
Mechanisms of Inflammasome Activation and Involvement in Liver Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
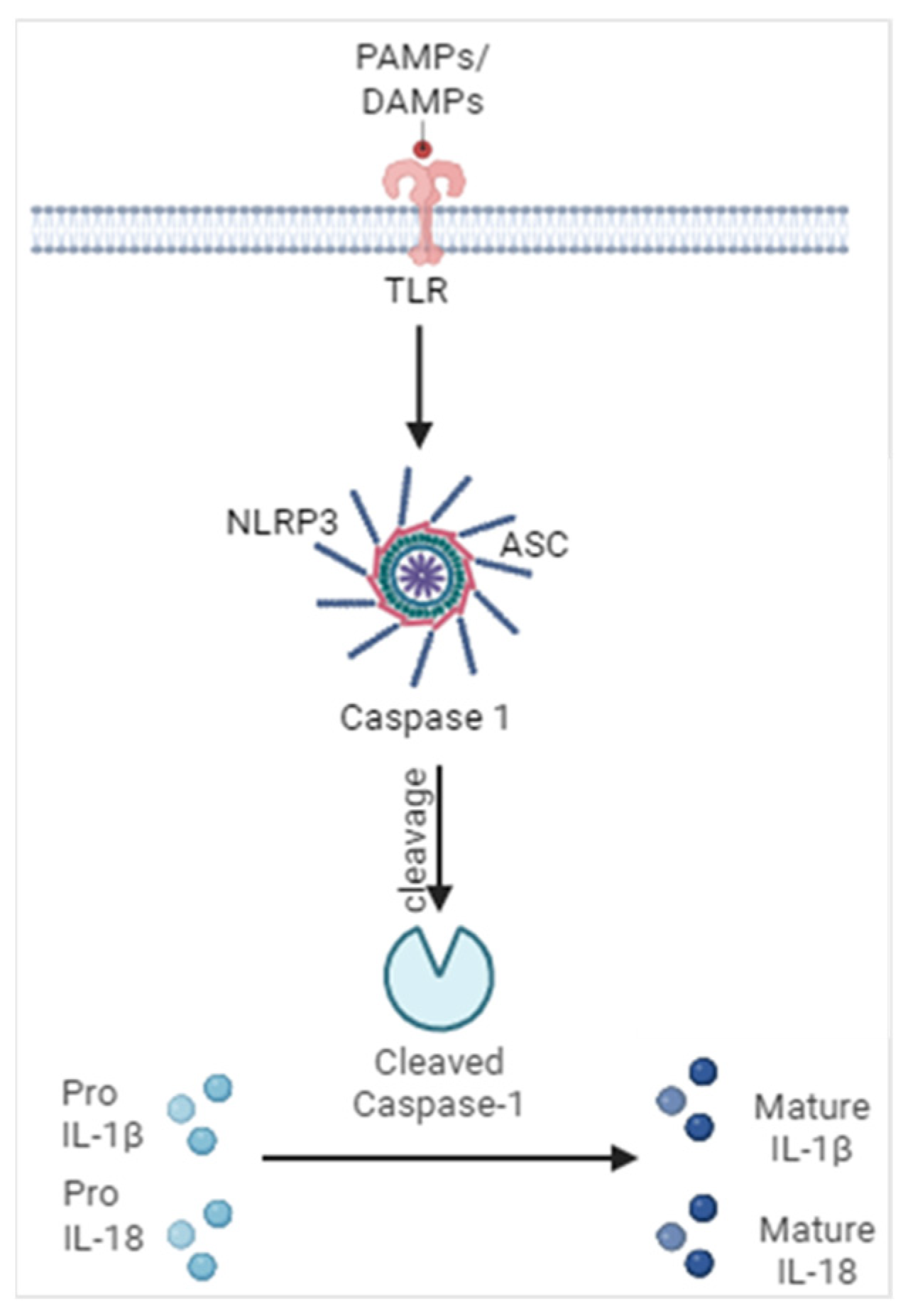
2. Mechanisms Leading to Inflammasome Activation
2.1. Endoplasmic Reticulum Stress (ER Stress)
2.2. Reactive Oxygen Species (ROS)
2.3. Ions and Inflammasomes
2.4. Proteases
2.5. Complement System
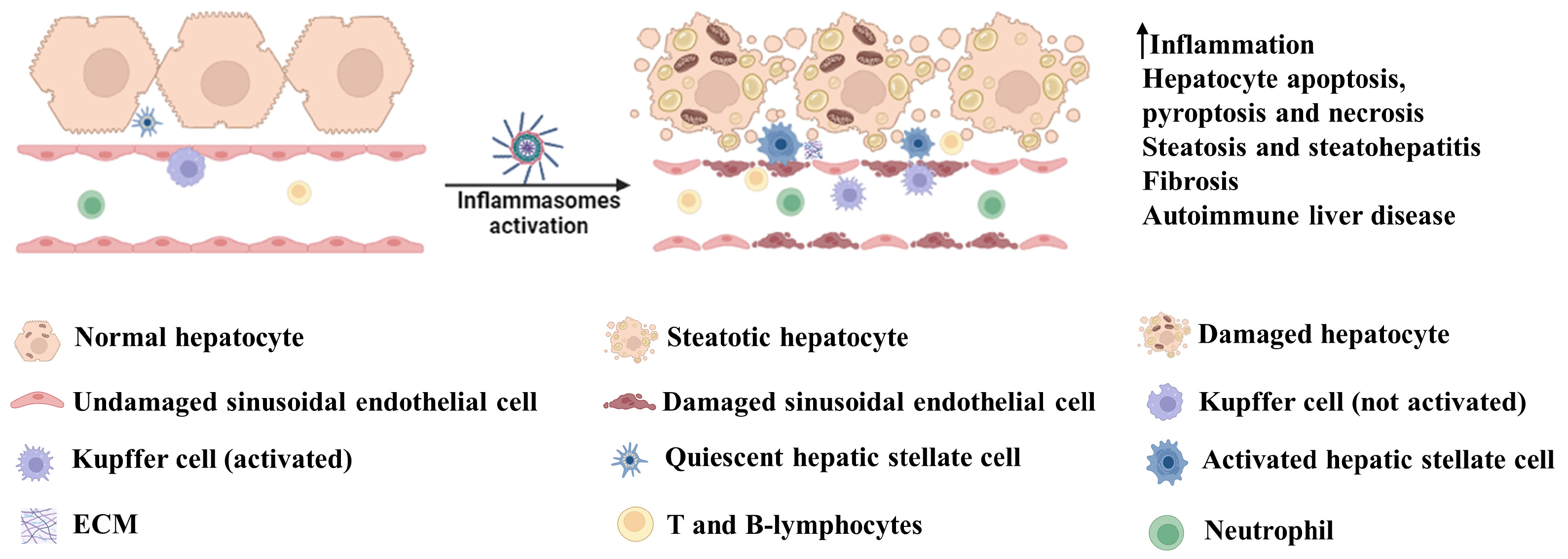
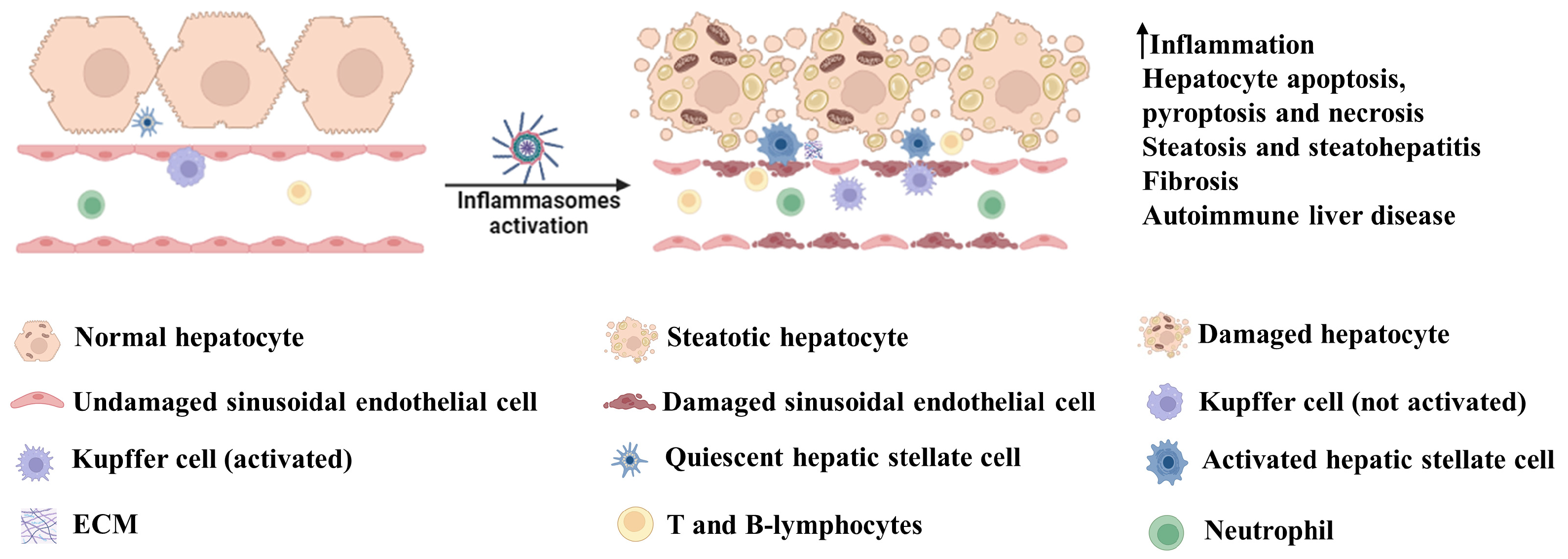
3. Involvement of Inflammasome Activation in Liver Diseases
3.1. Acetaminophen (APAP)-Induced Hepatotoxicity
3.2. High-Fat Diet-Induced Hepatotoxicity
3.3. Hormone-Induced Hepatotoxicity
3.4. Alcohol-Induced Hepatotoxicity
3.5. Endotoxin-Induced Hepatotoxicity
3.6. Viral Hepatitis
3.7. Autoimmune Liver Disease
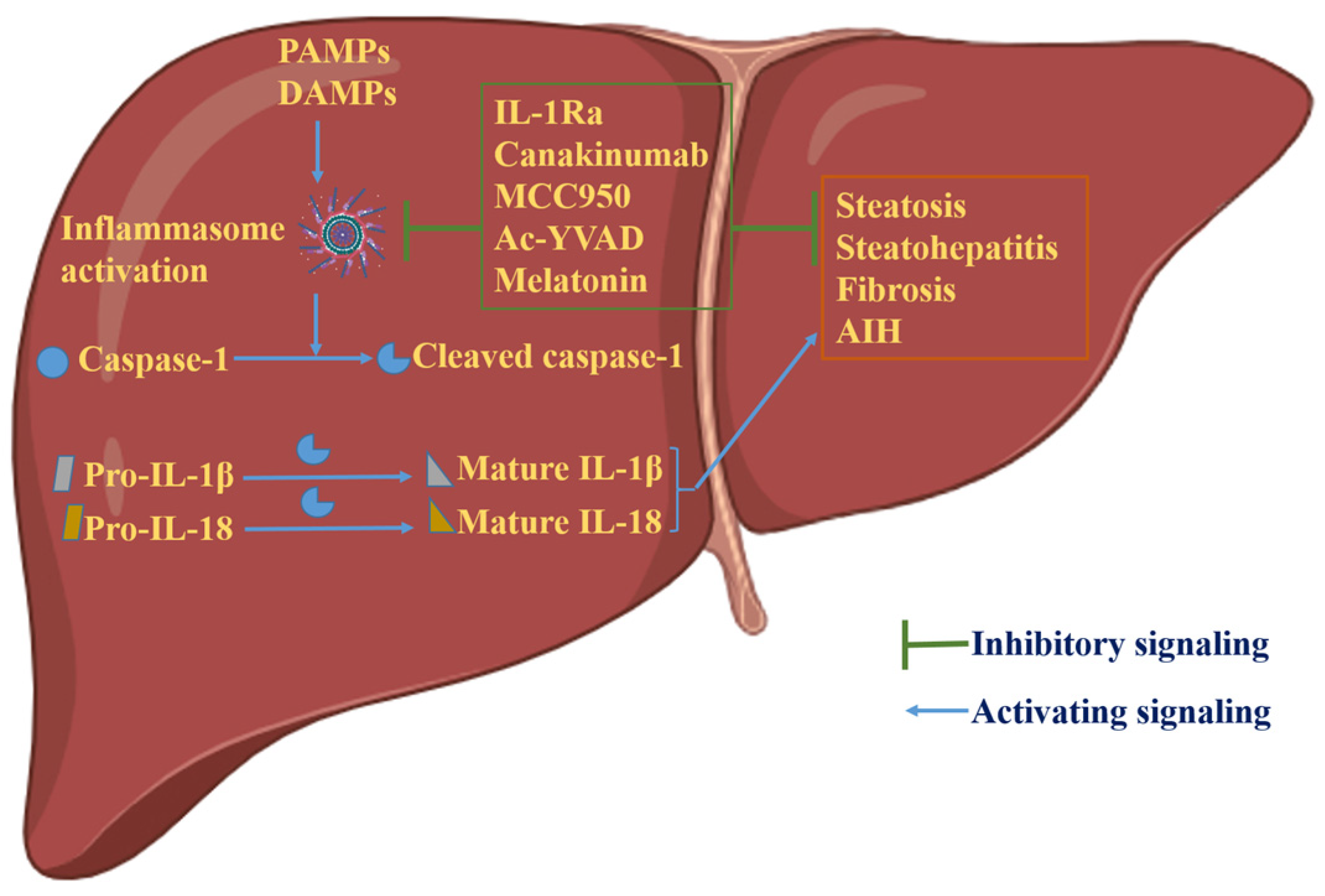
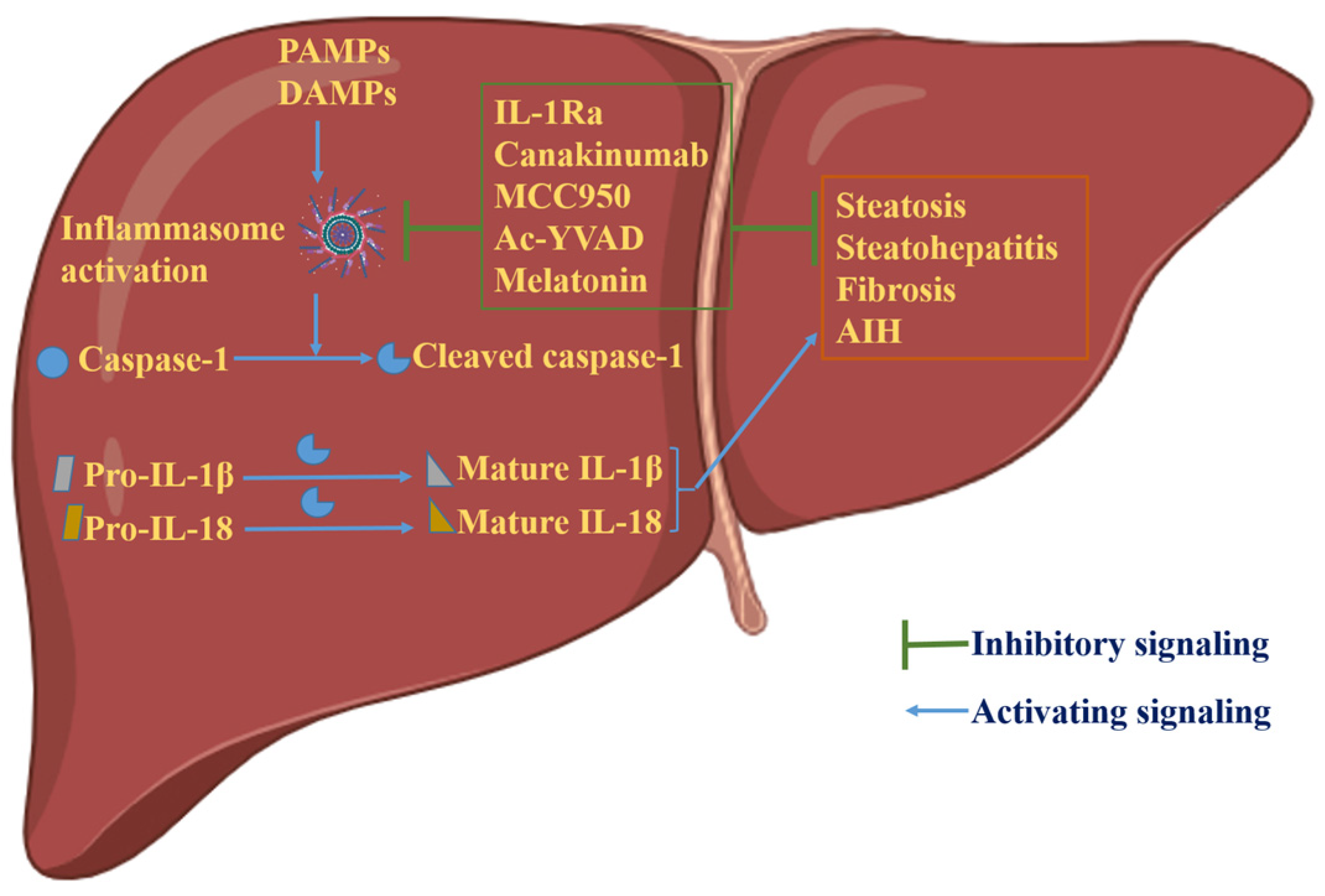
4. Inflammasome-Dependent Therapies in Liver Disease
4.1. Anakinra (IL-1Ra)
4.2. Canakinumab
4.3. MCC950
4.4. Ac-YVAD
4.5. Gasdermin D Inhibitors
4.6. IL-18 Inhibitors
4.7. Melatonin
5. Inflammasome Activation Is Not Always Detrimental
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AIM2 | Absent in melanoma 2 |
| ANA | Antinuclear antibodies |
| ASGPR | Asialogylcoprotein receptor |
| ASC | Apoptosis-associated speck like protein containing a CARD |
| cAMP | Cyclic adenosine monophosphate |
| CARD | Caspase activation and recruitment domain |
| DAMPs | Damage-Associated Molecular Patterns |
| FDA | Food and Drug Administration |
| IP3R | Inositol triphosphate receptor |
| LKM1 | Liver kidney microsomal antibody type 1 |
| LPS | Lipopolysaccharide |
| MAC | Membrane attack complex |
| NASH | Non-alcoholic steatohepatitis |
| NLRP3 | Nucleotide-binding domain, leucine-rich-containing family, pyrin domain-3 |
| PAMPs | Pathogen-Associated Molecular Patterns |
| PYD | Pyrin domain |
| ROS | Reactive oxygen species |
| RyR | Ryanodine receptor |
| TCE | Trichloroethane |
References
- Benseler, V.; Warren, A.; Vo, M.; Holz, L.E.; Tay, S.S.; Le Couteur, D.G.; Breen, E.; Allison, A.C.; van Rooijen, N.; McGuffog, C.; et al. Hepatocyte entry leads to degradation of autoreactive CD8 T cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16735–16740. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cytokine amplification and inhibition of immune and inflammatory responses. J. Viral Hepat. 1997, 4 (Suppl. S2), 6–15. [Google Scholar] [CrossRef] [PubMed]
- Andus, T.; Bauer, J.; Gerok, W. Effects of cytokines on the liver. Hepatology 1991, 13, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Xu, M.J.; Gao, B. Hepatocytes: A key cell type for innate immunity. Cell. Mol. Immunol. 2016, 13, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Zhang, Z.; Lin, F.; Zou, Z.-S.; Xu, R.-N.; Jin, L.; Fu, J.-L.; Shi, F.; Shi, M.; Wang, H.-F.; et al. Interleukin-17–producing CD4+ T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology 2010, 51, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 2011, 166, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.; Chang, S.H.; Martinez, G.J.; Yang, X.O.; Nurieva, R.; Kang, H.S.; Ma, L.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 2009, 30, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.; Mehal, W.Z.; Crispe, I.N. IL-18 augments perforin-dependent cytotoxicity of liver NK-T cells. J. Immunol. 1998, 161, 2217–2222. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.D.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Van Opdenbosch, N.; Gurung, P.; Vande Walle, L.; Fossoul, A.; Kanneganti, T.D.; Lamkanfi, M. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat. Commun 2014, 5, 3209. [Google Scholar] [CrossRef] [PubMed]
- Afonina, I.S.; Müller, C.; Martin, S.J.; Beyaert, R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity 2015, 42, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Xue, W.; Wei, H.; Fan, Q.; Li, X.; Qiu, Y.; Cui, D. Research Progress of Pyroptosis in Fatty Liver Disease. Int. J. Mol. Sci. 2023, 24, 13065. [Google Scholar] [CrossRef] [PubMed]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 2021, 74, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Ju, D. Inflammasome: A Double-Edged Sword in Liver Diseases. Front. Immunol. 2018, 9, 2201. [Google Scholar] [CrossRef]
- Schaffert, C.S.; Duryee, M.J.; Hunter, C.D.; Hamilton, B.C., 3rd; DeVeney, A.L.; Huerter, M.M.; Klassen, L.W.; Thiele, G.M. Alcohol metabolites and lipopolysaccharide: Roles in the development and/or progression of alcoholic liver disease. World J. Gastroenterol. 2009, 15, 1209–1218. [Google Scholar] [CrossRef]
- Tapia, V.S.; Daniels, M.J.D.; Palazón-Riquelme, P.; Dewhurst, M.; Luheshi, N.M.; Rivers-Auty, J.; Green, J.; Redondo-Castro, E.; Kaldis, P.; Lopez-Castejon, G.; et al. The three cytokines IL-1β, IL-18, and IL-1α share related but distinct secretory routes. J. Biol. Chem. 2019, 294, 8325–8335. [Google Scholar] [CrossRef]
- Charan, H.V.; Dwivedi, D.K.; Khan, S.; Jena, G. Mechanisms of NLRP3 inflammasome-mediated hepatic stellate cell activation: Therapeutic potential for liver fibrosis. Genes Dis. 2023, 10, 480–494. [Google Scholar] [CrossRef]
- Tan, Z.; Liu, Q.; Jiang, R.; Lv, L.; Shoto, S.S.; Maillet, I.; Quesniaux, V.; Tang, J.; Zhang, W.; Sun, B.; et al. Interleukin-33 drives hepatic fibrosis through activation of hepatic stellate cells. Cell. Mol. Immunol. 2018, 15, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Van Sweringen, H.L.; Quillin, R.C.; Schuster, R.; Blanchard, J.; Burns, J.M.; Tevar, A.D.; Edwards, M.J.; Lentsch, A.B. Interleukin-33 is hepatoprotective during liver ischemia/reperfusion in mice. Hepatology 2012, 56, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Volarevic, V.; Mitrovic, M.; Milovanovic, M.; Zelen, I.; Nikolic, I.; Mitrovic, S.; Pejnovic, N.; Arsenijevic, N.; Lukic, M.L. Protective role of IL-33/ST2 axis in Con A-induced hepatitis. J. Hepatol. 2012, 56, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J.P. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc. Natl. Acad. Sci. USA 2009, 106, 9021–9026. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Baird, T.D.; Zhou, D.; Palam, L.R.; Spandau, D.F.; Wek, R.C. Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J. Biol. Chem. 2010, 285, 33165–33174. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wen, Y.; Lv, L.L.; Liu, H.; Tang, R.N.; Ma, K.L.; Liu, B.C. Involvement of endoplasmic reticulum stress in angiotensin II-induced NLRP3 inflammasome activation in human renal proximal tubular cells in vitro. Acta Pharmacol. Sin. 2015, 36, 821–830. [Google Scholar] [CrossRef]
- Li, W.; Cao, T.; Luo, C.; Cai, J.; Zhou, X.; Xiao, X.; Liu, S. Crosstalk between ER stress, NLRP3 inflammasome, and inflammation. Appl. Microbiol. Biotechnol. 2020, 104, 6129–6140. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Kolliputi, N.; Shaik, R.S.; Waxman, A.B. The inflammasome mediates hyperoxia-induced alveolar cell permeability. J. Immunol. 2010, 184, 5819–5826. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Yi, T.; Chen, C. NF-κB-Gasdermin D (GSDMD) Axis Couples Oxidative Stress and NACHT, LRR and PYD Domains-Containing Protein 3 (NLRP3) Inflammasome-Mediated Cardiomyocyte Pyroptosis Following Myocardial Infarction. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 6044–6052. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process. Int. J. Mol. Sci. 2013, 14, 3834–3859. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.-g. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, K.; Zhang, K.; Zhou, F.; Zhu, L. Induction of oxidative and nitrosative stresses in human retinal pigment epithelial cells by all-trans-retinal. Exp. Cell Res. 2016, 348, 87–94. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Negash, A.A.; Olson, R.M.; Griffin, S.; Gale, M., Jr. Modulation of calcium signaling pathway by hepatitis C virus core protein stimulates NLRP3 inflammasome activation. PLOS Pathog. 2019, 15, e1007593. [Google Scholar] [CrossRef]
- Lee, G.-S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef]
- Yamashima, T.; Kohda, Y.; Tsuchiya, K.; Ueno, T.; Yamashita, J.; Yoshioka, T.; Kominami, E. Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA-074: A novel strategy for neuroprotection based on ‘calpain-cathepsin hypothesis’. Eur. J. Neurosci. 1998, 10, 1723–1733. [Google Scholar] [CrossRef]
- Liu, X.; Li, M.; Chen, Z.; Yu, Y.; Shi, H.; Yu, Y.; Wang, Y.; Chen, R. Mitochondrial calpain-1 activates NLRP3 inflammasome by cleaving ATP5A1 and inducing mitochondrial ROS in CVB3-induced myocarditis. 2022, 117, 40. [CrossRef]
- Chevriaux, A.; Pilot, T.; Derangère, V.; Simonin, H.; Martine, P.; Chalmin, F.; Ghiringhelli, F.; Rébé, C. Cathepsin B Is Required for NLRP3 Inflammasome Activation in Macrophages, Through NLRP3 Interaction. Front. Cell Dev. Biol. 2020, 8, 167. [Google Scholar] [CrossRef] [PubMed]
- Baral, A.; Park, P.-H. Leptin Induces Apoptotic and Pyroptotic Cell Death via NLRP3 Inflammasome Activation in Rat Hepatocytes. Int. J. Mol. Sci. 2021, 22, 12589. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Mo, S.T. Caspase-8 inactivation drives autophagy-dependent inflammasome activation in myeloid cells. Sci. Adv. 2022, 8, eabn9912. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Hughes, T.R.; Triantafilou, M.; Morgan, B.P. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci. 2013, 126, 2903–2913. [Google Scholar] [CrossRef] [PubMed]
- Samstad, E.O.; Niyonzima, N.; Nymo, S.; Aune, M.H.; Ryan, L.; Bakke, S.S.; Lappegård, K.T.; Brekke, O.L.; Lambris, J.D.; Damås, J.K.; et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J. Immunol. 2014, 192, 2837–2845. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, J.; Teng, Y.; Sun, H.; Tian, G.; He, L.; Li, P.; Chen, Y.; Guo, Y.; Li, J.; et al. Complement Receptor C5aR1 Inhibition Reduces Pyroptosis in hDPP4-Transgenic Mice Infected with MERS-CoV. Viruses 2019, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Mazaleuskaya, L.L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenetics Genom. 2015, 25, 416–426. [Google Scholar] [CrossRef] [PubMed]
- El-Hassan, H.; Anwar, K.; Macanas-Pirard, P.; Crabtree, M.; Chow, S.C.; Johnson, V.L.; Lee, P.C.; Hinton, R.H.; Price, S.C.; Kass, G.E. Involvement of mitochondria in acetaminophen-induced apoptosis and hepatic injury: Roles of cytochrome c, Bax, Bid, and caspases. Toxicol. Appl. Pharmacol. 2003, 191, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Kil, Y.-S.; Baral, A.; Jeong, B.-S.; Laatikainen, P.; Liu, Y.; Han, A.-R.; Hong, M.-J.; Kim, J.-B.; Choi, H.; Park, P.-H.; et al. Combining NMR and MS to Describe Pyrrole-2-Carbaldehydes in Wheat Bran of Radiation. J. Agric. Food Chem. 2022, 70, 13002–13014. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Jaeschke, H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J. Hepatol. 2017, 66, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Farhood, A.; Jaeschke, H. Role of caspase-1 and interleukin-1beta in acetaminophen-induced hepatic inflammation and liver injury. Toxicol. Appl. Pharmacol. 2010, 247, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Velázquez, K.T.; Enos, R.T.; Bader, J.E.; Sougiannis, A.T.; Carson, M.S.; Chatzistamou, I.; Carson, J.A.; Nagarkatti, P.S.; Nagarkatti, M.; Murphy, E.A. Prolonged high-fat-diet feeding promotes non-alcoholic fatty liver disease and alters gut microbiota in mice. World J. Hepatol. 2019, 11, 619–637. [Google Scholar] [CrossRef] [PubMed]
- Kamari, Y.; Shaish, A.; Vax, E.; Shemesh, S.; Kandel-Kfir, M.; Arbel, Y.; Olteanu, S.; Barshack, I.; Dotan, S.; Voronov, E.; et al. Lack of interleukin-1α or interleukin-1β inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J. Hepatol. 2011, 55, 1086–1094. [Google Scholar] [CrossRef]
- Gehrke, N.; Hofmann, L.J.; Straub, B.K.; Rühle, F. Hepatic interleukin-1 receptor type 1 signalling regulates insulin sensitivity in the early phases of nonalcoholic fatty liver disease. Clin. Transl. Med. 2022, 12, e1048. [Google Scholar] [CrossRef]
- Mirea, A.M.; Tack, C.J.; Chavakis, T.; Joosten, L.A.B.; Toonen, E.J.M. IL-1 Family Cytokine Pathways Underlying NAFLD: Towards New Treatment Strategies. Trends Mol. Med. 2018, 24, 458–471. [Google Scholar] [CrossRef]
- Vargas-Pozada, E.E.; Ramos-Tovar, E.; Rodriguez-Callejas, J.D.; Cardoso-Lezama, I.; Galindo-Gómez, S.; Gil-Becerril, K.; Vásquez-Garzón, V.R.; Arellanes-Robledo, J.; Tsutsumi, V.; Villa-Treviño, S.; et al. Activation of the NLRP3 inflammasome by CCl(4) exacerbates hepatopathogenic diet-induced experimental NASH. Ann. Hepatol. 2023, 28, 100780. [Google Scholar] [CrossRef]
- Xia, M.; Boini, K.M.; Abais, J.M.; Xu, M.; Zhang, Y.; Li, P.L. Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am. J. Pathol. 2014, 184, 1617–1628. [Google Scholar] [CrossRef]
- Koka, S.; Xia, M.; Zhang, C.; Zhang, Y.; Li, P.L.; Boini, K.M. Podocyte NLRP3 Inflammasome Activation and Formation by Adipokine Visfatin. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2019, 53, 355–365. [Google Scholar] [CrossRef]
- Ma, X.; Zhou, Y.; Qiao, B.; Jiang, S.; Shen, Q.; Han, Y.; Liu, A.; Chen, X.; Wei, L.; Zhou, L.; et al. Androgen aggravates liver fibrosis by activation of NLRP3 inflammasome in CCl(4)-induced liver injury mouse model. 2020, 318, E817–E829. [CrossRef]
- Zhang, L.L.; Huang, S.; Ma, X.X.; Zhang, W.Y.; Wang, D.; Jin, S.Y.; Zhang, Y.P.; Li, Y.; Li, X. Angiotensin(1-7) attenuated Angiotensin II-induced hepatocyte EMT by inhibiting NOX-derived H2O2-activated NLRP3 inflammasome/IL-1β/Smad circuit. Free Radic. Biol. Med. 2016, 97, 531–543. [Google Scholar] [CrossRef]
- Sheriff, L.; Lalor, P.F. The Impact of the NLRP3 Pathway in the Pathogenesis of Non-Alcoholic Fatty Liver Disease and Alcohol-Related Liver Disease. Livers 2021, 1, 68–81. [Google Scholar] [CrossRef]
- Purohit, V.; Bode, J.C.; Bode, C.; Brenner, D.A.; Choudhry, M.A.; Hamilton, F.; Kang, Y.J.; Keshavarzian, A.; Rao, R.; Sartor, R.B.; et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: Summary of a symposium. Alcohol 2008, 42, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Donohue, T.M., Jr. Alcohol-induced steatosis in liver cells. World J. Gastroenterol. 2007, 13, 4974–4978. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef]
- Ganz, M.; Csak, T.; Nath, B.; Szabo, G. Lipopolysaccharide induces and activates the Nalp3 inflammasome in the liver. World J. Gastroenterol. 2011, 17, 4772–4778. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Nishiguchi, S. Importance of Kupffer Cells in the Development of Acute Liver Injuries in Mice. Int. J. Mol. Sci. 2014, 15, 7711–7730. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, N.; Hövelmeyer, N.; Waisman, A.; Straub, B.K.; Weinmann-Menke, J.; Wörns, M.A.; Galle, P.R.; Schattenberg, J.M. Hepatocyte-specific deletion of IL1-RI attenuates liver injury by blocking IL-1 driven autoinflammation. J. Hepatol. 2018, 68, 986–995. [Google Scholar] [CrossRef]
- Budarina, N.A.; Belaia, O.F.; Chulanov, V.P.; Paĭmanov, N.V.; Pak, S.G. [Characteristics of cellular immunity in children with acute viral hepatitis A]. Ter. Arkhiv 2003, 75, 31–35. [Google Scholar]
- Pan, X.; Xu, H.; Zheng, C.; Li, M.; Zou, X.; Cao, H.; Xu, Q. Human hepatocytes express absent in melanoma 2 and respond to hepatitis B virus with interleukin-18 expression. Virus Genes 2016, 52, 445–452. [Google Scholar] [CrossRef]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.Y.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1β Production through the NLRP3 Inflammasome by Hepatic Macrophages Links Hepatitis C Virus Infection with Liver Inflammation and Disease. PLOS Pathog. 2013, 9, e1003330. [Google Scholar] [CrossRef]
- Li, Y.; Yu, P.; Kessler, A.L.; Shu, J.; Liu, X.; Liang, Z.; Liu, J.; Li, Y.; Li, P.; Wang, L.; et al. Hepatitis E virus infection activates NOD-like receptor family pyrin domain-containing 3 inflammasome antagonizing interferon response but therapeutically targetable. Hepatology 2022, 75, 196–212. [Google Scholar] [CrossRef]
- Zalinger, Z.B.; Elliott, R.; Weiss, S.R. Role of the inflammasome-related cytokines Il-1 and Il-18 during infection with murine coronavirus. J. Neurovirology 2017, 23, 845–854. [Google Scholar] [CrossRef]
- Chen, H.; Han, Z.; Fan, Y.; Chen, L.; Peng, F.; Cheng, X.; Wang, Y.; Su, J.; Li, D. CD4+ T-cell subsets in autoimmune hepatitis: A review. Hepatol. Commun. 2023, 7, e0269. [Google Scholar] [CrossRef] [PubMed]
- Floreani, A.; Restrepo-Jiménez, P.; Secchi, M.F.; De Martin, S.; Leung, P.S.C.; Krawitt, E.; Bowlus, C.L.; Gershwin, M.E.; Anaya, J.M. Etiopathogenesis of autoimmune hepatitis. J. Autoimmun. 2018, 95, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Zhang, X.; Wang, S.; Li, Y.; Fan, J.; Chen, W.; Zai, W.; Wang, S.; Wang, Y.; Chen, M.; et al. NOD-Like Receptor Protein 3 Inflammasome-Dependent IL-1β Accelerated ConA-Induced Hepatitis. Front. Immunol. 2018, 9, 758. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, G.; Liang, Y.; Du, X.; Boor, P.J.; Sun, J.; Khan, M.F. Redox regulation of hepatic NLRP3 inflammasome activation and immune dysregulation in trichloroethene-mediated autoimmunity. Free Radic. Biol. Med. 2019, 143, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Yang, G.; Chen, H.Y.; Hsu, D.K.; Tomilov, A.; Olson, K.A.; Dehnad, A.; Fish, S.R.; Cortopassi, G.; Zhao, B.; et al. Galectin-3 regulates inflammasome activation in cholestatic liver injury. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 4202–4213. [Google Scholar] [CrossRef] [PubMed]
- Arend, W.P. Interleukin-1 receptor antagonist. Adv. Immunol. 1993, 54, 167–227. [Google Scholar] [CrossRef]
- Mancini, R.; Benedetti, A.; Jezequel, A.M. An interleukin-1 receptor antagonist decreases fibrosis induced by dimethylnitrosamine in rat liver. Virchows Arch. 1994, 424, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.Z.; Xiang, D.; Xie, C.; Li, J.J.; Hu, J.J.; He, H.L.; Yuan, Y.S.; Gao, J.; Han, W.; Yu, Y. Protective effect of recombinant human IL-1Ra on CCl4-induced acute liver injury in mice. World J. Gastroenterol. 2010, 16, 2771–2779. [Google Scholar] [CrossRef]
- Hu, J.; Yan, D.; Gao, J.; Xu, C.; Yuan, Y.; Zhu, R.; Xiang, D.; Weng, S.; Han, W.; Zang, G.; et al. rhIL-1Ra reduces hepatocellular apoptosis in mice with acetaminophen-induced acute liver failure. Lab. Investig. 2010, 90, 1737–1746. [Google Scholar] [CrossRef]
- Szabo, G.; Mitchell, M. IL-1 receptor antagonist plus pentoxifylline and zinc for severe alcohol-associated hepatitis. Hepatology 2022, 76, 1058–1068. [Google Scholar] [CrossRef]
- Massaro, M.G.; Pompili, M.; Sicignano, L.L.; Pizzolante, F.; Verrecchia, E.; Vecchio, F.M.; Rigante, D.; Manna, R. Improvement of Liver Involvement in Familial Mediterranean Fever After the Introduction of Canakinumab: A Case Report. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020059. [Google Scholar] [CrossRef] [PubMed]
- Vergis, N.; Patel, V.C.; Bogdanowicz, K.; Czyzewska-Khan, J.; Keshinro, R.; Fiorentino, F.; Day, E.; Middleton, P.; Atkinson, S.; Cross, M.; et al. OS034—Il-1beta Signal Inhibition in acute alcoholic hepatitis: A multicentre, randomised, double-blind, placebo-controlled phase 2 trial of canakinumab therapy (ISAIAH). J. Hepatol. 2022, 77, S34–S35. [Google Scholar] [CrossRef]
- Vande Walle, L.; Stowe, I.B.; Šácha, P. MCC950/CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease-associated mutants for inflammasome inhibition. PLOS Biol. 2019, 17, e3000354. [Google Scholar] [CrossRef] [PubMed]
- Nizami, S.; Millar, V.; Arunasalam, K.; Zarganes-Tzitzikas, T.; Brough, D.; Tresadern, G.; Brennan, P.E.; Davis, J.B.; Ebner, D.; Di Daniel, E. A phenotypic high-content, high-throughput screen identifies inhibitors of NLRP3 inflammasome activation. Sci. Rep. 2021, 11, 15319. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Rouquet, N.; Pagès, J.C.; Molina, T.; Briand, P.; Joulin, V. ICE inhibitor YVADcmk is a potent therapeutic agent against in vivo liver apoptosis. Curr. Biol. 1996, 6, 1192–1195. [Google Scholar] [CrossRef]
- Morrison, M.C.; Mulder, P.; Salic, K.; Verheij, J.; Liang, W.; van Duyvenvoorde, W.; Menke, A.; Kooistra, T.; Kleemann, R.; Wielinga, P.Y. Intervention with a caspase-1 inhibitor reduces obesity-associated hyperinsulinemia, non-alcoholic steatohepatitis and hepatic fibrosis in LDLR−/−.Leiden mice. Int. J. Obes. 2016, 40, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Wang, Q.; Dong, Y.; Ma, W.; Zhang, Y.; Zhao, Y.; Bian, F.; Chen, L. High Glucose-Aggravated Hepatic Insulin Resistance: Role of the NLRP3 Inflammasome in Kupffer Cells. Obesity 2020, 28, 1270–1282. [Google Scholar] [CrossRef]
- Lei, Y.; Tang, L.; Chen, Q.; Wu, L.; He, W.; Tu, D.; Wang, S.; Chen, Y.; Liu, S.; Xie, Z.; et al. Disulfiram ameliorates nonalcoholic steatohepatitis by modulating the gut microbiota and bile acid metabolism. Nat. Commun. 2022, 13, 6862. [Google Scholar] [CrossRef]
- Sangineto, M.; Grabherr, F.; Adolph, T.E. Dimethyl fumarate ameliorates hepatic inflammation in alcohol related liver disease. Liver Int. 2020, 40, 1610–1619. [Google Scholar] [CrossRef]
- Xu, B.; Jiang, M.; Chu, Y.; Wang, W.; Chen, D.; Li, X.; Zhang, Z.; Zhang, D.; Fan, D.; Nie, Y.; et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2018, 68, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Faggioni, R.; Cattley, R.C.; Guo, J.; Flores, S.; Brown, H.; Qi, M.; Yin, S.; Hill, D.; Scully, S.; Chen, C.; et al. IL-18-binding protein protects against lipopolysaccharide-induced lethality and prevents the development of Fas/Fas ligand-mediated models of liver disease in mice. J. Immunol. 2001, 167, 5913–5920. [Google Scholar] [CrossRef] [PubMed]
- Knorr, J.; Kaufmann, B.; Inzaugarat, M.E.; Holtmann, T.M.; Geisler, L.; Hundertmark, J.; Kohlhepp, M.S.; Boosheri, L.M.; Chilin-Fuentes, D.R.; Birmingham, A. Interleukin-18 signaling promotes activation of hepatic stellate cells in mouse liver fibrosis. Hepatology 2023, 77, 1968–1982. [Google Scholar] [CrossRef]
- Cao, Z.; Fang, Y.; Lu, Y.; Tan, D.; Du, C.; Li, Y.; Ma, Q.; Yu, J.; Chen, M.; Zhou, C.; et al. Melatonin alleviates cadmium-induced liver injury by inhibiting the TXNIP-NLRP3 inflammasome. J. Pineal Res. 2017, 62, e12389. [Google Scholar] [CrossRef]
- Mansoori, A.; Salimi, Z.; Hosseini, S.A.; Hormoznejad, R.; Jafarirad, S.; Bahrami, M.; Asadi, M. The effect of melatonin supplementation on liver indices in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis of randomized clinical trials. Complement. Ther. Med. 2020, 52, 102398. [Google Scholar] [CrossRef]
- Fernández-Ortiz, M.; Sayed, R.K.A. Age and Chronodisruption in Mouse Heart: Effect of the NLRP3 Inflammasome and Melatonin Therapy. Int. J. Mol. Sci. 2022, 23, 6846. [Google Scholar] [CrossRef] [PubMed]
- Volt, H.; García, J.A.; Doerrier, C.; Díaz-Casado, M.E.; Guerra-Librero, A.; López, L.C.; Escames, G.; Tresguerres, J.A.; Acuña-Castroviejo, D. Same molecule but different expression: Aging and sepsis trigger NLRP3 inflammasome activation, a target of melatonin. J. Pineal Res. 2016, 60, 193–205. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Najafi, M. Anti-Inflammatory Activity of Melatonin: A Focus on the Role of NLRP3 Inflammasome. Inflammation 2021, 44, 1207–1222. [Google Scholar] [CrossRef] [PubMed]
- Rojas Márquez, J.D.; Ana, Y.; Baigorrí, R.E.; Stempin, C.C.; Cerban, F.M. Mammalian Target of Rapamycin Inhibition in Trypanosoma cruzi-Infected Macrophages Leads to an Intracellular Profile That Is Detrimental for Infection. Front. Immunol. 2018, 9, 313. [Google Scholar] [CrossRef]
- Sun, Q.; Gao, W.; Loughran, P.; Shapiro, R.; Fan, J.; Billiar, T.R.; Scott, M.J. Caspase 1 activation is protective against hepatocyte cell death by up-regulating beclin 1 protein and mitochondrial autophagy in the setting of redox stress. J. Biol. Chem. 2013, 288, 15947–15958. [Google Scholar] [CrossRef]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2014, 94, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Li, L.; Lu, P.; Li, X.; Tian, D.; Liu, M. NLRP6 exerts a protective role via NF-kB with involvement of CCL20 in a mouse model of alcoholic hepatitis. Biochem. Biophys. Res. Commun. 2020, 528, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liu, Q.; Tang, Q.; Jing, X.; Wu, T.; Zhang, J.; Zhang, G.; Zhou, J.; Zhang, Z.; Zhao, Y.; et al. Hepatocyte-specific deletion of Nlrp6 in mice exacerbates the development of non-alcoholic steatohepatitis. Free Radic. Biol. Med. 2021, 169, 110–121. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baral, A. Mechanisms of Inflammasome Activation and Involvement in Liver Disease. J. Mol. Pathol. 2024, 5, 171-186. https://doi.org/10.3390/jmp5020011
Baral A. Mechanisms of Inflammasome Activation and Involvement in Liver Disease. Journal of Molecular Pathology. 2024; 5(2):171-186. https://doi.org/10.3390/jmp5020011
Chicago/Turabian StyleBaral, Ananda. 2024. "Mechanisms of Inflammasome Activation and Involvement in Liver Disease" Journal of Molecular Pathology 5, no. 2: 171-186. https://doi.org/10.3390/jmp5020011
APA StyleBaral, A. (2024). Mechanisms of Inflammasome Activation and Involvement in Liver Disease. Journal of Molecular Pathology, 5(2), 171-186. https://doi.org/10.3390/jmp5020011