
Surgical Management of Intrathoracic Triton Tumors: Insights into Emerging Molecular and Epigenetic Mechanisms with a Case Series of Three Patients

 , , , , , ,
, , , , , ,  , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
Cases Enrollment and Ethical Consent
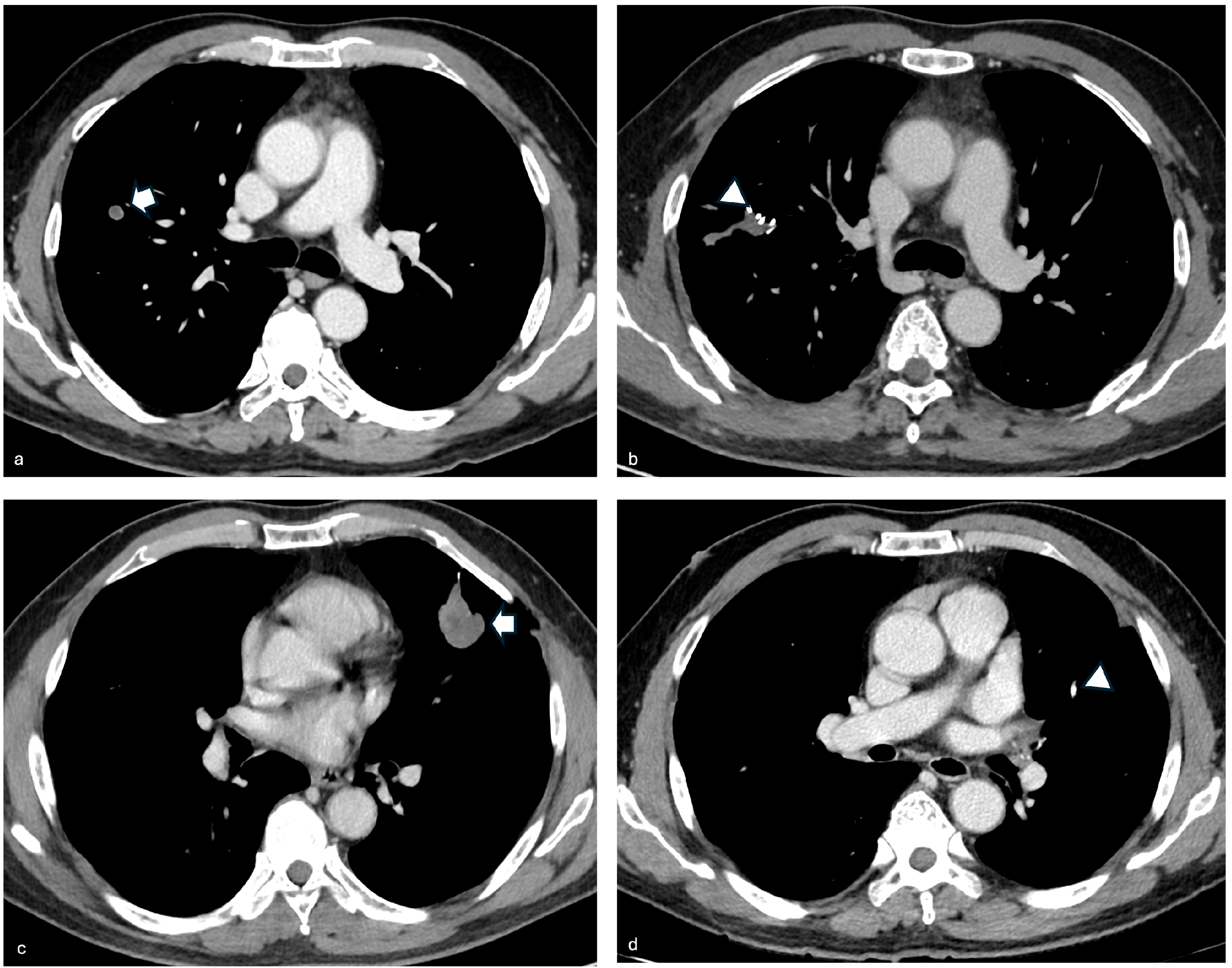
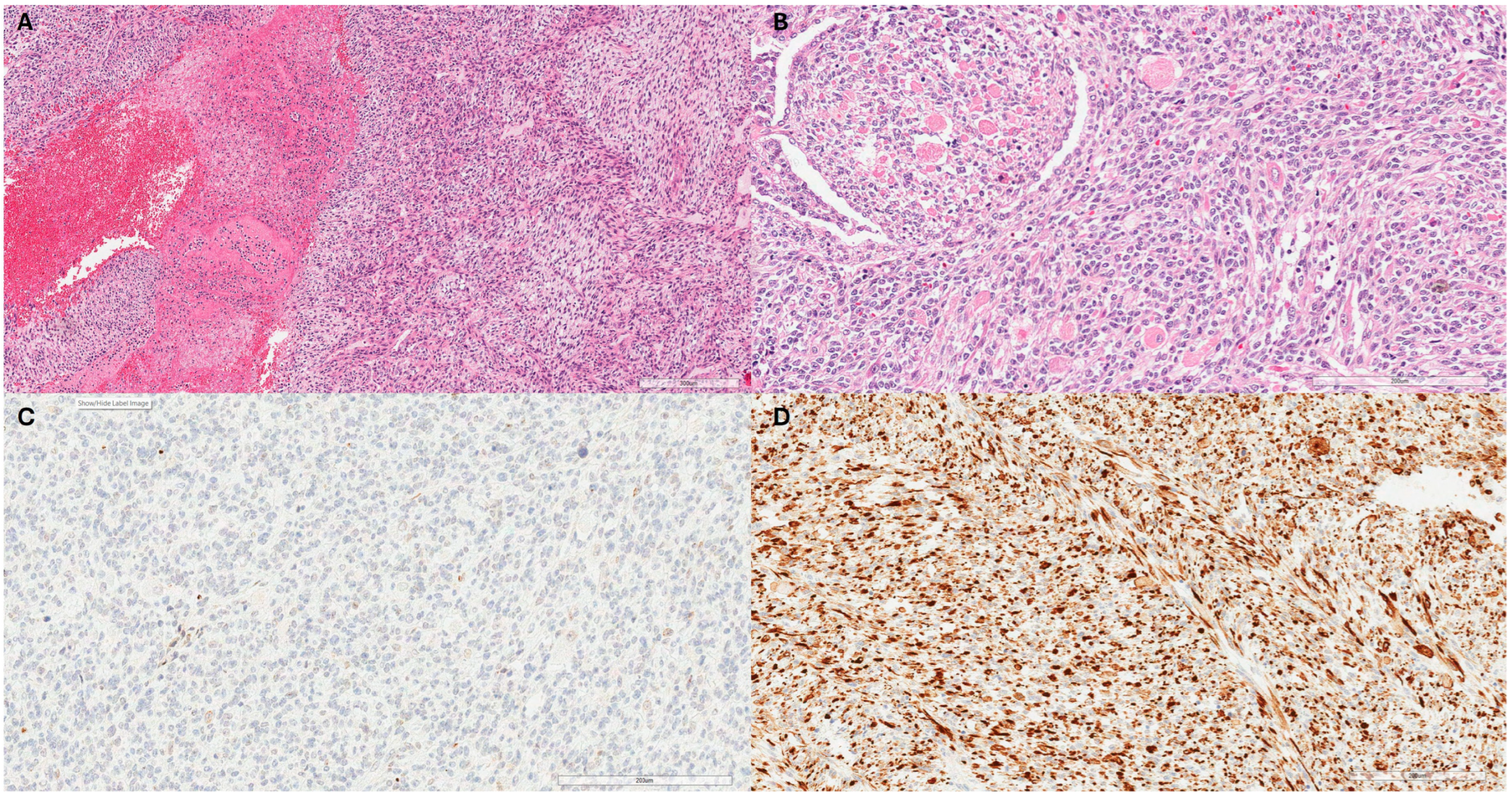
3. Case Series Presentation
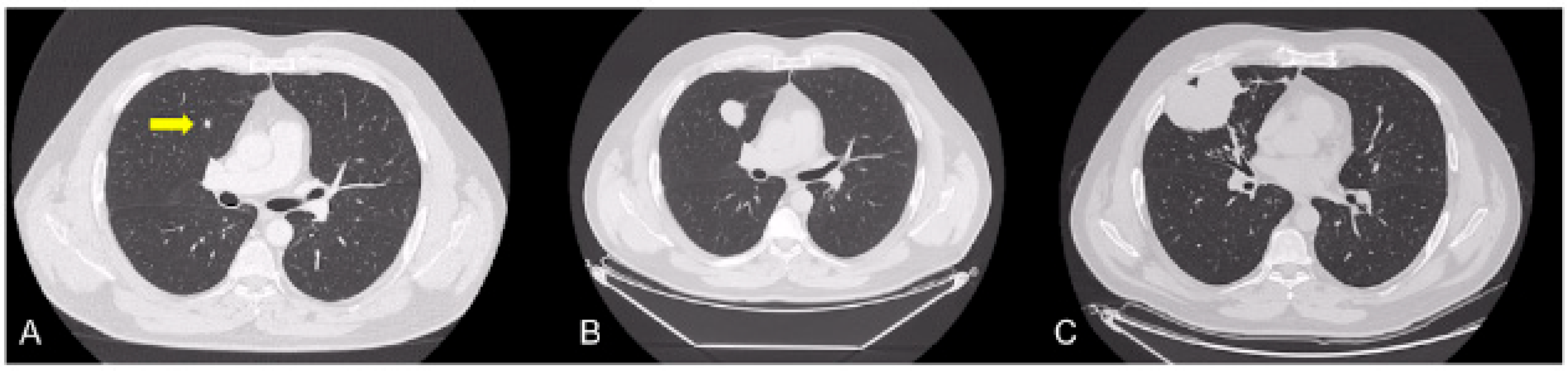
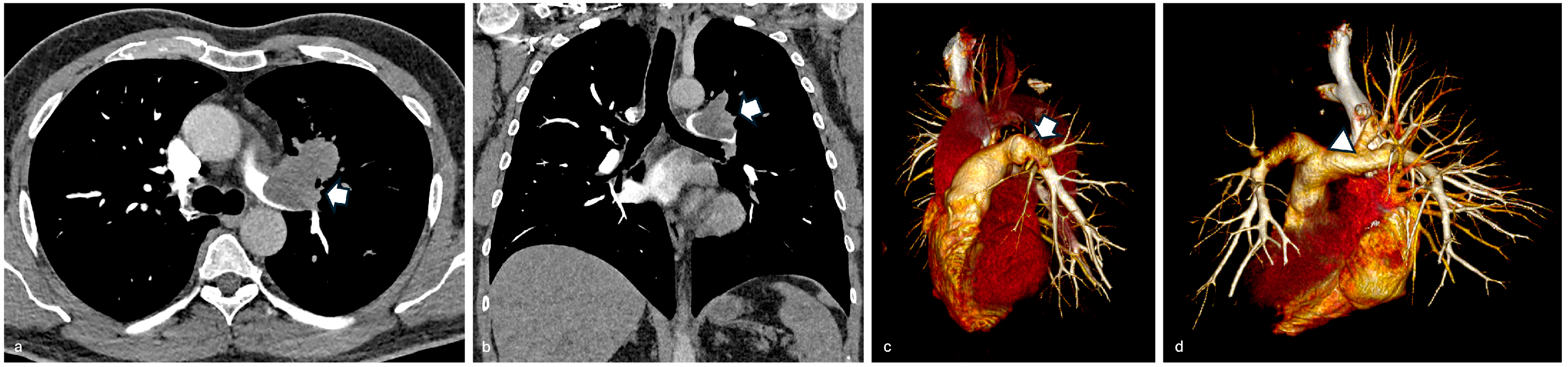
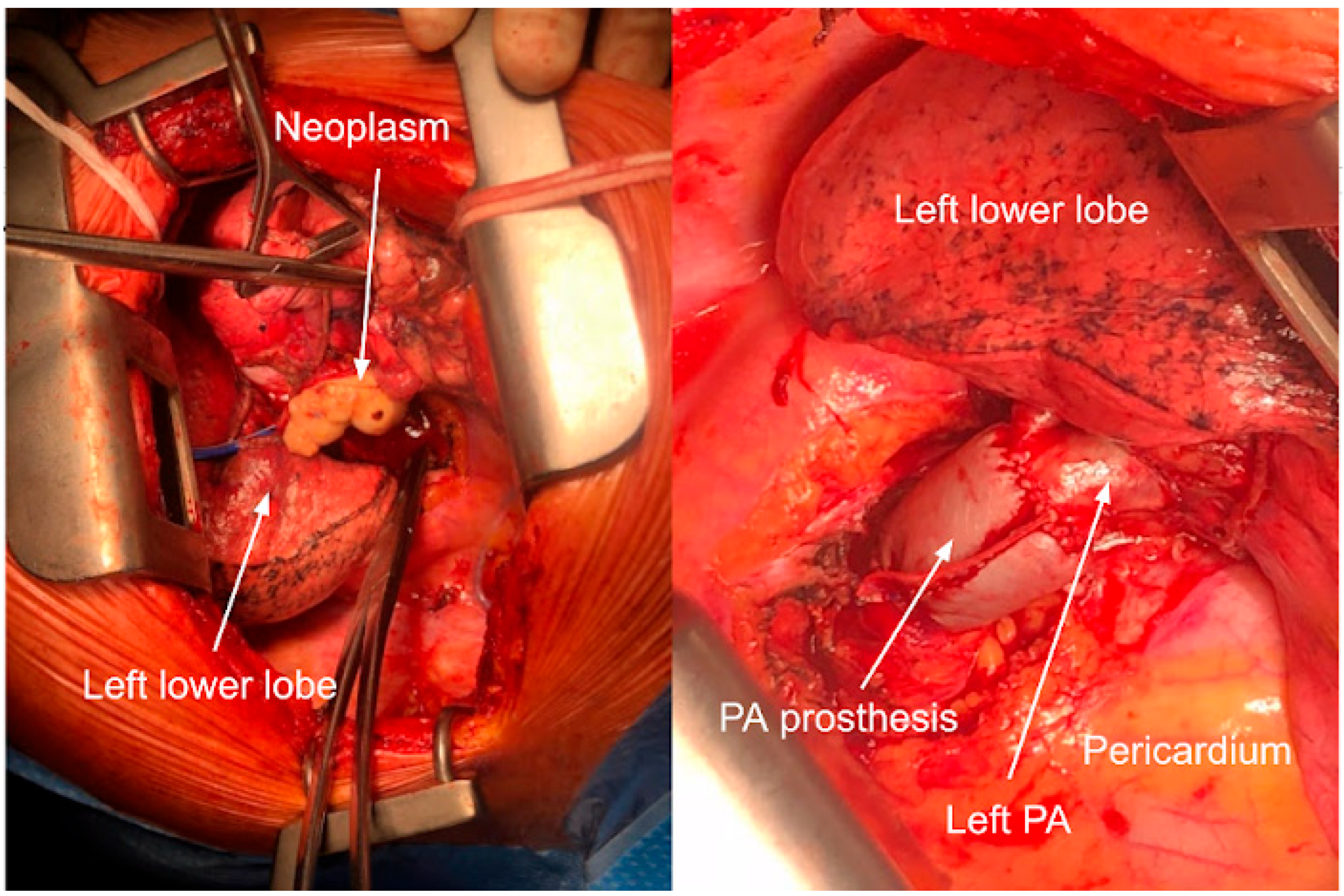
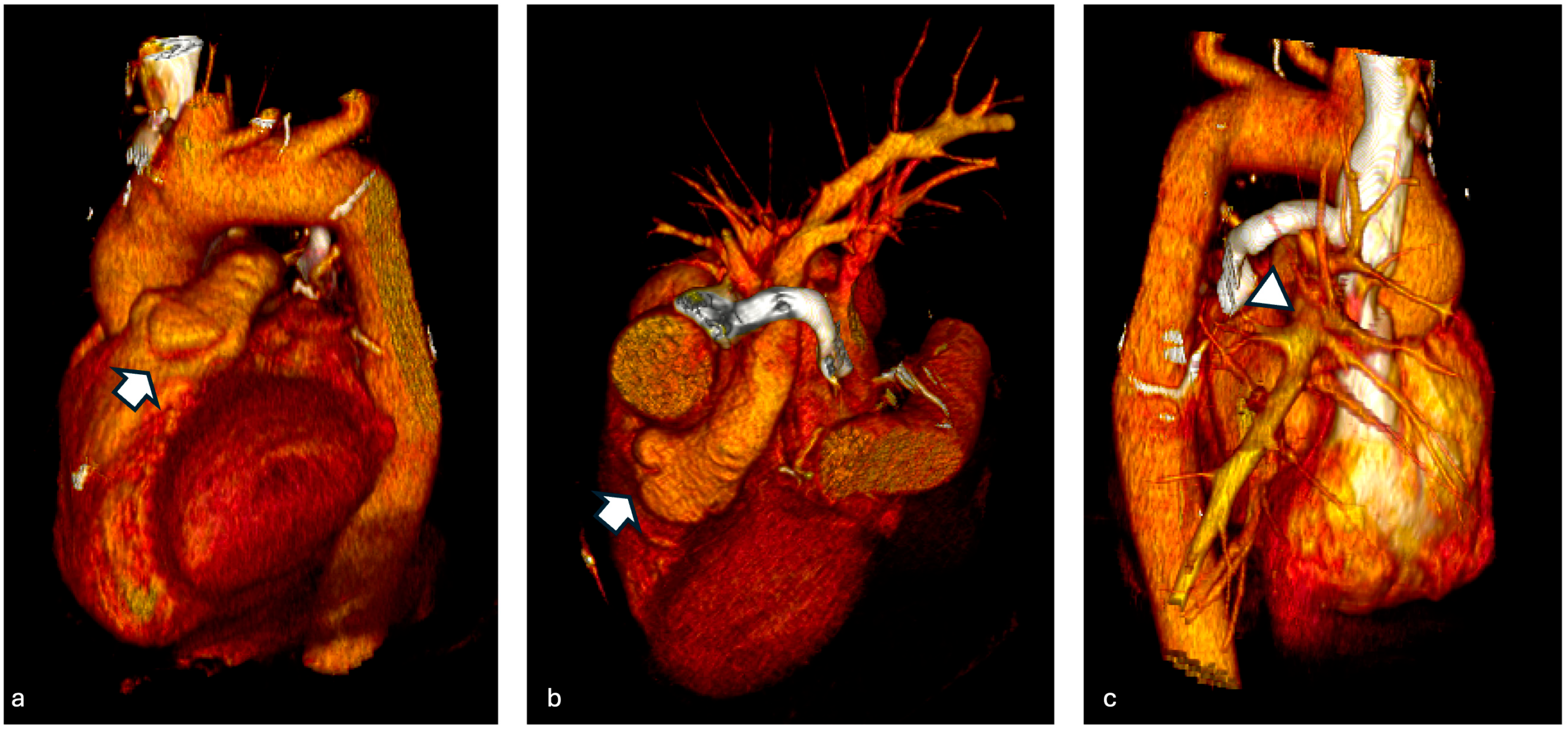
3.1. Case 1
3.2. Case 2
3.3. Case 3
4. Discussion
4.1. Molecular Scenario in MPNSTs and Recent Advances in Epigenome Investigations
4.2. Genetic Alterations in NF1-Associated and Sporadic MPNSTs
4.3. Genomic Instability and Wnt/β-Catenin Dysregulation
4.4. Non-Canonical Sonic Hedgehog (SHH) Signaling and Epigenetic Modifications
4.5. miRNA Dysregulation and Epigenetic Aberrations
4.6. Target Therapies, Pre-Clinical Experiments, and Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Definition of Malignant Triton Tumor—NCI Dictionary of Cancer Terms—NCI. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/malignant-triton-tumor (accessed on 27 December 2024).
- Victoria, L.; McCulloch, T.M.; Callaghan, E.J.; Bauman, N.M. Malignant Triton Tumor of the Head and Neck: A Case Report and Review of the Literature. Head Neck J. Sci. Spec. Head Neck 1999, 21, 663–670. [Google Scholar] [CrossRef]
- Stasik, C.J.; Tawfik, O. Malignant Peripheral Nerve Sheath Tumor with Rhabdomyosarcomatous Differentiation (Malignant Triton Tumor). Arch. Pathol. Lab. Med. 2006, 130, 1878–1881. [Google Scholar] [CrossRef] [PubMed]
- Locatelli: Formation de Membres Surnumeraires—Google Scholar. Available online: https://scholar.google.com/scholar_lookup?&title=Formation%20de%20membres%20surnum%C3%A9raires&journal=Assoc%20Anatomistes&volume=20&pages=279-82&publication_year=1925&author=Locatelli%2CP (accessed on 27 December 2024).
- Sharpe, J.C.; Young, R.H. Recklinghausen’s Neurofibromatosis: Clinical Manifestations in Thirty-One Cases. Arch. Intern. Med. 1937, 59, 299–328. [Google Scholar] [CrossRef]
- Woodruff, J.M.; Chernik, N.L.; Smith, M.C.; Millett, W.B.; Foote, F.W. Peripheral Nerve Tumors with Rhabdomyosarcomatous Differentiation (Malignant “Triton” Tumors). Cancer 1973, 32, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.M.N.; Lu, Q.R. Therapeutic Targets for Malignant Peripheral Nerve Sheath Tumors. Future Neurol. 2019, 14, FNL7. [Google Scholar] [CrossRef]
- Farid, M.; Demicco, E.G.; Garcia, R.; Ahn, L.; Merola, P.R.; Cioffi, A.; Maki, R.G. Malignant Peripheral Nerve Sheath Tumors. Oncologist 2014, 19, 193–201. [Google Scholar] [CrossRef]
- Somatilaka, B.N.; Sadek, A.; McKay, R.M.; Le, L.Q. Malignant Peripheral Nerve Sheath Tumor: Models, Biology, and Translation. Oncogene 2022, 41, 2405–2421. [Google Scholar] [CrossRef]
- Lee, K.S.; Im, J.G.; Kim, I.Y.; Kim, P.N.; Han, M.C.; Kim, C.W. Tumours Involving the Intrathoracic Vagus and Phrenic Nerves Demonstrated by Computed Tomography: Anatomical Features. Clin. Radiol. 1991, 44, 302–305. [Google Scholar] [CrossRef]
- Strauss, B.L.; Gutmann, D.H.; Dehner, L.P.; Hirbe, A.; Zhu, X.; Marley, E.F.; Liapis, H. Molecular Analysis of Malignant Triton Tumors. Hum. Pathol. 1999, 30, 984–988. [Google Scholar] [CrossRef]
- Thoenen, E.; Curl, A.; Iwakuma, T. TP53 in Bone and Soft Tissue Sarcomas. Pharmacol. Ther. 2019, 202, 149–164. [Google Scholar] [CrossRef]
- Nassif, E.F.; Auclin, E.; Bahleda, R.; Honoré, C.; Mir, O.; Dumont, S.; Mery, B.; Hodroj, K.; Brahmi, M.; Trédan, O.; et al. TP53 Mutation as a Prognostic and Predictive Marker in Sarcoma: Pooled Analysis of MOSCATO and ProfiLER Precision Medicine Trials. Cancers 2021, 13, 3362. [Google Scholar] [CrossRef] [PubMed]
- Magallón-Lorenz, M.; Terribas, E.; Ortega-Bertran, S.; Creus-Bachiller, E.; Fernández, M.; Requena, G.; Rosas, I.; Mazuelas, H.; Uriarte-Arrazola, I.; Negro, A.; et al. Deep Genomic Analysis of Malignant Peripheral Nerve Sheath Tumor Cell Lines Challenges Current Malignant Peripheral Nerve Sheath Tumor Diagnosis. iScience 2023, 26, 106096. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Zhou, H.; Dong, Y.; Alhaskawi, A.; Hasan Abdullah Ezzi, S.; Wang, Z.; Lai, J.; Goutham Kota, V.; Hasan Abdulla Hasan Abdulla, M.; Lu, H. Malignant Peripheral Nerve Sheath Tumors: Latest Concepts in Disease Pathogenesis and Clinical Management. Cancers 2023, 15, 1077. [Google Scholar] [CrossRef]
- Legius, E.; Marchuk, D.A.; Collins, F.S.; Glover, T.W. Somatic Deletion of the Neurofibromatosis Type 1 Gene in a Neurofibrosarcoma Supports a Tumour Suppressor Gene Hypothesis. Nat. Genet. 1993, 3, 122–126. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, Y.; Jones, S.; Sausen, M.; McMahon, K.; Sharma, R.; Wang, Q.; Belzberg, A.J.; Chaichana, K.; Gallia, G.L.; et al. Somatic Mutations of SUZ12 in Malignant Peripheral Nerve Sheath Tumors. Nat. Genet. 2014, 46, 1170–1172. [Google Scholar] [CrossRef]
- Upton, B.; Chu, Q.; Li, B.D.L. Li-Fraumeni Syndrome: The Genetics and Treatment Considerations for the Sarcoma and Associated Neoplasms. Surg. Oncol. Clin. N. Am. 2009, 18, 145–156, ix. [Google Scholar] [CrossRef] [PubMed]
- Nichols, K.E.; Malkin, D.; Garber, J.E.; Fraumeni, J.F.; Li, F.P. Germ-Line P53 Mutations Predispose to a Wide Spectrum of Early-Onset Cancers. Cancer Epidemiol. Biomark. Prev. 2001, 10, 83–87. [Google Scholar]
- Evans, D.G.; Burkitt-Wright, E.; Ealing, J.; Vassello, G.; Eelloo, J.; Lee, A. Six at Sixty. Malignant Peripheral Nerve Sheath Tumours in NF1: 20-Year Review of a Highly Cited Paper. J. Med. Genet. 2024, 61, 1132–1134. [Google Scholar] [CrossRef]
- Martinez-Font, E.; Pérez-Capó, M.; Vögler, O.; Martín-Broto, J.; Alemany, R.; Obrador-Hevia, A. WNT/β-Catenin Pathway in Soft Tissue Sarcomas: New Therapeutic Opportunities? Cancers 2021, 13, 5521. [Google Scholar] [CrossRef]
- Lesluyes, T.; Delespaul, L.; Coindre, J.-M.; Chibon, F. The CINSARC Signature as a Prognostic Marker for Clinical Outcome in Multiple Neoplasms. Sci. Rep. 2017, 7, 5480. [Google Scholar] [CrossRef]
- Hartmann, C. A Wnt Canon Orchestrating Osteoblastogenesis. Trends Cell Biol. 2006, 16, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Font, E.; Felipe-Abrio, I.; Calabuig-Fariñas, S.; Ramos, R.; Terrasa, J.; Vögler, O.; Alemany, R.; Martín-Broto, J.; Obrador-Hevia, A. Disruption of TCF/β-Catenin Binding Impairs Wnt Signaling and Induces Apoptosis in Soft Tissue Sarcoma Cells. Mol. Cancer Ther. 2017, 16, 1166–1176. [Google Scholar] [CrossRef] [PubMed]
- Kuhnen, C.; Herter, P.; Müller, O.; Muehlberger, T.; Krause, L.; Homann, H.; Steinau, H.U.; Müller, K.M. Beta-Catenin in Soft Tissue Sarcomas: Expression Is Related to Proliferative Activity in High-Grade Sarcomas. Mod. Pathol. 2000, 13, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Annavarapu, S.R.; Cialfi, S.; Dominici, C.; Kokai, G.K.; Uccini, S.; Ceccarelli, S.; McDowell, H.P.; Helliwell, T.R. Characterization of Wnt/β-Catenin Signaling in Rhabdomyosarcoma. Lab. Investig. 2013, 93, 1090–1099. [Google Scholar] [CrossRef]
- Duchartre, Y.; Kim, Y.-M.; Kahn, M. The Wnt Signaling Pathway in Cancer. Crit. Rev. Oncol. Hematol. 2016, 99, 141–149. [Google Scholar] [CrossRef]
- Reilly, K.M. Extending the Convergence of Canonical WNT Signaling and Classic Cancer Pathways for Treatment of Malignant Peripheral Nerve Sheath Tumors. Cancer Discov. 2013, 3, 610–612. [Google Scholar] [CrossRef]
- Ng, T.L.; Gown, A.M.; Barry, T.S.; Cheang, M.C.U.; Chan, A.K.W.; Turbin, D.A.; Hsu, F.D.; West, R.B.; Nielsen, T.O. Nuclear Beta-Catenin in Mesenchymal Tumors. Mod. Pathol. 2005, 18, 68–74. [Google Scholar] [CrossRef]
- Jeng, K.-S.; Chang, C.-F.; Lin, S.-S. Sonic Hedgehog Signaling in Organogenesis, Tumors, and Tumor Microenvironments. Int. J. Mol. Sci. 2020, 21, 758. [Google Scholar] [CrossRef]
- Banaszek, N.; Kurpiewska, D.; Kozak, K.; Rutkowski, P.; Sobczuk, P. Hedgehog Pathway in Sarcoma: From Preclinical Mechanism to Clinical Application. J. Cancer Res. Clin. Oncol. 2023, 149, 17635–17649. [Google Scholar] [CrossRef]
- Suppiah, S.; Mansouri, S.; Mamatjan, Y.; Liu, J.C.; Bhunia, M.M.; Patil, V.; Rath, P.; Mehani, B.; Heir, P.; Bunda, S.; et al. Multiplatform Molecular Profiling Uncovers Two Subgroups of Malignant Peripheral Nerve Sheath Tumors with Distinct Therapeutic Vulnerabilities. Nat. Commun. 2023, 14, 2696. [Google Scholar] [CrossRef]
- Amirnasr, A.; Verdijk, R.M.; van Kuijk, P.F.; Kartal, P.; Vriends, A.L.M.; French, P.J.; van Royen, M.E.; Taal, W.; Sleijfer, S.; Wiemer, E.A.C. Deregulated microRNAs in Neurofibromatosis Type 1 Derived Malignant Peripheral Nerve Sheath Tumors. Sci. Rep. 2020, 10, 2927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Murray, B.; Mo, G.; Shern, J.F. The Role of Polycomb Repressive Complex in Malignant Peripheral Nerve Sheath Tumor. Genes 2020, 11, 287. [Google Scholar] [CrossRef] [PubMed]
- González-Muñoz, T.; Kim, A.; Ratner, N.; Peinado, H. The Need for New Treatments Targeting MPNST: The Potential of Strategies Combining MEK Inhibitors with Antiangiogenic Agents. Clin. Cancer Res. 2022, 28, 3185–3195. [Google Scholar] [CrossRef] [PubMed]
- De Raedt, T.; Walton, Z.; Yecies, J.L.; Li, D.; Chen, Y.; Malone, C.F.; Maertens, O.; Jeong, S.M.; Bronson, R.T.; Lebleu, V.; et al. Exploiting Cancer Cell Vulnerabilities to Develop a Combination Therapy for Ras-Driven Tumors. Cancer Cell 2011, 20, 400–413. [Google Scholar] [CrossRef]
- Somaiah, N.; Van Tine, B.A.; Chmielowski, B.; Drabick, J.J.; Chawla, S.P.; McKean, M.; Spira, A.I.; O’Byrne, K.J.; Foresto, S.A.; Movva, S.; et al. A Phase 2 Study of Alrizomadlin, a Novel MDM2/P53 Inhibitor, in Combination with Pembrolizumab for Treatment of Patients with Malignant Peripheral Nerve Sheath Tumor (MPNST). J. Clin. Oncol. 2023, 41, e14627. [Google Scholar] [CrossRef]
- Deyle, D.R.; Escobar, D.Z.; Peng, K.-W.; Babovic-Vuksanovic, D. Oncolytic Measles Virus as a Novel Therapy for Malignant Peripheral Nerve Sheath Tumors. Gene 2015, 565, 140–145. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Age (Years) | Gender | Surgery | Chemotherapy | Radiotherapy | Outcome |
|---|---|---|---|---|---|---|
| Case 1 | 55 | Male | Yes (radical) | Etoposide + Epirubicin | Yes | Alive, no recurrence |
| Case 2 | 56 | Male | Yes (subtotal) | - | Yes | Deceased (18 months after surgery) |
| Case 3 | 56 | Male | Yes (subtotal) | VAC protocol | Yes | Alive, local recurrence |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonis, A.; Busetto, A.; Pezzuto, F.; Pagliarini, G.; Verzeletti, V.; Pezzella, M.; Cannone, G.; Faccioli, E.; Mammana, M.; Comacchio, G.M.; et al. Surgical Management of Intrathoracic Triton Tumors: Insights into Emerging Molecular and Epigenetic Mechanisms with a Case Series of Three Patients. J. Mol. Pathol. 2025, 6, 10. https://doi.org/10.3390/jmp6020010
Bonis A, Busetto A, Pezzuto F, Pagliarini G, Verzeletti V, Pezzella M, Cannone G, Faccioli E, Mammana M, Comacchio GM, et al. Surgical Management of Intrathoracic Triton Tumors: Insights into Emerging Molecular and Epigenetic Mechanisms with a Case Series of Three Patients. Journal of Molecular Pathology. 2025; 6(2):10. https://doi.org/10.3390/jmp6020010
Chicago/Turabian StyleBonis, Alessandro, Alberto Busetto, Federica Pezzuto, Giulia Pagliarini, Vincenzo Verzeletti, Mario Pezzella, Giorgio Cannone, Eleonora Faccioli, Marco Mammana, Giovanni Maria Comacchio, and et al. 2025. "Surgical Management of Intrathoracic Triton Tumors: Insights into Emerging Molecular and Epigenetic Mechanisms with a Case Series of Three Patients" Journal of Molecular Pathology 6, no. 2: 10. https://doi.org/10.3390/jmp6020010
APA StyleBonis, A., Busetto, A., Pezzuto, F., Pagliarini, G., Verzeletti, V., Pezzella, M., Cannone, G., Faccioli, E., Mammana, M., Comacchio, G. M., Rebusso, A., Schiavon, M., Giraudo, C., Calabrese, F., Dell’Amore, A., Nicotra, S., Dei Tos, A. P., & Rea, F. (2025). Surgical Management of Intrathoracic Triton Tumors: Insights into Emerging Molecular and Epigenetic Mechanisms with a Case Series of Three Patients. Journal of Molecular Pathology, 6(2), 10. https://doi.org/10.3390/jmp6020010