
Regulation of a Metabolic Gene Signature in Response to Respiratory Viruses and Type I Interferon Signaling

Abstract
1. Introduction
2. Materials and Methods
Gene Expression Datasets and Bioinformatics
3. Results
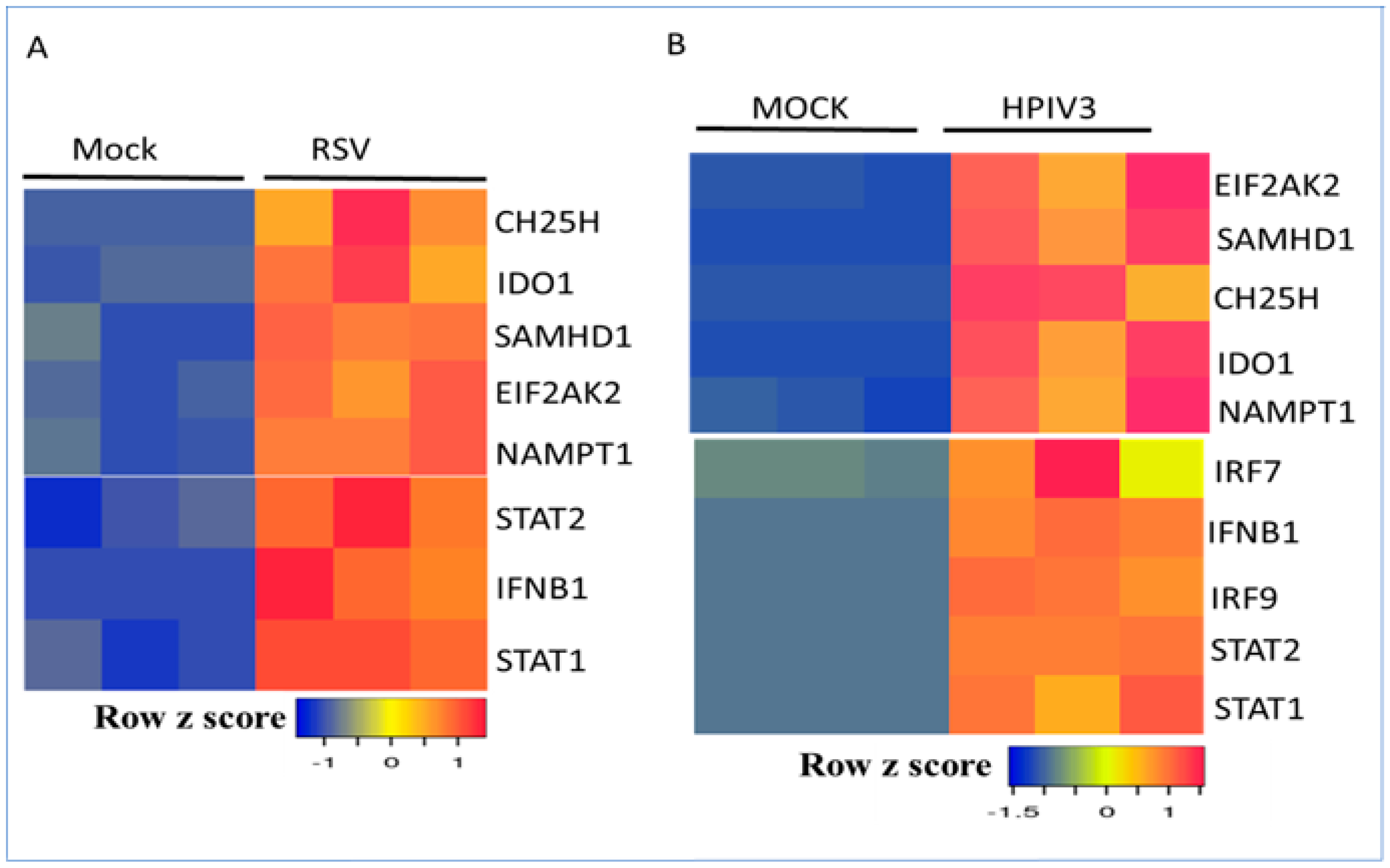
3.1. Profiling a Metabolic Gene Expression Signature in Response to Respiratory Viruses and Type I Interferon Signaling
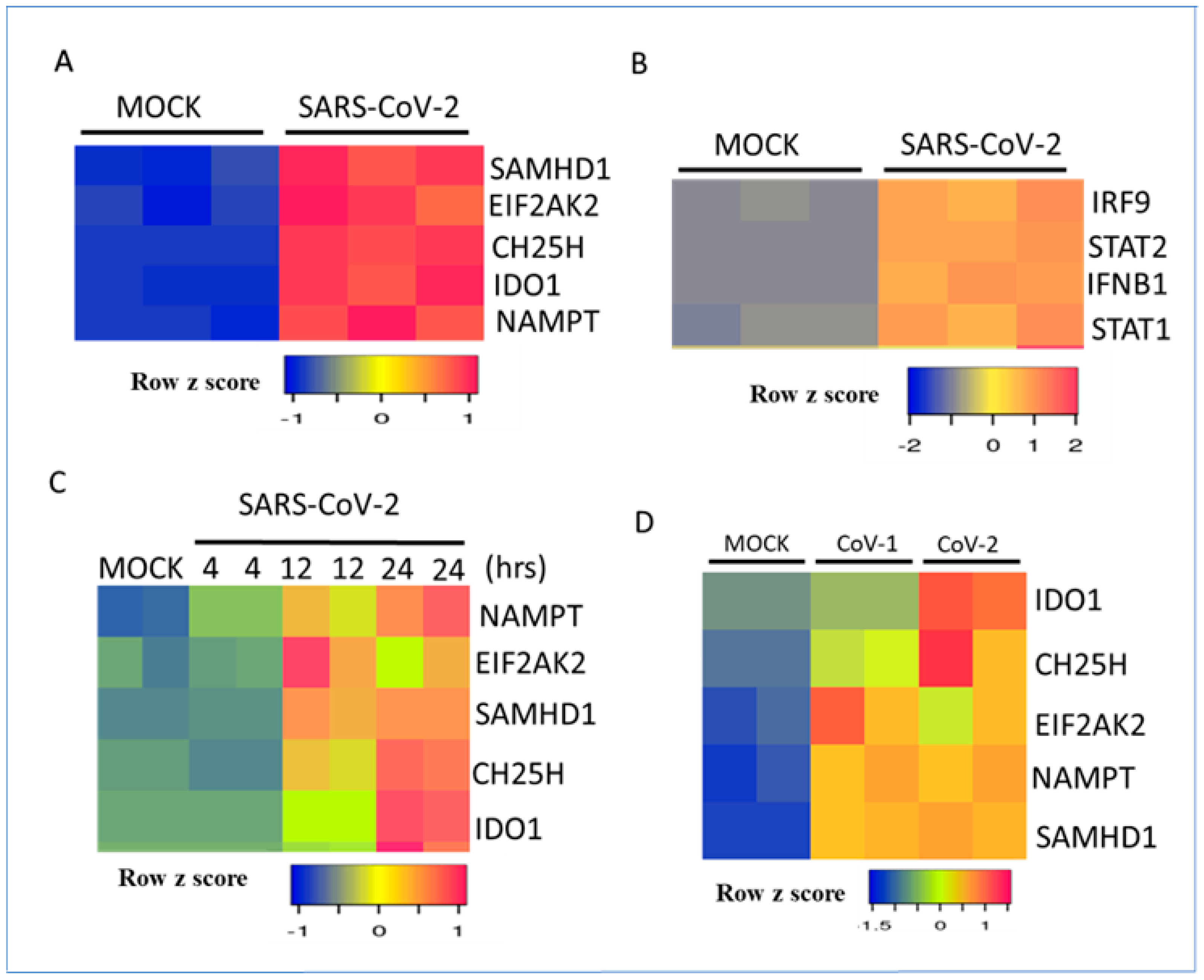
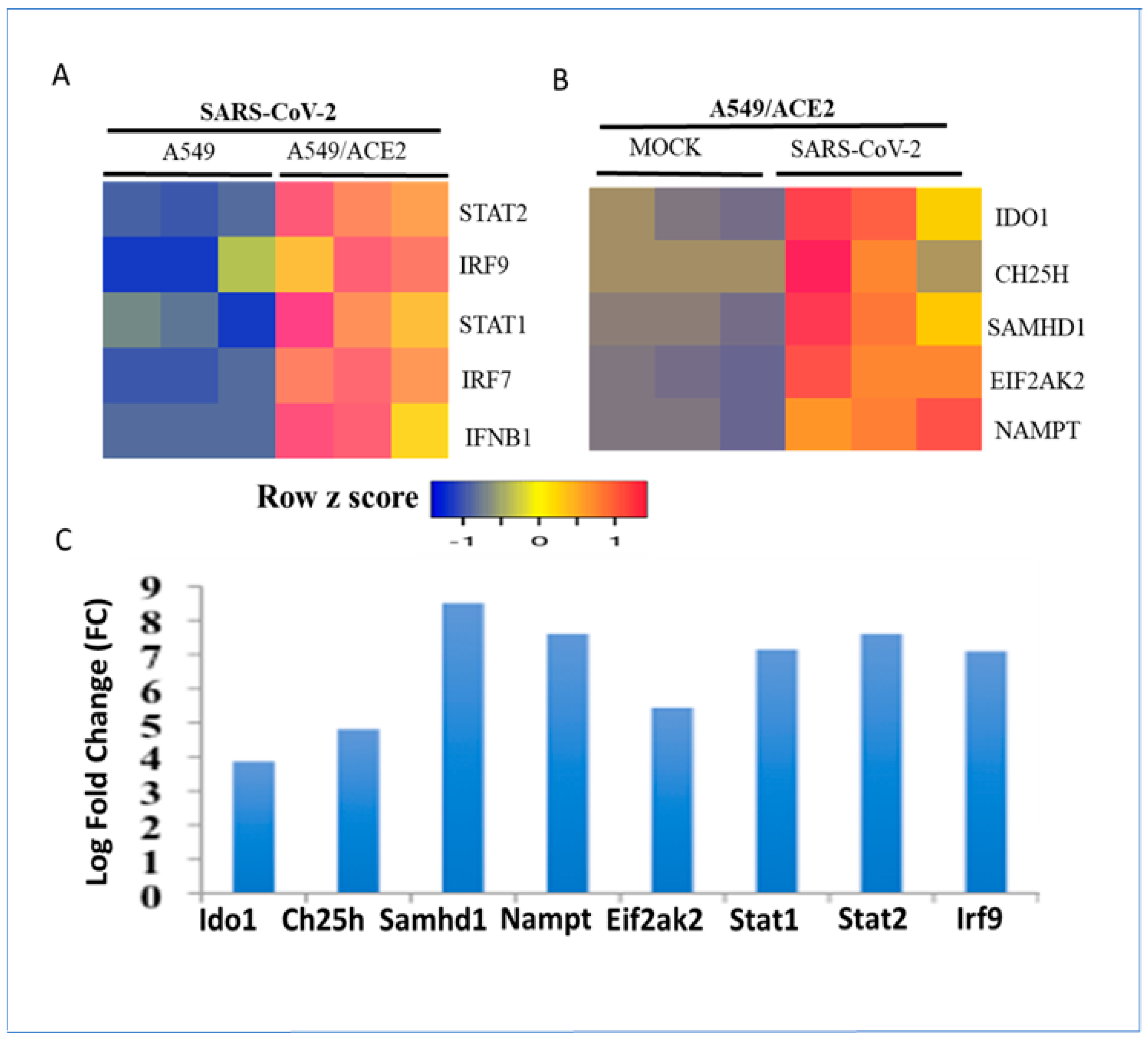
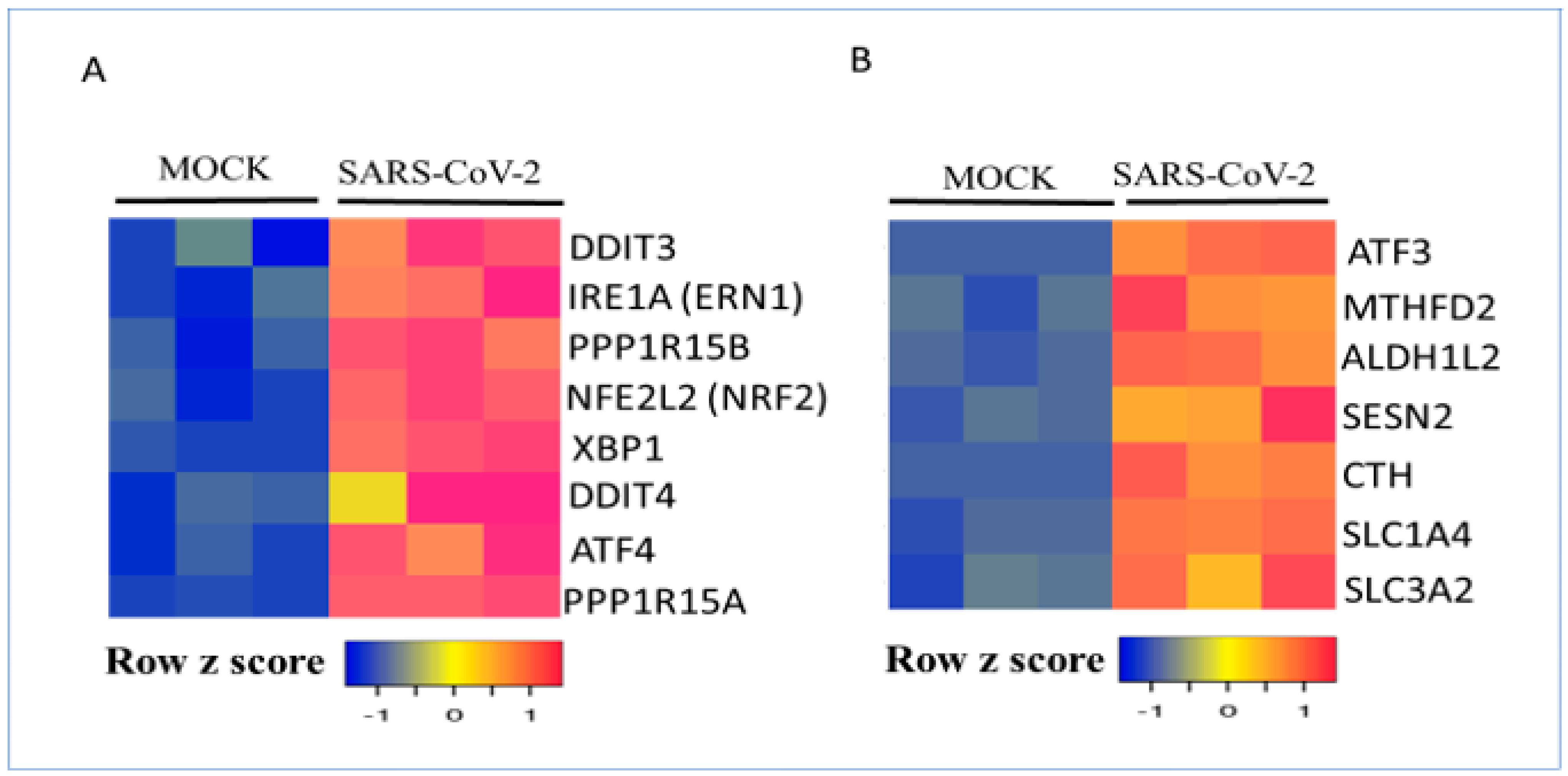
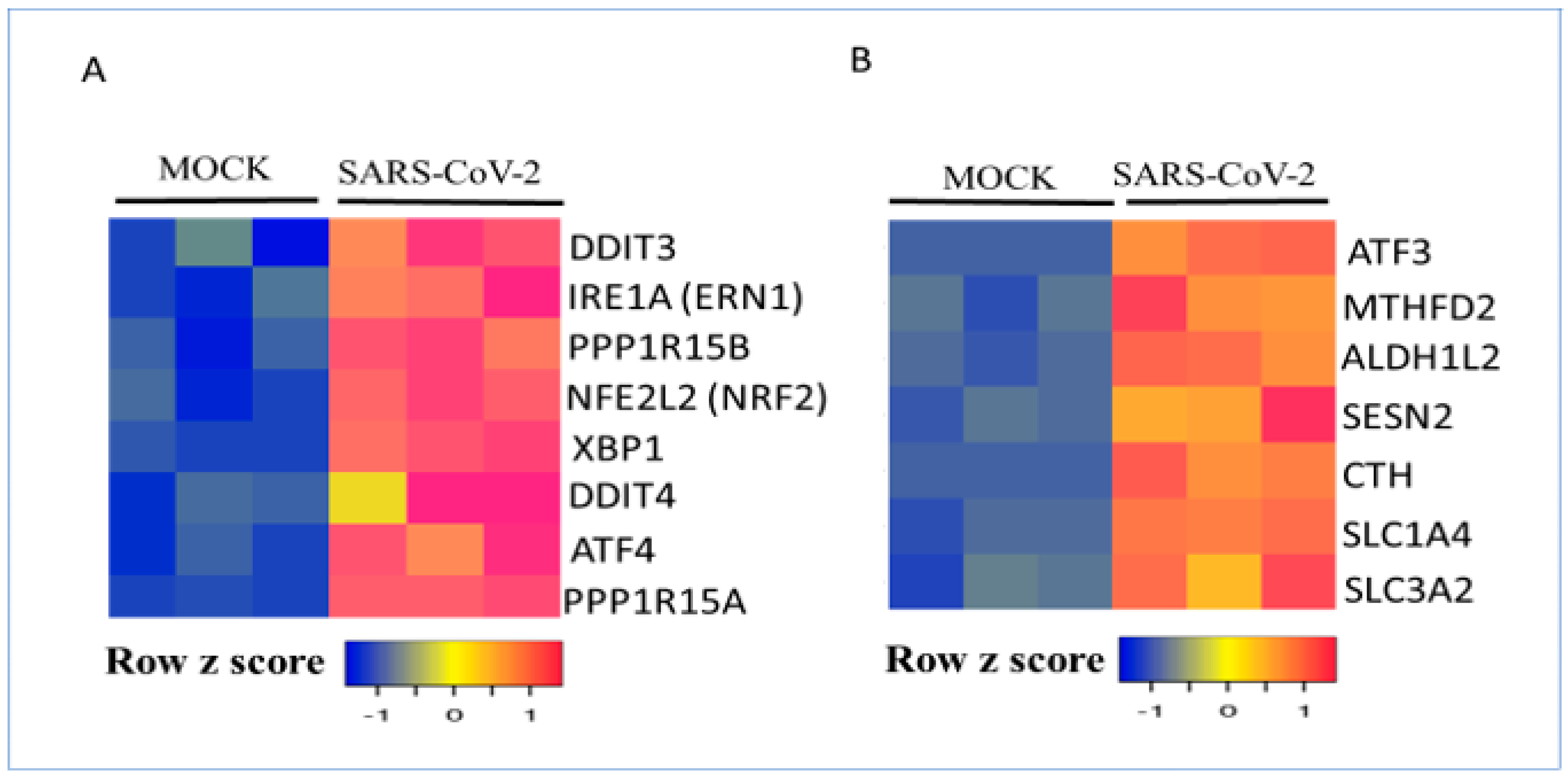
3.2. Regulation of Metabolic Gene Signature Expression by Coronaviruses
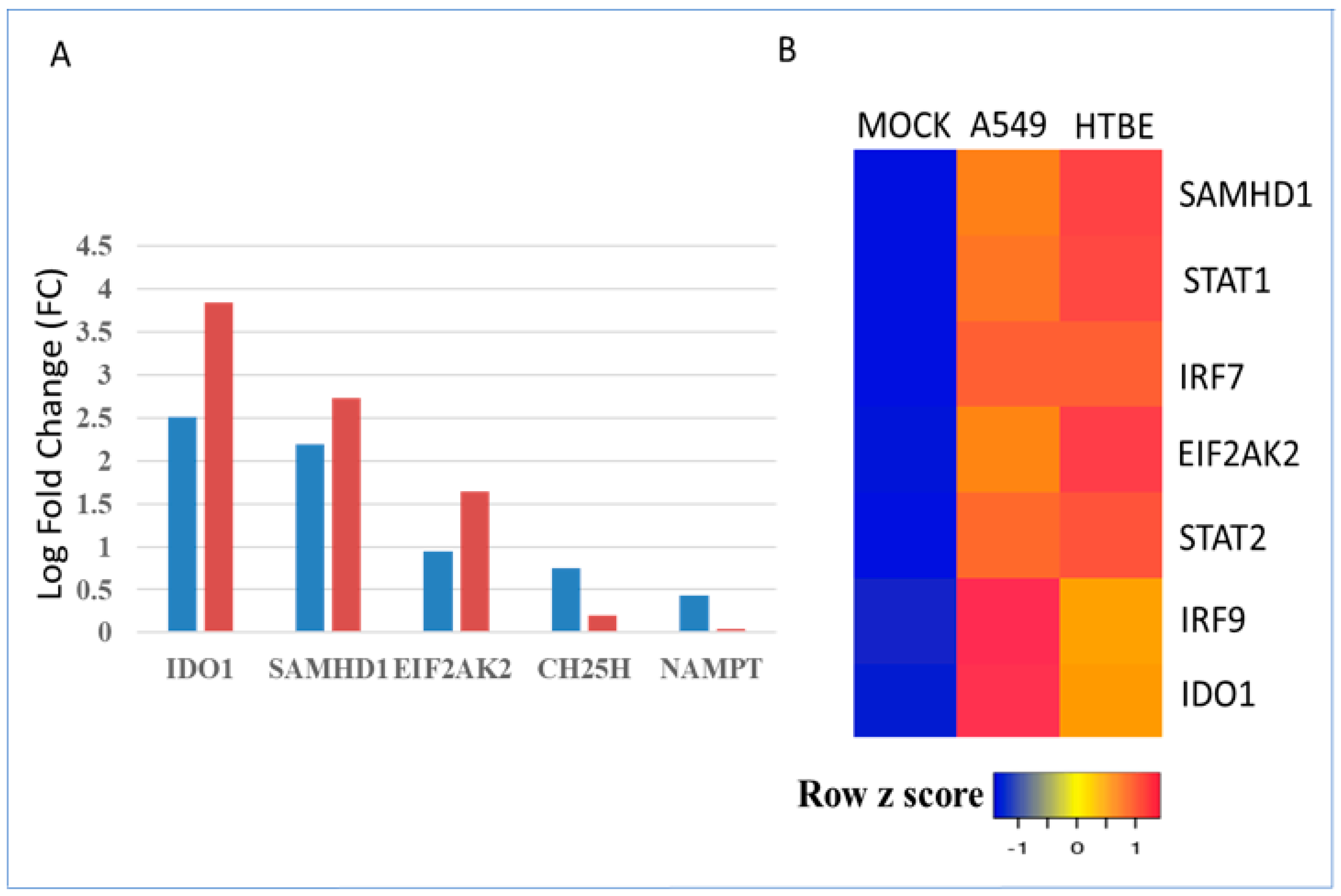
3.3. Regulation of Metabolic Gene Signature Expression by Influenza Viruses
3.4. Regulation of Metabolic Gene Signature by Double-Stranded RNA and EIF2AK2 In Vivo
3.5. Regulation of Metabolic Gene Signature by Type I Interferons in Human Lung Epithelial Cells
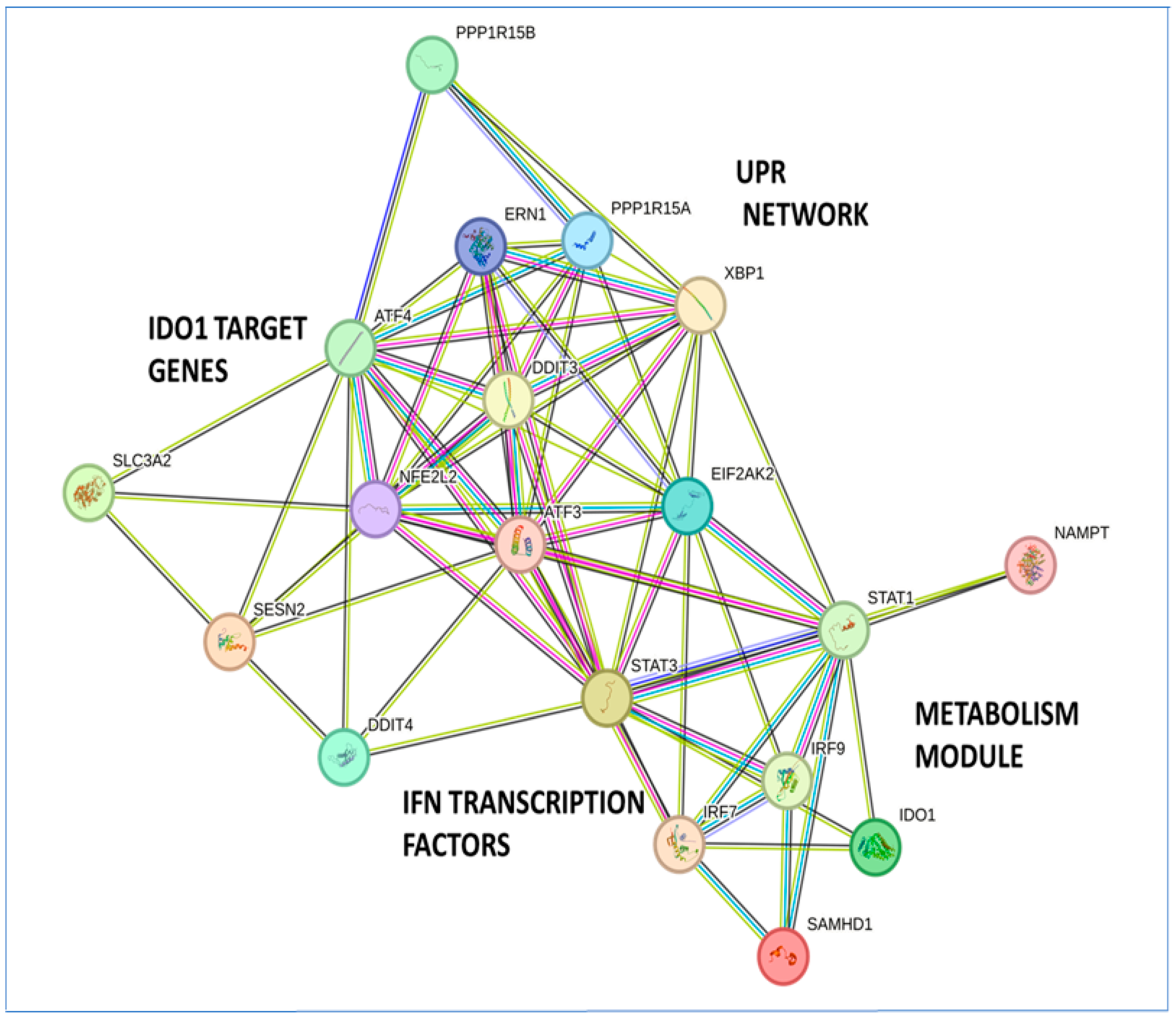
3.6. Visualization of the Protein Interaction Network of the Metabolic Gene Signature in the STRING Database
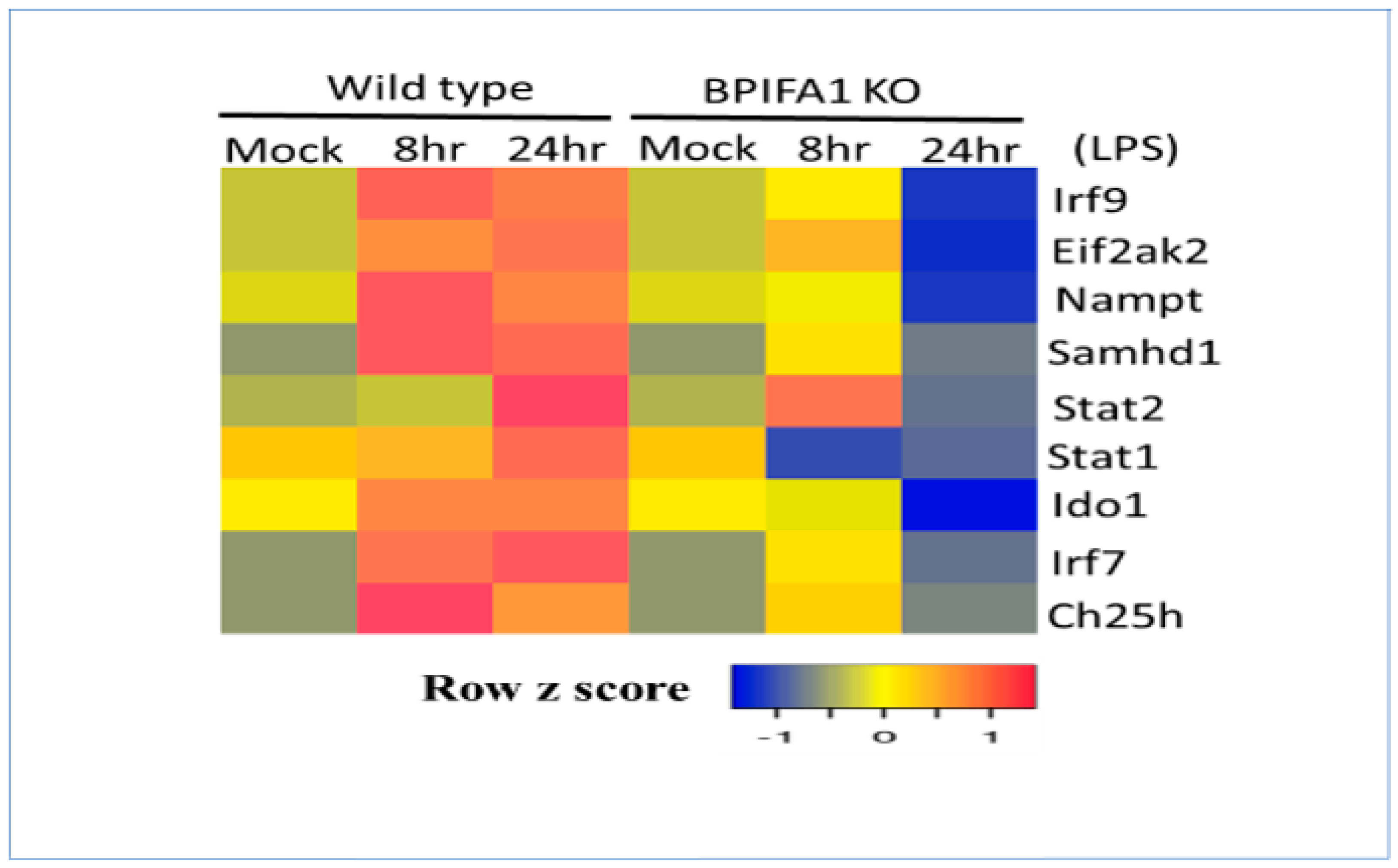
3.7. Lipopolysaccharide (LPS) Regulation of Metabolic Gene Signature in Acute Lung Injury (ALI)
3.8. Role of Metabolic Gene Signature in Integrated Stress Response (ISR)
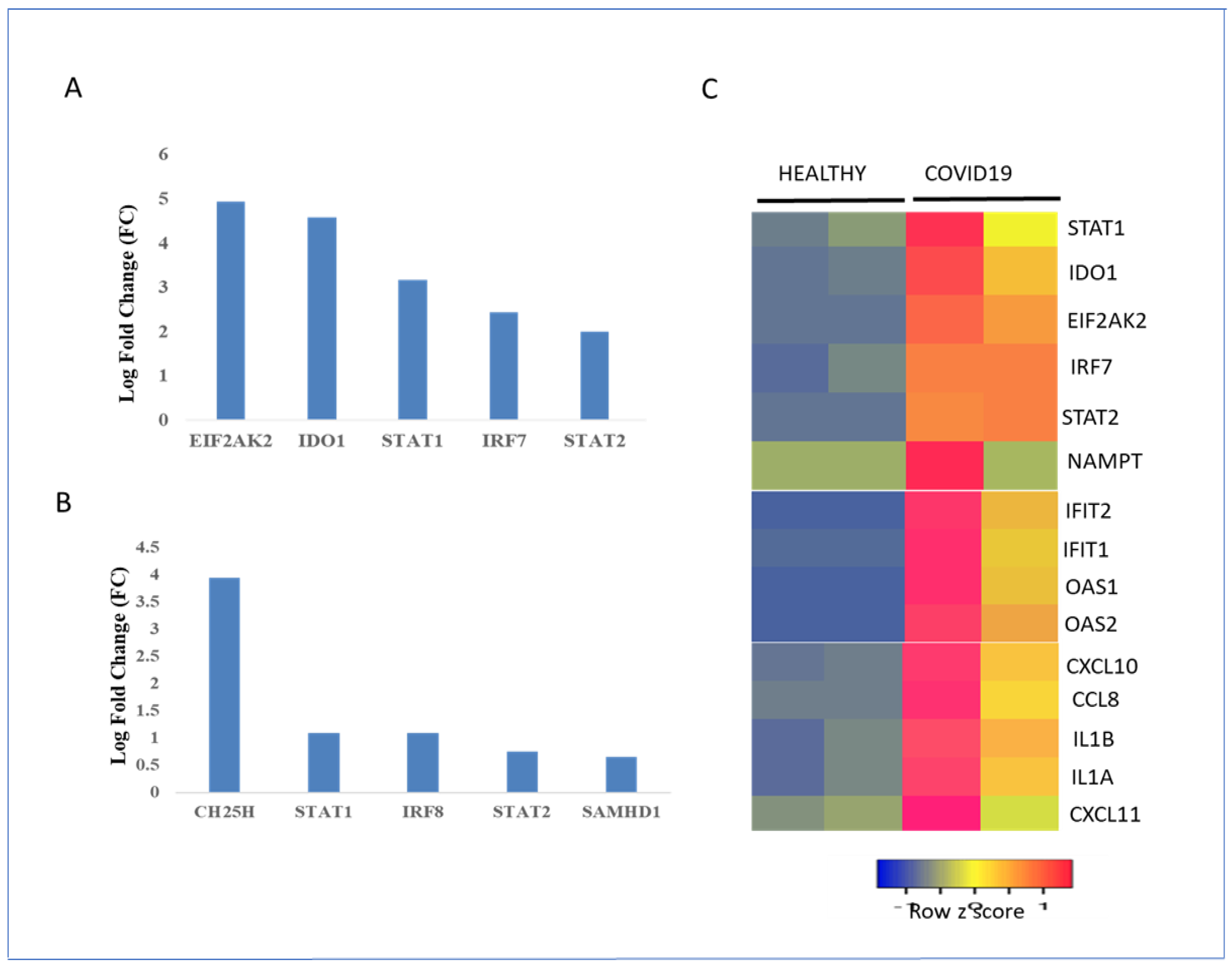
3.9. Regulation of the Metabolic Gene Signature in Healthy and COVID-19 Patients
4. Discussion
5. Conclusions
- This metabolic gene signature could potentially serve as a biomarker for the early detection of respiratory pathogens.
- By tracking changes in the metabolic gene signature by RT-PCR, clinicians could monitor respiratory disease severity and the response to therapy.
- Screening for chemical inhibitors of the metabolic gene signature could lead to novel therapeutic drugs. Further research is required to test the presence of similar metabolic gene signatures in other bacterial, viral, and inflammatory diseases where type I interferon signaling plays a critical role.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kohlmeier, J.E.; Woodland, D.L. Immunity to respiratory viruses. Annu. Rev. Immunol. 2009, 27, 61–82. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Kerr, I.M.; Williams, B.R.; Silverman, R.H.; Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.Y.; Danielson, M.L.; Town, M.; Derado, G.; Greenlund, K.J.; Kirley, P.D.; Alden, N.B.; Yousey-Hindes, K.; Anderson, E.J.; Ryan, P.A.; et al. COVID-NET Surveillance Team. Risk Factors for Coronavirus Disease 2019 (COVID-19)-Associated Hospitalization: COVID-19-Associated Hospitalization Surveillance Network and Behavioral Risk Factor Surveillance System. Clin. Infect. Dis. 2021, 72, e695–e703. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, G.; Tonetti, T.; Protti, A.; Langer, T.; Girardis, M.; Bellani, G.; Laffey, J.; Carrafiello, G.; Carsana, L.; Rizzuto, C.; et al. Pathophysiology of COVID-19-associated acute respiratory distress syndrome: A multicentre prospective observational study. Lancet Respir. Med. 2020, 8, 1201–1208. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Xu, J.; Xu, X.; Jiang, L.; Dua, K.; Hansbro, P.M.; Liu, G. SARS-CoV-2 induces transcriptional signatures in human lung epithelial cells that promote lung fibrosis. Respir. Res. 2020, 21, 182. [Google Scholar] [CrossRef]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Münch, C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef]
- O’Meara, M.J.; Guo, J.Z.; Swaney, D.L.; Tummino, T.A.; Hüttenhain, R. A SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug-Repurposing. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ziegler, C.G.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef]
- Delorey, T.M.; Ziegler, C.G.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Å.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Stefanoni, D.; Reisz, J.A.; Nemkov, T.; Bertolone, L.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hansen, K.C.; Hod, E.A.; et al. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight 2020, 5, e140327. [Google Scholar] [CrossRef]
- Lionetto, L.; Ulivieri, M.; Capi, M.; De Bernardini, D.; Fazio, F.; Petrucca, A.; Pomes, L.M.; De Luca, O.; Gentile, G.; Casolla, B.; et al. Increased kynurenine-to-tryptophan ratio in the serum of patients infected with SARS-CoV2: An observational cohort study. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166042. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. Int. Rev. Cell Mol. Biol. 2018, 336, 175–203. [Google Scholar] [PubMed]
- O’Neill, L.A.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Rudiansyah, M.; Jasim, S.A.; Mohammad Pour, Z.G.; Athar, S.S.; Jeda, A.S.; Doewes, R.I.; Jalil, A.T.; Bokov, D.O.; Mustafa, Y.F.; Noroozbeygi, M.; et al. Coronavirus disease 2019 (COVID-19) update: From metabolic reprogramming to immunometabolism. J. Med. Virol. 2022, 94, 4611–4627. [Google Scholar] [CrossRef]
- Ryu, K.W.; Nandu, T.; Kim, J.; Challa, S.; DeBerardinis, R.J.; Kraus, W.L. Metabolic regulation of transcription through compartmentalized NAD+ biosynthesis. Science 2018, 360, aa5780. [Google Scholar] [CrossRef]
- Raniga, K.; Liang, C. Interferons: Reprogramming the Metabolic Network against Viral Infection. Viruses 2018, 10, 36. [Google Scholar] [CrossRef]
- Phipps, S.; Lam, C.E.; Mahalingam, S.; Newhouse, M.; Ramirez, R.; Rosenberg, H.F.; Foster, P.S.; Matthaei, K.I. Eosinophils contribute to innate antiviral immunity and promote clearance of respiratory syncytial virus. Blood 2007, 110, 1578–1586. [Google Scholar] [CrossRef]
- Bajwa, G.; DeBerardinis, R.J.; Shao, B.; Hall, B.; Farrar, J.D.; Gill, M.A. Cutting Edge: Critical Role of Glycolysis in Human Plasmacytoid Dendritic Cell Antiviral Responses. J. Immunol. 2016, 196, 2004–2009. [Google Scholar] [CrossRef]
- Wu, D.; Sanin, D.E.; Everts, B.; Chen, Q.; Qiu, J.; Buck, M.D.; Patterson, A.; Smith, A.M.; Chang, C.H.; Liu, Z.; et al. Type 1 Interferons Induce Changes in Core Metabolism that Are Critical for Immune Function. Immunity 2016, 44, 1325–1336. [Google Scholar] [CrossRef]
- Smith, T.; Rohaim, M.A.; Munir, M. Mapping Molecular Gene Signatures Mediated by SARS-COV-2 and Large-Scale and Genome-Wide Transcriptomics Comparative Analysis among Respiratory Viruses of Medical Importance. Mol. Cell. Probes 2022, 64, 101820. [Google Scholar] [CrossRef]
- Bucasas, K.L.; Mian, A.I.; Demmler-Harrison, G.J.; Caviness, A.C.; Piedra, P.A.; Franco, L.M.; Shaw, C.A.; Zhai, Y.; Wang, X.; Bray, M.S.; et al. Global gene expression profiling in infants with acute respiratory syncytial virus bronchiolitis demonstrates systemic activation of interferon signaling networks. Pediatr. Infect. Dis. J. 2013, 32, e68–e76. [Google Scholar] [CrossRef]
- Liu, H.L.; Yeh, I.J.; Phan, N.N.; Wu, Y.H.; Yen, M.C.; Hung, J.H.; Chiao, C.C.; Chen, C.F.; Sun, Z.; Jiang, J.Z.; et al. Gene signatures of SARS-CoV/SARS-CoV-2-infected ferret lungs in short- and long-term models. Infect. Genet. Evol. 2020, 85, 104438. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Scalambra, L.; Triboli, L.; Ray, F.; Giorgi, F.M. Gene regulatory network inference resources: A practical overview. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194430. [Google Scholar] [CrossRef] [PubMed]
- Ramana, C.V. Insights into functional connectivity in mammalian signal transduction pathways by pairwise comparison of protein interaction partners of critical signaling hubs. Biomol. Concepts 2022, 13, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Ramana, C.V.; Das, B. Profiling transcription factor sub-networks in type I interferon signaling and in response to SARS-CoV-2 infection. Comput. Math. Biophys 2021, 9, 273–288. [Google Scholar] [CrossRef]
- Nguyen, L.C.; Renner, D.M.; Silva, D.; Yang, D.; Parenti, N.; Medina, K.M.; Nicolaescu, V.; Gula, H.; Drayman, N.; Valdespino, A.; et al. SARS-CoV2 diverges from other betacoronaviruses in only partially activating the IRE1a/XBP1 ER stress pathway in human lung-derive cells. mBio 2022, e0241522. [Google Scholar] [CrossRef] [PubMed]
- Wyler, E.; Mösbauer, K.; Franke, V.; Diag, A.; Gottula, L.T.; Arsiè, R.; Klironomos, F.; Koppstein, D.; Hönzke, K.; Ayoub, S.; et al. Transcriptomic profiling of SARS-CoV-2 infected human cell lines identifies HSP90 as target for COVID-19 therapy. iScience 2021, 24, 102151. [Google Scholar] [CrossRef] [PubMed]
- Daamen, A.R.; Bachali, P.; Owen, K.A.; Kingsmore, K.M.; Hubbard, E.L.; Labonte, A.C.; Robl, R.; Shrotri, S.; Grammer, A.C.; Lipsky, P.E. Comprehensive transcriptomic analysis of COVID-19 blood, lung, and airway. Sci. Rep. 2021, 11, 7052. [Google Scholar] [CrossRef]
- Walters, K.A.; D’Agnillo, F.; Sheng, Z.M.; Kindrachuk, J.; Schwartzman, L.M.; Kuestner, R.E.; Chertow, D.S.; Golding, B.T.; Taubenberger, J.K.; Kash, J.C. 1918 pandemic influenza virus and Streptococcus pneumoniae co-infection results in activation of coagulation and widespread pulmonary thrombosis in mice and humans. J. Pathol. 2016, 238, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.; Sridhar, S.; Peng, R.; Phillips, J.E.; Cohn, R.G.; Burns, L.; Woods, J.; Ramanujam, M.; Loubeau, M.; Tyagi, G.; et al. Double-stranded RNA induces molecular and inflammatory signatures that are directly relevant to COPD. Mucosal. Immunol. 2013, 6, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Minor, R.A.; Linnmon, G.V.; Miller-DeGraff, L.; Dixon, D.; Andrews, D.M.; Kaufman, R.J.; Imani, F. Double-stranded RNA-activated protein kinase regulates early innate immune responses during respiratory syncytial virus infection. J. Interferon Cytokine Res. 2010, 30, 263–272. [Google Scholar] [CrossRef]
- Britto, C.J.; Niu, N.; Khanal, S.; Huleihel, L.; Herazo-Maya, J.D.; Thompson, A.; Sauler, M.; Slade, M.D.; Sharma, L.; Dela Cruz, C.S.; et al. BPIFA1 regulates lung neutrophil recruitment and interferon signaling during acute inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L321–L333. [Google Scholar] [CrossRef] [PubMed]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web- enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017. Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Henrickson, K.J. Parainfluenza viruses. Clin. Microbiol. Rev. 2003, 16, 242–264. [Google Scholar] [CrossRef]
- Blanc, M.; Hsieh, W.Y.; Robertson, K.A.; Kropp, K.A.; Forster, T.; Shui, G.; Lacaze, P.; Watterson, S.; Griffiths, S.J.; Spann, N.J.; et al. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 2013, 38, 106–118. [Google Scholar] [CrossRef]
- Garcia-Ortega, M.B.; Lopez, G.J.; Jimenez, G.; Garcia-Garcia, J.A.; Conde, V.; Boulaiz, H.; Carrillo, E.; Perán, M.; Marchal, J.A.; Garcia, M.A. Clinical and therapeutic potential of protein kinase PKR in cancer and metabolism. Expert. Rev. Mol. Med. 2017, 19, e9. [Google Scholar] [CrossRef]
- Deutschmann, J.; Gramberg, T. SAMHD1 … and Viral Ways around It. Viruses 2021, 13, 395. [Google Scholar] [CrossRef]
- Dantoft, W.; Robertson, K.A.; Watkins, W.J.; Strobl, B.; Ghazal, P. Metabolic Regulators Nampt and Sirt6 Serially Participate in the Macrophage Interferon Antiviral Cascade. Front. Microbiol. 2019, 10, 355. [Google Scholar] [CrossRef]
- Fiore, A.; Zeitler, L.; Russier, M.; Groß, A.; Hiller, M.K.; Parker, J.L.; Stier, L.; Köcher, T.; Newstead, S.; Murray, P.J. Kynurenine importation by SLC7A11 propagates anti-ferroptotic signaling. Mol. Cell 2022, 82, 920–932.e7. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Chiou, J.; Poirion, O.B.; Buchanan, J.; Valdez, M.J.; Verheyden, J.M.; Hou, X.; Kudtarkar, P.; Narendra, S.; Newsome, J.M.; et al. Single-cell multi omic profiling of human lungs reveals cell-type-specific and age-dynamic control of SARS-CoV2 host genes. eLife 2020, 9, e62522. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, V.; Ravà, M.; Marotta, D.; Di Lucia, P.; Laura, C.; Sala, E.; Grillo, M.; Bono, E.; Giustini, L.; Perucchini, C.; et al. Administration of aerosolized SARS-CoV-2 to K18-hACE2 mice uncouples respiratory infection from fatal neuroinvasion. Sci. Immunol. 2022, 7, eabl9929. [Google Scholar] [CrossRef] [PubMed]
- Michael, P.; Brabant, D.; Bleiblo, F.; Ramana, C.V.; Rutherford, M.; Khurana, S.; Tai, T.C.; Kumar, A.; Kumar, A. Influenza A induced cellular signal transduction pathways. J. Thorac. Dis. 2013, 5 (Suppl. 2), S132–S141. [Google Scholar] [PubMed]
- Kash, J.C.; Tumpey, T.M.; Proll, S.C.; Carter, V.; Perwitasari, O.; Thomas, M.J.; Basler, C.F.; Palese, P.; Taubenberger, J.K.; García-Sastre, A.; et al. Genomic analysis of increased host immune response and cell death responses induced by 1918 influenza virus. Nature 2008, 443, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef]
- Han, C.W.; Jeong, M.S.; Jang, S.B. Structure and Function of the Influenza A Virus Non-Structural Protein 1. J. Microbiol. Biotechnol. 2019, 29, 1184–1192. [Google Scholar] [CrossRef]
- Bleiblo, F.; Michael, P.; Brabant, D.; Ramana, C.V.; Tai, T.; Saleh, M.; Parrillo, J.E.; Kumar, A.; Kumar, A. JAK kinases are required for the bacterial RNA and poly I:C induced tyrosine phosphorylation of PKR. Int. J. Clin. Exp. Med. 2013, 6, 16–25. [Google Scholar]
- Dalet, A.; Gatti, E.; Pierre, P. Integration of PKR-dependent translation inhibition with innate immunity is required for a coordinated anti-viral response. FEBS Lett. 2015, 589, 1539–1545. [Google Scholar] [CrossRef]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef]
- Kerr, C.H.; Skinnider, M.A.; Andrews, D.D.T.; Madero, A.M.; Chan, Q.W.T.; Stacey, R.G.; Stoynov, N.; Jan, E.; Foster, L.J. Dynamic rewiring of the human interactome by interferon signaling. Genome Biol. 2020, 21, 140. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Sharma, S.D.; Kumar, A.; Ende, Z.; Mishina, M.; Wang, Y.; Falls, Z.; Samudrala, R.; Pohl, J.; Knight, P.R.; et al. Antiviral Approaches against Influenza Virus. Clin. Microbiol. Rev. 2023, 36, e0004022. [Google Scholar] [CrossRef] [PubMed]
- Hanada, S.; Pirzadeh, M.; Carver, K.Y.; Deng, J.C. Respiratory Viral Infection-Induced Microbiome Alterations and Secondary Bacterial Pneumonia. Front. Immunol. 2018, 9, 2640. [Google Scholar] [CrossRef] [PubMed]
- Zamyatina, A.; Heine, H. Lipopolysaccharide Recognition in the Crossroads of TLR4 and Caspase-4/11 Mediated Inflammatory Pathways. Front. Immunol. 2020, 11, 585146. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. LPS in microbial pathogenesis: Promise and fulfillment. J. Endotoxin Res. 2002, 8, 329–335. [Google Scholar] [CrossRef]
- Czerkies, M.; Korwek, Z.; Prus, W.; Kochańczyk, M.; Jaruszewicz-Błońska, J.; Tudelska, K.; Błoński, S.; Kimmel, M.; Brasier, A.R.; Lipniacki, T. Cell fate in antiviral response arises in the crosstalk of IRF, NF-κB and JAK/STAT pathways. Nat. Commun. 2018, 9, 493. [Google Scholar] [CrossRef]
- Liu, S.Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Valverde-Estrella, L.; López-Serrat, M.; Sánchez-Sànchez, G.; Vico, T.; Lloberas, J.; Celada, A. Induction of Samhd1 by interferon gamma and lipopolysaccharide in murine macrophages requires IRF1. Eur. J. Immunol. 2020, 50, 1321–1334. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Bonifati, S.; Qin, Z.; St Gelais, C.; Kodigepalli, K.M.; Barrett, B.S.; Kim, S.H.; Antonucci, J.M.; Ladner, K.J.; Buzovetsky, O.; et al. SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-κB and interferon pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E3798–E3807. [Google Scholar] [CrossRef]
- Tsou, Y.A.; Tung, M.C.; Alexander, K.A.; Chang, W.D.; Tsai, M.H.; Chen, H.L.; Chen, C.M. The Role of BPIFA1 in Upper Airway Microbial Infections and Correlated Diseases. BioMed Res. Int. 2018, 2018, 2021890. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, Z.; Li, Y.; Li, Y. The Regulation of Integrated Stress Response Signaling Pathway on Viral Infection and Viral Antagonism. Front. Microbiol. 2022, 12, 814635. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.C.; Yang, D.; Nicolaescu, V.; Best, T.J.; Gula, H.; Saxena, D.; Gabbard, J.D.; Chen, S.N.; Ohtsuki, T.; Friesen, J.B.; et al. Cannabidiol inhibits SARS-CoV-2 replication through induction of the host ER stress and innate immune responses. Sci. Adv. 2022, 8, eabi6110. [Google Scholar] [CrossRef]
- Stone, T.W.; Williams, R.O. Interactions of IDO and the Kynurenine Pathway with Cell Transduction Systems and Metabolism at the Inflammation-Cancer Interface. Cancers 2023, 15, 2895. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Wang, T. Metabolic Reprogramming in COVID-19. Int. J. Mol. Sci. 2021, 22, 11475. [Google Scholar] [CrossRef]
- Li, H.; Li, X.; Wu, Q.; Wang, X.; Qin, Z.; Wang, Y.; He, Y.; Wu, Q.; Li, L.; Chen, H. Plasma proteomic and metabolomic characterization of COVID-19 survivors 6 months after discharge. Cell Death Dis. 2022, 13, 235. [Google Scholar] [CrossRef]
- Costantini, S.; Madonna, G.; Di Gennaro, E.; Capone, F.; Bagnara, P.; Capone, M.; Sale, S.; Nicastro, C.; Atripaldi, L.; Fiorentino, G.; et al. New Insights into the Identification of Metabolites and Cytokines Predictive of Outcome for Patients with Severe SARS-CoV-2 Infection Showed Similarity with Cancer. Int. J. Mol. Sci. 2023, 24, 4922. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef] [PubMed]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef] [PubMed]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Arunachalam, P.S.; Wimmers, F.; Mok, C.K.P.; Perera, R.A.; Scott, M.; Hagan, T.; Sigal, N.; Feng, Y.; Bristow, L.; Tak-Yin Tsang, O.; et al. Systems biological assessment of immunity tomild versus severe COVID-19 infection in humans. Science 2020, 369, 1210–1220. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Miao, V.N.; Owings, A.H.; Navia, A.W.; Tang, Y.; Bromley, J.D.; Lotfy, P.; Sloan, M.; Laird, H.; Williams, H.B.; et al. Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19. Cell 2021, 184, 4713–4733.e22. [Google Scholar] [CrossRef]
- Kumar, A.; Yang, Y.L.; Flati, V.; Der, S.; Kadereit, S.; Deb, A.; Haque, J.; Reis, L.; Weissmann, C.; Williams, B.R. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: Role of IRF-1 and NF-kappaB. EMBO J. 1997, 16, 406–416. [Google Scholar] [CrossRef]
- Bleiblo, F.; Michael, P.; Brabant, D.; Ramana, C.V.; Tai, T.; Saleh, M.; Parrillo, J.E.; Kumar, A.; Kumar, A. Bacterial RNA induces myocyte cellular dysfunction through the activation of PKR. J. Thorac. Dis. 2012, 4, 114–125. [Google Scholar]
- Anderson, B.R.; Muramatsu, H.; Nallagatla, S.R.; Bevilacqua, P.C.; Sansing, L.H.; Weissman, D.; Karikó, K. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 2010, 38, 5884–5892. [Google Scholar] [CrossRef]
- Hull, C.M.; Bevilacqua, P.C. Discriminating Self and Non-Self by RNA: Roles for RNA Structure, Misfolding, and Modification in Regulating the Innate Immune Sensor PKR. Acc. Chem. Res. 2016, 49, 1242–1249. [Google Scholar] [CrossRef]
- Cao, Q.; Liu, Z.; Xiong, Y.; Zhong, Z.; Ye, Q. Multiple Roles of 25-Hydroxycholesterol in Lipid Metabolism, Antivirus Process, Inflammatory Response, and Cell Survival. Oxid. Med. Cell Longev. 2020, 2020, 8893305. [Google Scholar] [CrossRef] [PubMed]
- Gold, E.S.; Diercks, A.H.; Podolsky, I.; Podyminogin, R.L.; Askovich, P.S.; Treuting, P.M.; Aderem, A. 25-Hydroxycholesterol acts as an amplifier of inflammatory signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 10666–10671. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Scott, A.L. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J. Leukoc. Biol. 2010, 88, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Russo, L.; Muir, L.; Geletka, L.; Delproposto, J.; Baker, N.; Flesher, C.; O’Rourke, R.; Lumeng, C.N. Cholesterol 25-hydroxylase (CH25H) as a promoter of adipose tissue inflammation in obesity and diabetes. Mol. Metab. 2020, 39, 100983. [Google Scholar] [CrossRef]
- Xu, Y.; Nasri, M.; Dannenmann, B.; Mir, P.; Zahabi, A.; Welte, K.; Morishima, T.; Skokowa, J. NAMPT/SIRT2-mediated inhibition of the p53-p21 signaling pathway is indispensable for maintenance and hematopoietic differentiation of human iPS cells. Stem Cell Res. Ther. 2021, 12, 112. [Google Scholar] [CrossRef]
- Liu, T.F.; Yoza, B.K.; El Gazzar, M.; Vachharajani, V.T.; McCall, C.E. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J. Biol. Chem. 2011, 286, 9856–9864. [Google Scholar] [CrossRef]
- Zhai, L.; Ladomersky, E.; Lenzen, A.; Nguyen, B.; Patel, R.; Lauing, K.L.; Wu, M.; Wainwright, D.A. IDO1 in cancer: A Gemini of immune checkpoints. Cell Mol. Immunol. 2018, 15, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.M.; Zhou, L.; Cui, J.; Li, L.; Wu, N.; Yu, A.; Poddar, S.; Liang, K.; Abt, E.R.; Kim, S.; et al. NAD+ depletion by type I interferon signaling sensitizes pancreatic cancer cells to NAMPT inhibition. Proc. Natl. Acad. Sci. USA 2021, 118, e2012469118. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Interferome | Interferome | Sub-Cellular | Transcription |
|---|---|---|---|---|---|
| (IFN-a/b) | (IFN-g) | Location | Regulation | ||
| Nucleotide Metabolism | |||||
| SAMHD1 | SAM-domain and HD-domain containing protein | + | + | Nucleus, Cytosol | IRF7, NF-κB, NAD+ levels |
| NAMPT | Nicotinamide Phosphoribosyltransferase | + | + | Nucleus, Cytosol | NAD+ levels |
| Lipid/cholesterol metabolism | |||||
| CH25H | Cholesterol 25-Hydroxylase | + | + | Cytosol, ER | Oxysterol production |
| Prorein/Aminoacid metabolism | |||||
| EIF2AK2 | Eukaryotic initation factor 2 alpha kinase 2 | + | + | Cytosol, Nucleus | NFkB, IRF1, STAT1 |
| IDO1 | Indolaemine 2,3-Dioxygenase1 | + | + | Cytosol, Nucleus | ATF4, NRF2, ATF3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramana, C.V. Regulation of a Metabolic Gene Signature in Response to Respiratory Viruses and Type I Interferon Signaling. J. Mol. Pathol. 2024, 5, 133-152. https://doi.org/10.3390/jmp5010009
Ramana CV. Regulation of a Metabolic Gene Signature in Response to Respiratory Viruses and Type I Interferon Signaling. Journal of Molecular Pathology. 2024; 5(1):133-152. https://doi.org/10.3390/jmp5010009
Chicago/Turabian StyleRamana, Chilakamarti V. 2024. "Regulation of a Metabolic Gene Signature in Response to Respiratory Viruses and Type I Interferon Signaling" Journal of Molecular Pathology 5, no. 1: 133-152. https://doi.org/10.3390/jmp5010009
APA StyleRamana, C. V. (2024). Regulation of a Metabolic Gene Signature in Response to Respiratory Viruses and Type I Interferon Signaling. Journal of Molecular Pathology, 5(1), 133-152. https://doi.org/10.3390/jmp5010009