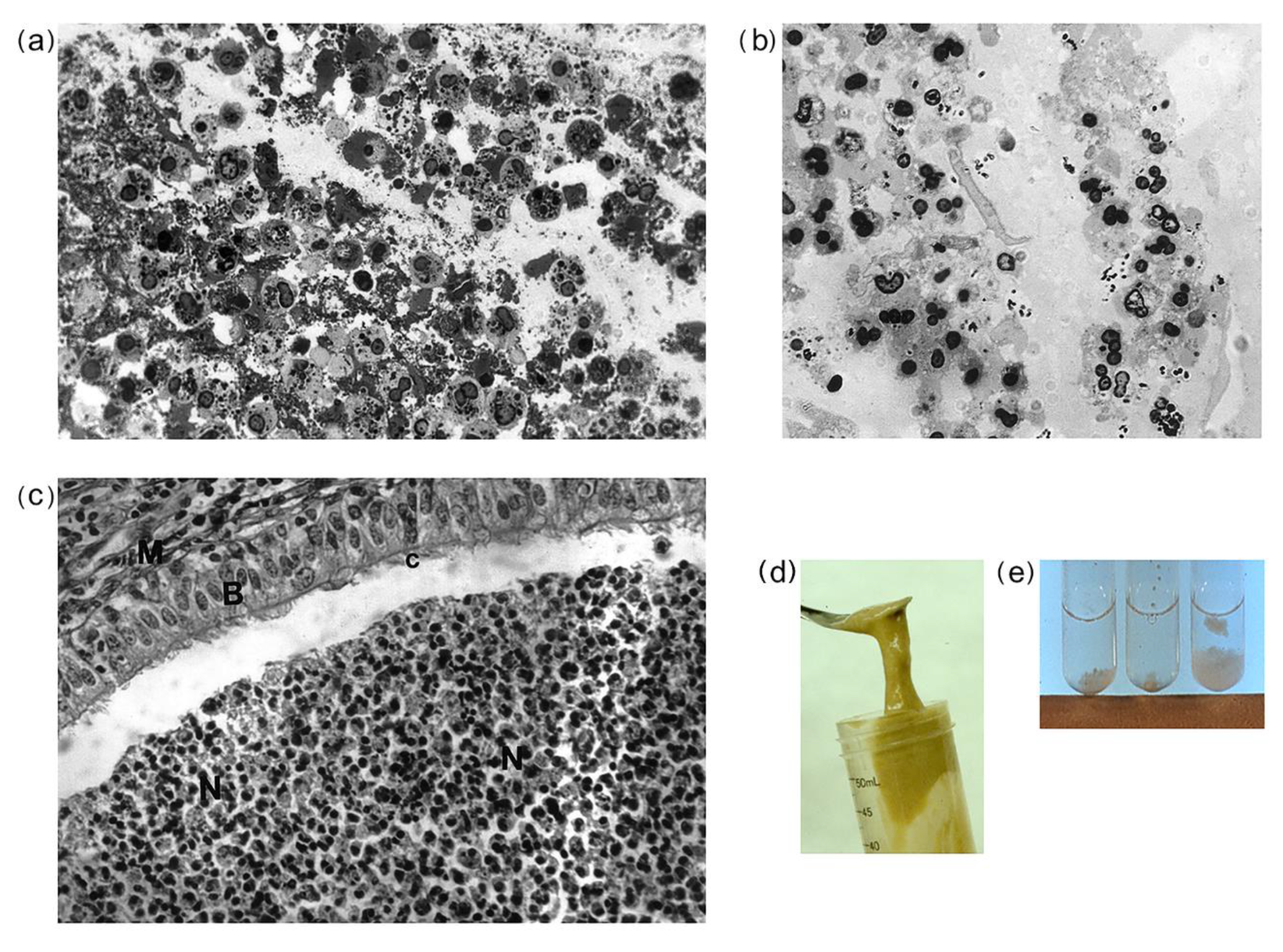
3.1. Morphological Observations of CF and Non-CF Sputa, and CF Bronchial Plugs
3.2. Chemical Results
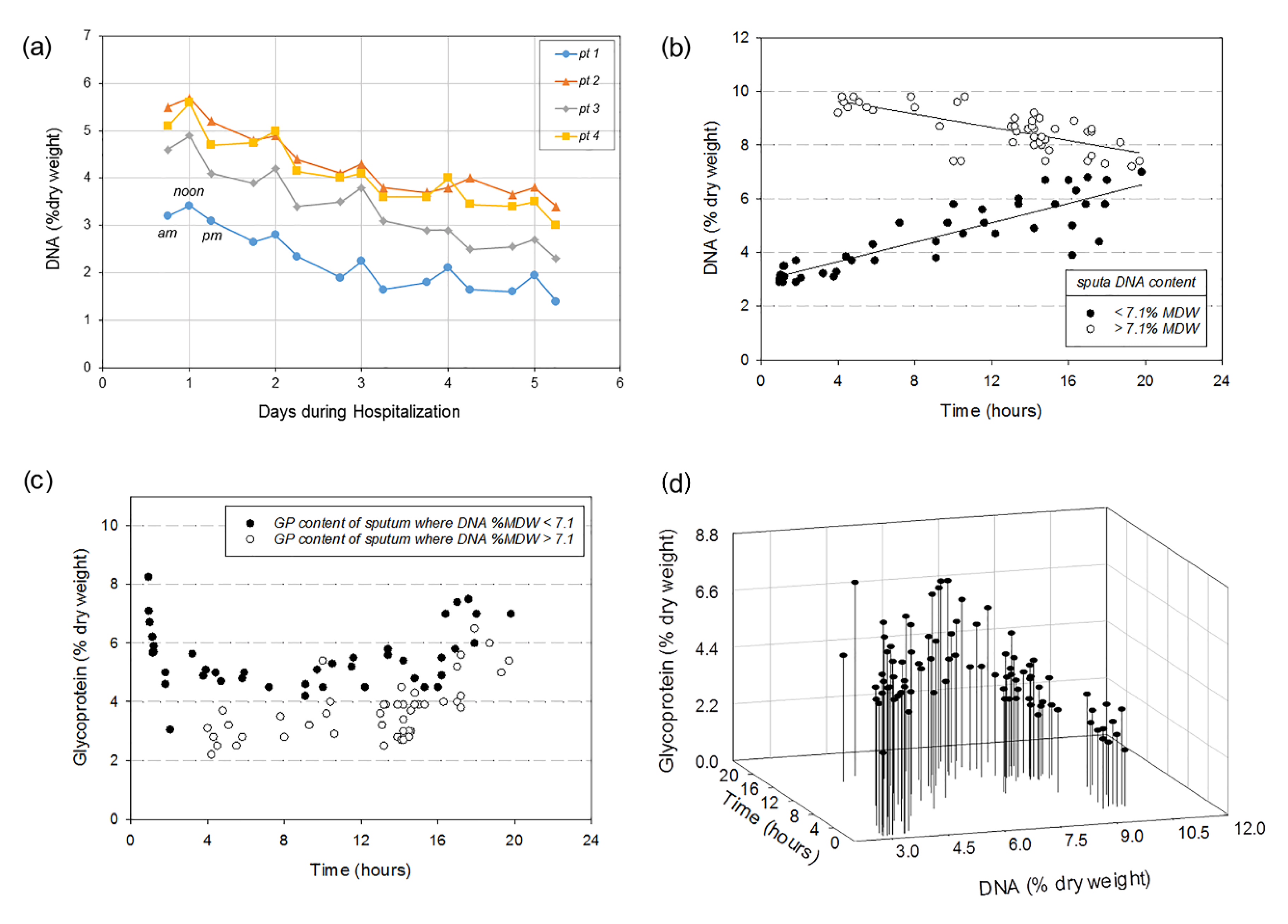
3.3. Within-Specimen DNA Content Variability for CF and Purulent Non-CF Sputa
3.4. Individuals’ CF Sputum DNA Content over Several Days of Treatment
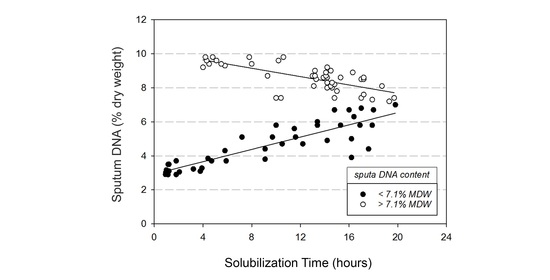
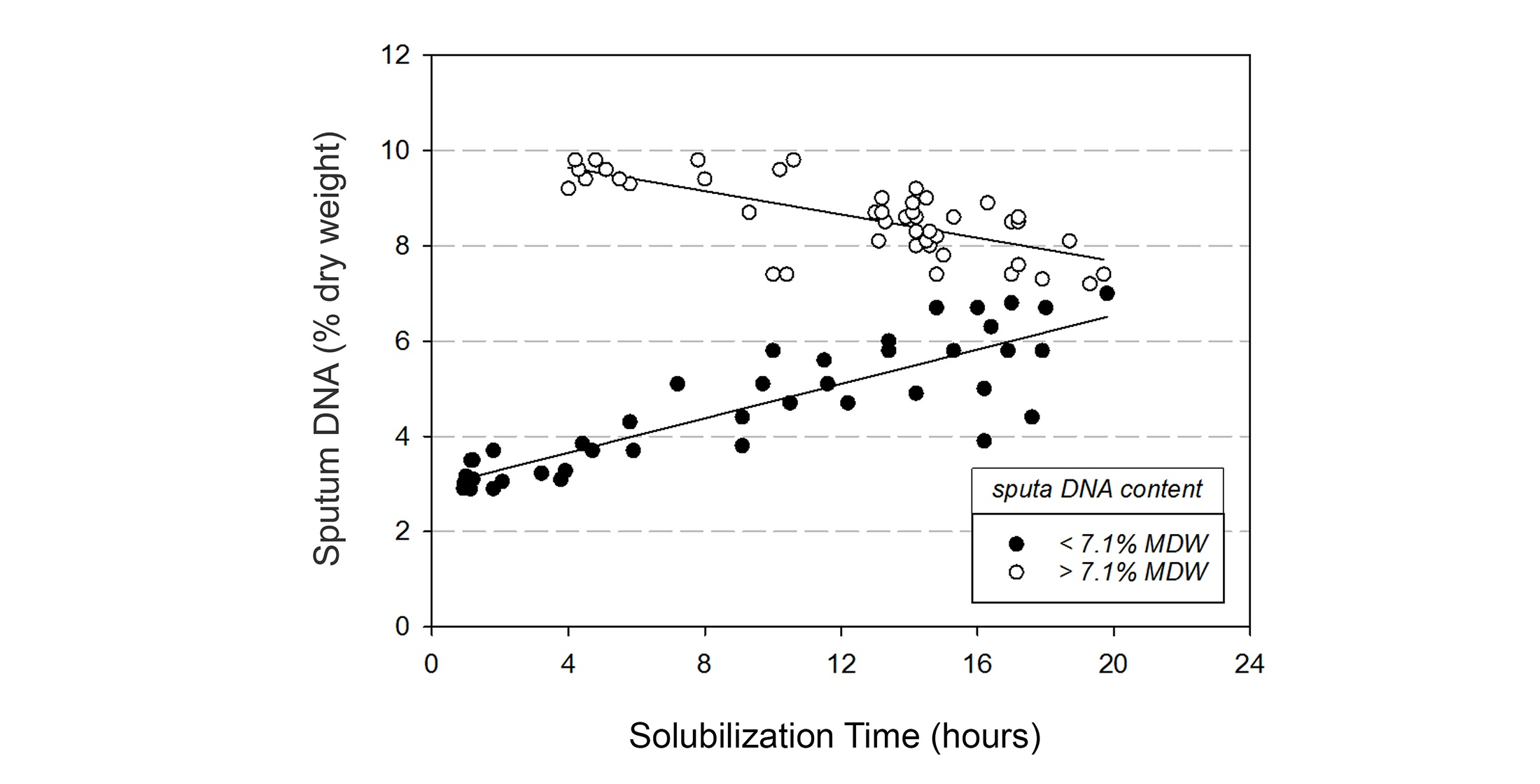
3.5. Chemical Solubilization Studies
4. Discussion
4.1. CF Sputa Visual and Compositional Characteristics
4.2. Variable DNA and Glycoprotein Content Affect Sputum Solubilization
4.3. Potential Contributors to CF Mucus DNA Heterogeneity and Related Solubility
4.4. Potential of CF Mucus Character to Promote Bacterial Adaptation and Survival
4.5. Obstructive CF Mucus Potential to Increase Risk and Time of Exposure to Cytotoxic Agents
4.6. Obstructive CF Mucus Potential to Limit Access and Effectiveness of Beneficial Agents
4.7. CF Mucus Content as Potential Guide to Enhanced Airway Therapeutics Interventions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Puchelle, E.; Bajolet, O.; Abély, M. Airway mucus in cystic fibrosis. Paediatr. Respir. Rev. 2002, 3, 115–119. [Google Scholar] [CrossRef]
- Mawhinney, T.P.; Chance, D.L.; Waters, J.K.; Mossine, V.V.; He, S.; Cassity, N.A. Characterization of blood group antigen-containing oligosaccharides isolated from human respiratory mucous glycoproteins. In Recent Developments in Carbohydrate Research; Pandalai, S.G., Ed.; Transworld Research Network: Kerala, India, 2003; Volume 1, pp. 1–25. [Google Scholar]
- Döring, G.; Flume, P.; Heijerman, H.; Elborn, J.S. Treatment of lung infection in patients with cystic fibrosis: Current and future strategies. J. Cyst. Fibros. 2012, 11, 461–479. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.B.; Markovetz, M.R.; Ehre, C. Mucus, mucins, and cystic fibrosis. Pediatr. Pulmonol. 2019, 54, S84–S96. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef]
- De Boeck, K. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr. 2020, 109, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, V.; Packer, N.H.; Thaysen-Andersen, M. Host mucin glycosylation plays a role in bacterial adhesion in lungs of individuals with cystic fibrosis. Expert Rev. Respir. Med. 2013, 7, 553–576. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.M.; Liu, J.; Stein, S.R.; Stefanski, C.D.; Strubberg, A.M.; Clarke, L.L. Cellular chloride and bicarbonate retention alters intracellular pH regulation in Cftr KO crypt epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 310, G70–G80. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Schreiber, R.; Hadorn, H.B. Bicarbonate in cystic fibrosis. J. Cyst. Fibros. 2017, 16, 653–662. [Google Scholar] [CrossRef]
- Quinton, P.M. Both ways at once: Keeping small airways clean. Physiology 2017, 32, 380–390. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Matthews, L.W.; Spector, S.; Lemm, J.; Potter, J.L. Studies on pulmonary secretions. I. The over-all chemical composition of pulmonary secretions from patients with cystic fibrosis, bronchiectasis, and laryngectomy. Am. Rev. Respir. Dis. 1963, 88, 199–204. [Google Scholar] [PubMed]
- Boat, T.F.; Cheng, P.W.; Iyer, R.N.; Carlson, D.M.; Polony, I. Mucous glycoproteins of non-purulent tracheobronchial secretions and sputum of patients with bronchitis and cystic fibrosis. Arch. Biochem. Biophys. 1976, 177, 95–104. [Google Scholar] [CrossRef]
- King, M. Is cystic fibrosis mucus abnormal? Pediatr. Res. 1981, 15, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Davril, M.; DeGroote, S.; Humbert, P.; Galabert, C.; Dumur, V.; Lafitte, J.-J.; Lamblin, G.; Roussel, P. The sialylation of bronchial mucins secreted by patients suffering from cystic fibrosis or from chronic bronchitis is related to the severity of airway infection. Glycobiology 1999, 9, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Thornton, D.J. From Mucins to Mucus: Toward a More Coherent Understanding of This Essential Barrier. Proc. Am. Thorac. Soc. 2004, 1, 54–61. [Google Scholar] [CrossRef]
- Hill, D.B.; Long, R.F.; Kissner, W.J.; Atieh, E.; Garbarine, I.C.; Markovetz, M.R.; Fontana, N.C.; Christy, M.; Habibpour, M.; Tarran, R.; et al. Pathological mucus and impaired mucus clearance in cystic fibrosis patients result from increased concentration, not altered pH. Eur. Respir. J. 2018, 52, 1801297. [Google Scholar] [CrossRef]
- Esther, C.R., Jr.; Muhlebach, M.S.; Ehre, C.; Hill, D.B.; Wolfgang, M.C.; Kesimer, M.; Ramsey, K.A.; Markovetz, M.R.; Garbarine, I.C.; Forest, M.G.; et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci. Transl. Med. 2019, 11, eaav3488. [Google Scholar] [CrossRef]
- Mawhinney, T.P.; Adelstein, E.; Morris, D.A.; Mawhinney, A.M.; Barbero, G.J. Structure determination of five sulfated oligosaccharides derived from tracheobronchial mucus glycoproteins. J. Biol. Chem. 1987, 262, 2994–3001. [Google Scholar]
- Mawhinney, T.P.; Adelstein, E.; Gayer, D.A.; Landrum, D.C.; Barbero, G.J. Structural analysis of monosulfated side-chain oligosaccharides isolated from human tracheobronchial mucous glycoproteins. Carbohydr. Res. 1992, 223, 187–207. [Google Scholar] [CrossRef]
- Mawhinney, T.P.; Landrum, D.C.; Gayer, D.A.; Barbero, G.J. Sulfated sialyl-oligosaccharides derived from tracheobronchial mucous glycoproteins of a patient suffering from cystic fibrosis. Carbohydr. Res. 1992, 235, 179–197. [Google Scholar] [CrossRef]
- Chance, D.L.; Mawhinney, T.P. Disulfated oligosaccharides derived from tracheobronchial mucous glycoproteins of a patient suffering from cystic fibrosis. Carbohydr. Res. 1996, 295, 157–177. [Google Scholar] [CrossRef]
- Chance, D.L.; Mawhinney, T.P. Carbohydrate sulfation effects on growth of Pseudomonas aeruginosa. Microbiology 2000, 146, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Lamblin, G.; DeGroote, S.; Perini, J.M.; Delmotte, P.; Scharfman, A.; Davril, M.; Lo-Guidice, J.M.; Houdret, N.; Dumur, V.; Klein, A.; et al. Human airway mucin glycosylation: A combinatory of carbohydrate determinants which vary in cystic fibrosis. Glycoconj. J. 2001, 18, 661–684. [Google Scholar] [CrossRef] [PubMed]
- DeGroote, S.; Maes, E.; Humbert, P.; Delmotte, P.; Lamblin, G.; Roussel, P. Sulfated oligosaccharides isolated from the respiratory mucins of a secretor patient suffering from chronic bronchitis. Biochimie 2003, 85, 369–379. [Google Scholar] [CrossRef]
- Mawhinney, T.P.; Chance, D.L. Structural analysis of sulfated oligosaccharides possessing (α1-2)-fucosyl residues derived from cystic fibrosis tracheobronchial mucous glycoproteins. In Recent Developments in Carbohydrate Research; Pandalai, S.G., Ed.; Transworld Research Network: Kerala, India, 2005; Volume 2, pp. 67–92. [Google Scholar]
- Ostedgaard, L.S.; Meyerholz, D.K.; Chen, J.-H.; Pezzulo, A.A.; Karp, P.H.; Rokhlina, T.; Ernst, S.E.; Hanfland, R.A.; Reznikov, L.; Ludwig, P.S.; et al. The DeltaF508 mutation causes CFTR misprocessing and cystic fibrosis-like disease in pigs. Sci. Transl. Med. 2011, 3, 74ra24. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.X.; Ostedgaard, L.S.; Hoegger, M.J.; Moninger, T.O.; Karp, P.H.; McMenimen, J.D.; Choudhury, B.; Varki, A.; Stoltz, D.A.; Welsh, M.J. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J. Clin. Investig. 2016, 126, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Luan, X.; Belev, G.; Tam, J.S.; Jagadeeshan, S.; Hassan, N.; Gioino, P.; Grishchenko, N.; Huang, Y.; Carmalt, J.L.; Duke, T.; et al. Cystic fibrosis swine fail to secrete airway surface liquid in response to inhalation of pathogens. Nat. Commun. 2017, 8, 786. [Google Scholar] [CrossRef]
- Ermund, A.; Trillo-Muyo, S.; Hansson, G.C. Assembly, release, and transport of airway mucins in pigs and humans. Ann. Am. Thorac. Soc. 2018, 15, S159–S163. [Google Scholar] [CrossRef]
- Ehre, C.; Rushton, Z.L.; Wang, B.; Hothem, L.N.; Morrison, C.B.; Fontana, N.C.; Markovetz, M.R.; DeLion, M.F.; Kato, T.; Villalon, D.; et al. An improved inhaled mucolytic to treat airway muco-obstructive diseases. Am. J. Respir. Crit. Care Med. 2019, 199, 171–180. [Google Scholar] [CrossRef]
- Fernandez-Petty, C.M.; Hughes, G.W.; Bowers, H.L.; Watson, J.D.; Rosen, B.H.; Townsend, S.M.; Santos, C.; Ridley, C.E.; Chu, K.K.; Birket, S.E.; et al. A glycopolymer improves vascoelasticity and mucociliary transport of abnormal cystic fibrosis mucus. JCI Insight 2019, 4, e125954. [Google Scholar] [CrossRef]
- Kim, M.D.; Baumlin, N.; Yoshida, M.; Polineni, D.; Salathe, S.F.; David, J.K.; Peloquin, C.A.; Wanner, A.; Dennis, J.S.; Sailland, J.; et al. Losartan rescues inflammation-related mucociliary dysfunction in relevant models of cystic fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation Website, Drug Development Pipeline—Clinical Trials Tool. Available online: https://www.cff.org/trials/pipeline (accessed on 8 September 2020).
- 2018 Cystic Fibrosis Foundation Patient Registry Highlights. Available online: https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2018-Cystic-Fibrosis-Foundation-Patient-Registry-Highlights.pdf (accessed on 8 September 2020).
- Stanford, G.E.; Dave, K.; Simmonds, N.J. Pulmonary exacerbations in adults with cystic fibrosis—A grown-up issue in a changing CF landscape. Chest 2020. [Google Scholar] [CrossRef] [PubMed]
- King, M.; Dasgupta, B.; Tomkiewicz, R.P.; Brown, N.E. Rheology of cystic fibrosis sputum after in vitro treatment with hypertonic saline alone and in combination with recombinant human deoxyribonuclease, I. Am. J. Respir. Crit. Care Med. 1997, 156, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, B.; Brown, N.E.; King, M. Effects of sputum oscillations and rhDNase in vitro: A combined approach to treat cystic fibrosis lung disease. Pediatr. Pulmonol. 1998, 26, 250–255. [Google Scholar] [CrossRef]
- Rubin, B.K. Mucus, phlegm, and sputum in cystic fibrosis. Respir. Care 2009, 54, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Kreda, S.M.; Davis, C.W.; Rose, M.C. CFTR, mucins, and mucus obstruction in cystic fibrosis. Cold Spring Harb. Perspect. Med. 2012, 2, a009589. [Google Scholar] [CrossRef]
- Daviskas, E.; Rubin, B.K. Effect of inhaled dry powder mannitol on mucus and its clearance. Expert Rev. Respir. Med. 2013, 7, 65–75. [Google Scholar] [CrossRef]
- Ehre, C.; Ridley, C.; Thornton, D.J. Cystic fibrosis: An inherited disease affecting mucin-producing organs. Int. J. Biochem. Cell Biol. 2014, 52, 136–145. [Google Scholar] [CrossRef]
- Restrepo, M.I.; Keyt, H.; Reyes, L.F. Aerosolized antibiotics. Respir. Care 2015, 60, 762–773. [Google Scholar] [CrossRef]
- Tay, G.T.; Reid, D.W.; Bell, S.C. Inhaled antibiotics in cystic fibrosis (CF) and non-CF bronchiectasis. Semin. Respir. Crit. Care Med. 2015, 36, 267–286. [Google Scholar] [CrossRef]
- Yang, C.L.; Montgomery, M. Dornase alfa for cystic fibrosis. Cochrane Database Syst. Rev. 2018, 9, CD001127. [Google Scholar] [CrossRef] [PubMed]
- Demouveaux, B.; Gouyer, V.; Gottrand, F.; Narita, T.; Desseyn, J.-L. Gel-forming mucin interactome drives mucus viscoelasticity. Adv. Colloid Interface Sci. 2018, 252, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Ali, Z.S.; Sweezey, N.B.; Grasemann, H.; Palaniyar, N. Progression of cystic fibrosis lung disease from childhood to adulthood: Neutrophils, neutrophil extracellular trap (NET) formation, and NET degradation. Genes 2019, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.C.; Guo, S.; Huang, L.; Kerem, B.; Oren, Y.S.; Walker, A.J.; Hart, S.L. New approaches to genetic therapies for cystic fibrosis. J. Cyst. Fibros. 2020, 19, S54–S59. [Google Scholar] [CrossRef]
- Nair, G.B.; Ilowite, J.S. Pharmacologic agents for mucus clearance in bronchiectasis. Clin. Chest Med. 2012, 33, 363–370. [Google Scholar] [CrossRef]
- Leal, J.; Smyth, H.D.C.; Ghosh, D. Physicochemical properties of mucus and their impact on transmucosal drug delivery. Int. J. Pharm. 2017, 532, 555–572. [Google Scholar] [CrossRef]
- Hill, D.B.; Vasquez, P.A.; Mellnik, J.; McKinley, S.A.; Vose, A.; Mu, F.; Henderson, A.G.; Donaldson, S.H.; Alexis, N.E.; Boucher, R.C.; et al. A biophysical basis for mucus solids concentration as a candidate biomarker for airways disease. PLoS ONE 2014, 9, e87681. [Google Scholar] [CrossRef]
- Rubin, B.K. Mucus structure and properties in cystic fibrosis. Paediatr. Respir. Rev. 2007, 8, 4–7. [Google Scholar] [CrossRef]
- Duncan, G.A.; Jung, J.; Hanes, J.; Suk, J.S. The mucus barrier to inhaled gene therapy. Mol. Ther. 2016, 24, 2043–2053. [Google Scholar] [CrossRef]
- Thomen, R.P.; Walkup, L.L.; Roach, D.J.; Cleveland, Z.I.; Clancy, J.P.; Woods, J.C. Hyperpolarized 129Xe for investigation of mild cystic fibrosis lung disease in pediatric patients. J. Cyst. Fibros. 2017, 16, 275–282. [Google Scholar] [CrossRef]
- Adewale, A.T.; Falk Libby, E.; Fu, L.; Lenzie, A.; Boitet, E.R.; Birket, S.E.; Petty, C.F.; Johns, J.D.; Mazur, M.; Tearney, G.J.; et al. Novel therapy of bicarbonate, glutathione, and ascorbic acid improves cystic fibrosis mucus transport. Am. J. Respir. Cell Mol. Biol. 2020, 63, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, M.; Luo, Y.; Deng, L.; Hu, Z.; Song, Y. Determination of rheology and surface tension of airway surface liquid: A review of clinical relevance and measurement techniques. Respir. Res. 2019, 20, 274. [Google Scholar] [CrossRef] [PubMed]
- Chance, D.L.; Mawhinney, T.P. Using negative staining TEM to study structure/function relationships of cystic fibrosis host-adapted opportunistic pathogen Pseudomonas aeruginosa. Microsc. Microanal. 2017, 23, 1354–1355. [Google Scholar] [CrossRef]
- Chance, D.L.; Mossine, V.; Waters, J.; Wang, W.; Mawhinney, T.P. Evaluating the heterogeneity of Pseudomonas aeruginosa collection for application to in vitro research studies. Pediatr. Pulmonol. 2018, 53, 298. [Google Scholar] [CrossRef]
- Mossine, V.V.; Waters, J.K.; Chance, D.L.; Mawhinney, T.P. Transient proteotoxicity of bacterial virulence factor pyocyanin in renal tubular epithelial cells induces ER-related vacuolation and can be efficiently modulated by iron chelators. Toxicol. Sci. 2016, 154, 403–415. [Google Scholar] [CrossRef][Green Version]
- Mossine, V.V.; Chance, D.L.; Waters, J.K.; Mawhinney, T.P. Interaction of bacterial phenazines with colistimethate in bronchial epithelial cells. Antimicrob. Agents Chemother. 2018, 62, e02349-17. [Google Scholar] [CrossRef]
- Mawhinney, T.P.; Mossine, V.V.; Chance, D.L.; Waters, J.K. Cytotoxic interactions between Pseudomonas aeruginosa virulence factors and metal-based antimicrobials in vitro. FASEB J. 2019, 33, 662. [Google Scholar] [CrossRef]
- Cerriotti, G. A microchemical determination of deoxyribonucleic acid. J. Biol. Chem. 1952, 198, 297–303. [Google Scholar]
- Kissane, J.M.; Robins, E. The fluorometric measurement of deoxyribonucleic acid in animal tissues with special reference to the central nervous system. J. Biol. Chem. 1958, 233, 184–188. [Google Scholar]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Rouser, G.; Kritchevsky, G.; Yamamoto, A. Column chromatographic and associated procedures for separation and determination of phosphatides and glycolipids. Lipid Chromatograph. Anal. 1967, 1, 99–162. [Google Scholar]
- Poorthuis, B.J.; Yazaki, P.J.; Hostetler, K.Y. An improved two dimensional thin-layer chromatography system for the separation of phosphatidylglycerol and its derivatives. J. Lipid Res. 1976, 17, 433–437. [Google Scholar] [PubMed]
- Lowry, R.R.; Tinsley, I.J. A simple, sensitive method for lipid phosphorus. Lipids 1974, 9, 491–492. [Google Scholar] [CrossRef] [PubMed]
- Mawhinney, T.P.; Feather, M.S.; Barbero, G.J.; Martinez, J. The rapid, quantitative determination of neutral sugars (as aldononitrile acetates) and amino sugars (as O-methyloxime acetates) in glycoproteins by gas-liquid chromatography. Anal. Biochem. 1980, 101, 112–117. [Google Scholar] [CrossRef]
- Warren, L. The thiobarbituric acid assay of sialic acids. J. Biol. Chem. 1959, 234, 1971–1975. [Google Scholar] [PubMed]
- Mawhinney, T.P.; Madson, M.A.; Rice, R.H.; Feather, M.S.; Barbero, G.J. Gas-liquid chromatography and mass-spectral analysis of per-O-trimethylsilyl acyclic ketoxime derivatives of neuraminic acid. Carbohydr. Res. 1982, 104, 169–181. [Google Scholar] [CrossRef]
- Millonig, G. Advantages of a phosphate buffer for OsO4 solutions in fixation. J. Appl. Phys. 1961, 32, 1–5. [Google Scholar]
- Luft, J.H. Improvements in epoxy resin embedding methods. J. Biophys. Biochem. Cytol. 1961, 9, 409–414. [Google Scholar] [CrossRef]
- Reynolds, E.S. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 1963, 17, 208–212. [Google Scholar] [CrossRef]
- Chance, D.; Mawhinney, T. Cystic fibrosis sputum reflections from the pre-modulator age. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Birket, S.E.; Davis, J.M.; Fernandez, C.M.; Tuggle, K.L.; Oden, A.M.; Chu, K.K.; Tearney, G.J.; Fanucchi, M.V.; Sorscher, E.J.; Rowe, S.M. Development of an airway mucus defect in the cystic fibrosis rat. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Conod, E.J.; Conover, J.H.; Hirschhorn, K. Demonstration of human leukocyte degranulation induced by sera from homozygotes and heterozygotes for cystic fibrosis. Pediatr. Res. 1975, 9, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.B.; Markovetz, M.R.; Kissner, W.; Sears, P.; Ostrowski, L. Transition of CF mucus from mucin to DNA dominance: Therapeutic consequences. Pediatr. Pulmonol. 2019, 54, S159–S160. [Google Scholar] [CrossRef]
- Shelley, S.A.; Balis, J.U.; Paciga, J.E.; Espinoza, C.G.; Richman, A.V. Biochemical composition of adult human lung surfactant. Lung 1982, 160, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Zahm, J.-M.; Galabert, C.; Chaffin, A.; Chazalette, J.-P.; Grosskopf, C.; Puchelle, E. Improvement of cystic fibrosis airway mucus transportability by recombinant human DNase is related to changes in phospholipid profile. Am. J. Respir. Crit. Care Med. 1998, 157, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Bansil, R.; Turner, B.S. The biology of mucus: Composition, synthesis and organization. Adv. Drug Deliv. Rev. 2018, 124, 3–15. [Google Scholar] [CrossRef]
- Tomaiuolo, G.; Rusciano, G.; Caserta, S.; Carciati, A.; Carnovale, V.; Abete, P.; Sasso, A.; Guido, S. A new method to improve the clinical evaluation of cystic fibrosis patients by mucus viscoelastic properties. PLoS ONE 2014, 9, e82297. [Google Scholar] [CrossRef]
- Ma, J.T.; Tang, C.; Kang, L.; Voynow, J.A.; Rubin, B.K. Cystic fibrosis sputum rheology correlates with both acute and longitudinal changes in lung function. Chest 2018, 154, 370–377. [Google Scholar] [CrossRef]
- Rancourt, R.C.; Tai, S.; King, M.; Heltshe, S.L.; Penvari, C.; Accurso, F.J.; White, C.W. Thioredoxin liquefies and decreases the viscoelasticity of cystic fibrosis sputum. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L931–L938. [Google Scholar] [CrossRef][Green Version]
- Law, S.M.; Gray, R.D. Neutrophil extracellular traps and the dysfunctional innate immune response of cystic fibrosis lung disease: A review. J. Inflamm. 2017, 14, 1–8. [Google Scholar] [CrossRef]
- Gray, R.D.; Hardisty, G.; Regan, K.H.; Smith, M.; Robb, C.T.; Duffin, R.; Mackellar, A.; Felton, J.M.; Paemka, L.; Mccullagh, B.N.; et al. Delayed neutrophil apoptosis enhances NET formation in cystic fibrosis. Thorax 2018, 73, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Hoenderdos, K.; Lodge, K.M.; Hirst, R.A.; Chen, C.; Palazzo, S.G.C.; Emerenciana, A.; Summers, C.; Angyal, A.; Porter, L.; Juss, J.K.; et al. Hypoxia upregulates neutrophil degranulation and potential for tissue injury. Thorax 2016, 71, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Winstanley, C.; O’Brien, S.; Brockhurst, M.A. Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microbiol. 2016, 24, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Zanin, M.; Baviskar, P.; Webster, R.; Webby, R. The interaction between respiratory pathogens and mucus. Cell Host Microbe 2016, 19, 159–168. [Google Scholar] [CrossRef]
- Koo, H.; Allan, R.N.; Howlin, R.P.; Stoodley, P.; Hall-Stoodley, L. Targeting microbial biofilms: Current and prospective therapeutic strategies. Nat. Rev. Genet. 2017, 15, 740–755. [Google Scholar] [CrossRef] [PubMed]
- Secor, P.R.; Michaels, L.A.; Ratjen, A.; Jennings, L.K.; Singh, P.K. Entropically driven aggregation of bacteria by host polymers promotes antibiotic tolerance in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2018, 115, 10780–10785. [Google Scholar] [CrossRef]
- Waters, V.J.; Kidd, T.J.; Canton, R.; Ekkelenkamp, M.B.; Johansen, H.K.; Lipuma, J.J.; Bell, S.C.; Elborn, J.S.; Flume, P.A.; VanDevanter, D.R.; et al. Reconciling antimicrobial susceptibility testing and clinical response in antimicrobial treatment of chronic cystic fibrosis lung infections. Clin. Infect. Dis. 2019, 69, 1812–1816. [Google Scholar] [CrossRef]
- Woo, T.E.; Duong, J.; Jervis, N.M.; Rabin, H.R.; Parkins, M.D.; Storey, D.G. Virulence adaptations of Pseudomonas aeruginosa isolated from patients with non-cystic fibrosis bronchiectasis. Microbiology 2016, 162, 2126–2135. [Google Scholar] [CrossRef]
- Harrison, F.; McNally, A.; Da Silva, A.C.; Heeb, S.; Diggle, S.P. Optimised chronic infection models demonstrate that siderophore ‘cheating’ in Pseudomonas aeruginosa is context specific. ISME J. 2017, 11, 2492–2509. [Google Scholar] [CrossRef]
- Lema, G.; Dryja, D.; Vargas, I.; Enhorning, G. Pseudomonas aeruginosa from patients with cystic fibrosis affects function of pulmonary surfactant. Pediatr. Res. 2000, 47, 121. [Google Scholar] [CrossRef][Green Version]
- D’Andrea, M.M.; Fraziano, M.; Thaller, M.C.; Rossolini, G.M. The urgent need for novel antimicrobial agents and strategies to fight antibiotic resistance. Antibiotics 2019, 8, 254. [Google Scholar] [CrossRef] [PubMed]
- Wark, P.; Cookson, K.; Thiruchelvam, T.; Brannan, J.D.; Dorahy, D.J. Lumacaftor/ Ivacaftor improves exercise tolerance in patients with cystic fibrosis and severe airflow obstruction. BMC Pulm. Med. 2019, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wark, P.; McDonald, V.M. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database Syst. Rev. 2018, 9, CD001506. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Constituent | Non-Purulent a (n = 213) | Purulent (n = 322) | Cystic Fibrosis (n = 468) |
|---|---|---|---|
| Water (mg/mL) | 959 ± 15 | 939 ± 17 | 862 ± 35 d |
| Macromolecular dry weight (MDW, mg/mL) | |||
| 43 ± 2.4 | 61 ± 7.6 d | 98 ± 13.4 d | |
| Protein, mg/mL | 13.6 ± 1.2 | 19.1 ± 4.9 d | 42.2 ± 19.4 d |
| Lipid, mg/mL | 10.9 ± 0.6 | 19.0 ± 1.5 d | 33.5 ± 4.1 d |
| Carbohydrate, mg/mL | 8.8 ± 1.1 | 12.4 ± 1.6 d | 14.6 ± 2.5 |
| DNA, mg/mL | 0.006 ± 0.003 | 0.88 ± 0.26 d | 2.88 ± 1.36 d |
| Protein % MDW | 31.6% | 31.3% | 43.1% |
| Lipid % MDW | 25.3% | 31.1% | 34.2% |
| CHO % MDW | 20.5% | 20.3% | 14.9% |
| DNA % MDW | <0.025% | 1.44% | 2.94% |
| (0.014% estimate) | |||
| Survey Sputa Compositional Data as Component Average % MDW e | |||
 | |||
| DNA % MDW of Sputa Evaluated for Intra-Specimen Heterogeneity f | |||
| DNA % MDW +/−S.D. | DNA % MDW range | ||
| Purulent non-CF (n = 5) | 1.4 +/−0.6 | 0.54–2.15 | |
| Cystic Fibrosis (n = 12 | 3.9 +/−2.5 | 0.95–8.7 | |
| DNA and Mucous Glycoprotein % MDW of CF Sputa Evaluated for Effects on Sputum Solubility g | |||
| CF Sputa (n = 87) | % MDW +/−S.D. | % MDW range | |
| DNA % MDW | 6.7 +/−2.2 | 2.9–9.8 | |
| GP % MDW | 4.6 +/−1.4 | 2.2–8.3 | |
| DNA + GP % MDW | 11.3 +/−1.7 | 6.1–14.2 | |
| Purulent Non-CF | Cystic Fibrosis | ||||
|---|---|---|---|---|---|
| Total DNA | Range | % Variation | Total DNA | Range | % Variation |
| 0.54 | 0.46–0.59 | 14.8 | 0.95 | 0.09–1.93 | 103.2 |
| 1.13 | 1.02–1.31 | 9.7 | 1.18 | 0.56–3.07 | 160.2 |
| 1.3 | 1.10–1.47 | 13.1 | 1.27 | 0.48–3.19 | 151.2 |
| 1.82 | 1.63–2.09 | 14.8 | 1.97 | 0.35–5.13 | 160.4 |
| 2.15 | 1.84–2.23 | 3.7 | 2.54 | 0.74–6.22 | 144.9 |
| 2.88 | 0.63–5.65 | 96.2 | |||
| 3.46 | 0.87–6.33 | 82.9 | |||
| 4.72 | 1.17–7.49 | 58.7 | |||
| 5.35 | 3.45–6.32 | 18.1 | |||
| 6.51 | 4.72–7.83 | 20.3 | |||
| 7.13 | 6.33–7.95 | 11.5 | |||
| 8.7 | 7.81–9.05 | 4.0 | |||
| range of % variation | range of % variation | ||||
| 3.7–14.8% | 4.0–160% | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chance, D.L.; Mawhinney, T.P. Observations of, and Insights into, Cystic Fibrosis Mucus Heterogeneity in the Pre-Modulator Era: Sputum Characteristics, DNA and Glycoprotein Content, and Solubilization Time. J. Respir. 2021, 1, 8-29. https://doi.org/10.3390/jor1010002
Chance DL, Mawhinney TP. Observations of, and Insights into, Cystic Fibrosis Mucus Heterogeneity in the Pre-Modulator Era: Sputum Characteristics, DNA and Glycoprotein Content, and Solubilization Time. Journal of Respiration. 2021; 1(1):8-29. https://doi.org/10.3390/jor1010002
Chicago/Turabian StyleChance, Deborah L., and Thomas P. Mawhinney. 2021. "Observations of, and Insights into, Cystic Fibrosis Mucus Heterogeneity in the Pre-Modulator Era: Sputum Characteristics, DNA and Glycoprotein Content, and Solubilization Time" Journal of Respiration 1, no. 1: 8-29. https://doi.org/10.3390/jor1010002
APA StyleChance, D. L., & Mawhinney, T. P. (2021). Observations of, and Insights into, Cystic Fibrosis Mucus Heterogeneity in the Pre-Modulator Era: Sputum Characteristics, DNA and Glycoprotein Content, and Solubilization Time. Journal of Respiration, 1(1), 8-29. https://doi.org/10.3390/jor1010002