Abstract
Type 2 diabetes mellitus (T2DM) accounts for one-sixth of deaths globally, whereas cancer is the second leading cause of death in the U.S. T2DM is a known risk factor for many cancers. Reactive oxygen species (ROS)-altered metabolic and signaling pathways link T2DM to cancer. These reprogrammed metabolic and signaling pathways contribute to diabetic complications, impact the redox balance (oxidative stress), and have differential roles in the early and late stages of cancer. A respiratory chain that is highly reduced (as under hyperglycemic conditions) or if reduced cofactors accumulate, ROS are greatly elevated. ROS may cause mutations in mitochondrial DNA (mtDNA) that result in further ROS elevations. The amplification of ROS results in the activation of PKC, an overarching signaling pathway that activates MAPK with a subsequent regulation in several factors that result in pathophysiological manifestations of T2DM and cancer. An upregulation in PKC leads to a deregulation in NF-kß, which regulates the PKB/P13/Akt pathway and orchestrates the cell survival, growth, proliferation, and glucose metabolism manifested in cancer. It also affects Insulin Receptor Substrate (IRS-1), decreasing insulin-stimulated glucose transport and glucose uptake, disrupting subsequent cell signaling pathways contributing to the development of T2DM. Dyslipidemia is a hallmark of T2DM and cancer. ROS-induced lipid peroxidation leads to systemic inflammation, producing inflammatory prostaglandins, cytokines, and chemokines that result in tumor proliferation, rapid tumor growth, and modulation of immunity. The dual role of ROS in the early and late stages of cancer makes antioxidant therapy precarious and may be responsible for controversial results. A system that delivers an antioxidant directly to mitochondria may be useful in inhibiting the formation of ROS early during the pre-diabetic stage, whereas antioxidant therapy must be halted in later stages to retard metastasis.
1. Introduction
Diabetes mellitus (DM) accounted for 6.7 million deaths globally in 2021 and was the sixth leading cause of death worldwide [1,2]. Thirty-seven million people in the US suffer from DM. The annual cost incurred by this disease in the US is approximately USD 379 billion [3]. The American Diabetes Association lists three main types of diabetes—Type 1 results from an autoimmune destruction of beta-cells in the pancreas that produce insulin. Type 1 DM requires insulin therapy for survival. Type 2 DM (T2DM) results from an insulin secretory defect and insulin resistance or insensitivity and accounts for about 90–95% of people in the US with DM [4,5]. T2DM is the fastest growing disease globally and the mortality rate is expected to rise nearly 10-fold by the year 2030 [1]. Type 3 is gestational diabetes that develops in pregnant women who have not been previously diagnosed as diabetic, and while this type of DM usually subsides after the birth of the baby, both mother and child are predisposed to T2DM later in life [4].
Although cancer death rates have declined from a peak in 1991 through 2018, due largely to reductions in smoking, improved disease management, and breakthroughs in treatment and therapies, cancer remains the second leading cause of death in the US with over 600,000 cancer deaths projected in 2021 [6,7,8]. On a global basis, approximately 22 million new cancer cases are projected in 2030—based on population ageing and growth. This represents a 54% increase from the 14.1 million new cases in 2012 [9]. According to WHO, cancer is a major leading cause of death globally, accounting for 1 in 6 deaths in 2018 [10]. The economic and societal burden of cancer is great, ranging from 1.8% of gross domestic product (GDP) in the US in 2017 to 1.07% of GDP for the European Union [11].
T2DM has been associated with increased risks for various cancers [12] Among the cancers in which an increased risk with T2DM have been reported are pancreatic [13], colorectal [14], hepatocellular carcinoma (liver) [15], bladder [16], breast (postmenopausal) [17,18], endometrial [19], and non-Hodgkin’s lymphoma [20]. And although an inverse association was observed for prostate cancer [21], T2DM did not reduce aggressive forms of the cancer, and mortality rates were higher in diabetic patients [22]. T2DM is associated with a 23% increased risk in prostate cancer death and a 25% increased risk in all-cause mortality [23].
Obesity and sedentary lifestyle are risk factors shared by both T2DM and 13 types of cancer [24]. A report stating that 77% of pediatric T2DM patients exhibit obesity does not portend well for future cancer risk [25]. Obesity in individuals afflicted with insulin resistance (T2DM) leads to a hyperglycemic condition linked to higher risk of cancer and in which up to 10-fold higher levels of oxygen radicals (ROS) may be produced as a result of ab increased flux in glucose through normal metabolic pathways [26,27,28]. As a result, a number of metabolic and signaling pathways are activated that contribute to diabetic complications, impact the redox balance (oxidative stress), and have differential roles in the early and late stages of cancer [28,29]. It is the intent of this review to examine the link of these ROS-altered pathways in T2DM to cancer expression.
2. Oxidative Stress and Reactive Oxygen Species (ROS)
2.1. Background
Major atmospheric oxygenation events, occurring about 2.4 to 1.8 billion years ago and again, 800–500 million years ago, resulted in fundamental redox evolutions that impacted the elemental composition of the biosphere [30]. It is believed that the first ROS appeared on Earth with the first atmospheric oxygen, about 2.4–3.8 billion years ago [31]. Indeed, ROS have played a pivotal role in the evolutionary reactions of life.
The concept of “Oxidative Stress” has evolved from early studies in which it was observed that, in the presence of oxygen (O2), the lethal dose of X-ray radiation was about one-third of that required to produce an equivalent lethality under anoxic conditions [32]. This so-called “oxygen effect” was the result of radicals from O2 and hydrogen peroxide (H2O2) that attacked DNA [33], although ROS also attack proteins and lipids and are implicated in the causal etiology of T2DM [28], and cancer [29].
Oxidative stress, as a consequence of excessive ROS production, is characterized by an imbalance between the generation and removal of these species. A redox imbalance results in insulin resistance, beta-cell dysfunction, and impaired glucose intolerance, which are found in T2DM [34]. ROS may be generated exogenously and endogenously. Exogenous sources of ROS may be air pollutants, metals, asbestos, tobacco, or radiation, including UV [27,34,35]. Oxidative stress can arise from excess food intake, and it has been shown that an intake of excessive calories can lead to a 5–10-fold increase in ROS, resulting from a loss of normal respiratory chain regulation [27,36]. Redox imbalance has been shown to be one of the most fundamental reasons for cancer development, progression, and metastasis [37]. Among the mechanisms that exhibit potential for creating a prooxidant state, or oxidative stress, are the modulation of the cytochrome electron transport chain (ETC); inhibitors of antioxidant defenses; membrane active agents; and inflammatory agents [38]. All are interdependent manifestations of T2DM that link to cancer.
2.2. ROS and Endogenous Reactive Species
Molecular oxygen, polyunsaturated fatty acids, sulfhydryl compounds, quinones, and related compounds that can easily transfer single electrons tend to be the major endogenous sources of reactive species [38,39]. The primary ROS, superoxide anion [28], is formed by a univalent reduction in molecular oxygen. A superoxide anion is reduced to hydrogen peroxide (H2O2). Normally, the subsequent reaction yielding the highly damaging hydroxyl radical (•OH) proceeds slowly unless catalyzed by a heavy ion, e.g., iron. Iron–sulfur clusters are found in the mitochondrial complexes that facilitate the transfer of electrons in the Electron Transport Chain (ETC).
2.3. Antioxidant Defense in Maintaining Redox Balance
Redox imbalance is a ROS-initiated manifestation of T2DM. Although ROS are generated in normal euglycemic, insulin-regulated glucose metabolism, the levels are controlled by factors that regulate cellular respiration. These factors include NAD-linked substrates, succinate, oxygen, and antioxidant enzymes that maintain the redox balance. The major endogenous antioxidant enzyme, superoxide dismutase (SOD), is involved in the dismutation of the superoxide anion to H2O2. Hydrogen peroxide is reduced to water by catalase and glutathione peroxidase. The oxidized glutathione (GSSG) is re-reduced to GSH by glutathione reductase in the presence of NADPH [35,39]. These reactions are depicted in Figure 1. Overwhelmingly, this natural defense system occurs under hyperglycemic conditions; when the respiratory chain is highly reduced, reduced cofactors accumulate, and up to 10% of the respiratory oxygen consumed is lost as ROS [27]. An important alternative path to superoxide anion formation is through formation of singlet oxygen (1O2). Singlet oxygen is not a free radical, albeit it is an excited state species created by absorption of energy that lifts an electron to a higher energy orbit. It is highly reactive and produces biological effects similar to other ROS [40]. Evidence supports the thesis that singlet oxygen is the initial oxidant formed in the peroxidative steps of prostaglandin synthesis [41].
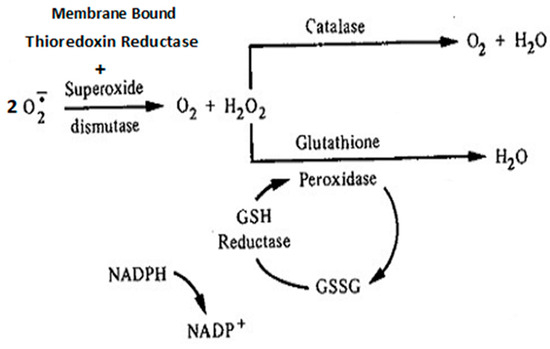
Figure 1.
Simplified schema of major antioxidant enzymes that maintain redox balance. These antioxidant defense enzymes include epidermal membrane-bound thioredoxin reductase and endogenous superoxide dismutase—both are involved in the dismutation of superoxide anions to hydrogen peroxide. The latter may be reduced to H2O and O2 by either catalase or glutathione peroxidase.
Antioxidant supplementation, used to maintain or restore the redox balance and prevent cancer, has largely either failed or been controversial [42,43,44]. Some antioxidants, e.g., ascorbic, and uric acids, promote autoxidation by reducing oxygen activators i.e., transition metals and quinones, resulting in free radical formation, thus acting as a pro-oxidant. Ascorbic acid, in the presence of a metal complex and oxygen (as might occur in the respiratory ETC), can undergo a redox-induced homolysis that leads to the formation of perhydroxyl radical or superoxide anion [45]. An adverse side effect of high vitamin C supplement intake has been associated with an increased risk of cardiovascular disease (CVD) mortality in diabetic postmenopausal women [46]. A water-soluble analog of vitamin E, Trolox, has been shown to increase melanoma metastasis in experimental animals [47]. A small clinical study, in which subjects were supplemented with vitamin E, found that the antioxidant significantly reduced skin malondialdehyde levels but did not affect other measures of oxidative stress in human skin [48].
Resveratrol, (3,4′,5-trihydroxystilbene) is a polyphenol derived primarily from red grape skin, peanuts, and berries that has shown some promise as an anti-cancer agent [49]. The polyphenol was shown to have beneficial effects on T2DM, e.g., increased insulin sensitivity, decreased blood glucose levels, and the positive regulation of several other biomarkers. However, the effects on cancer were dependent upon the type of cancer. It produced beneficial effects on breast cancer while causing severe, adverse effects in multiple myeloma patients. The polyphenol affects all carcinogenic stages, i.e., initiation, promotion, and progression, as well as induces the apoptotic pathway [50]. Specific flavonoids, secondary polyphenol metabolites, have been shown to have beneficial effects on T2DM by targeting cellular signaling pathways in specific tissues, influencing ß-cell function, insulin sensitivity, glucose metabolism, and lipid profile [50]. Other phytochemicals, e.g., those found in green tea, have been shown to have beneficial effects on certain human cancers, although conflicting results have also been reported [51,52]. Other flavonoids that have antioxidant properties and have been shown to have anti-neoplastic properties include curcumin (found in turmeric) and Epigallocatechin gallate (GCG, found in green tea) [53,54]. Dietary supplementation in clinical trials with Resveratrol have, generally, been ambiguous and some even detrimental [52].
ß-carotene (a tetraterpenoid, C40 compound consisting of eight isoprenoid residues) is known to quench singlet oxygen and to have strong antioxidant activity. Indeed, increased ß-carotene intake has been inversely associated with T2DM risk [55]. A study in which a diet quality score, plasma carotenoids (both total and ß-carotene), and lipid peroxidation were employed to monitor oxidative stress found that ß-carotene significantly reduced lipid peroxidation, associated with the oxidative status [56]. ß-carotene has been shown to have effects on adipogenesis, lipolysis, insulin resistance and obesity—all hallmarks of metabolic diseases and T2DM [57,58,59]. A seminal 1981 study found that individuals that consumed greater quantities of green, leafy, and yellow vegetables exhibited a lower risk for cancer, and it was proposed that ß-carotene, because of its singlet oxygen quenching and antioxidant activity, might be responsible [60]. Epidemiological studies found inverse relationships between cancer risks and ß-carotene intake and blood levels. However, clinical trials failed to support these findings and ß-carotene supplementation was found to actually increase lung cancer by 18% [43]. The conflicting clinical data for several types of cancers have been previously reviewed [43]. Experimentally, dietarily supplemented ß-carotene was shown to provide photoprotection against UV-induced skin tumors [61]. However, in 1998, a study reported that dietarily supplemented ß-carotene exacerbated UV-carcinogenesis [62]. A closed-formula diet was employed in the studies in which photoprotection occurred, whereas a semi-defined diet was employed in the study in which the exacerbation of carcinogenesis occurred [63]. A dietary study in which various levels of ß-carotene and vitamins C and E were added to the semi-defined diet failed to ameliorate the exacerbating effect of the carotenoid [64]. It became apparent that other dietary components present in the closed-formula diet (other carotenoids, their isomers, or other phytochemicals) not present in the semi-defined diet were responsible for the potentiation of the carcinogenic response to ß-carotene. The carotenoid has been shown to exhibit either limited antioxidant protection or to behave as a pro-oxidant under oxidative stress conditions [65], and several pathways have been proposed through which these responses might occur [44].
A synthetic phenol, 2,6-di-tert-butyl-p-cresol, or butylated hydroxytoluene (BHT), was shown to provide dramatic photoprotection against UV carcinogenesis [66,67]. The mechanism of the action of BHT is compatible to action mediated via UV dose diminution, perhaps by altering the keratin cross-links of the stratum corneum [67,68,69]. As with the ingestion of any chemical agent, other physiological effects are induced, and BHT induces hepatomegaly, accompanied with an induction of hepatic Phases I and II microsomal detoxification enzymes (mixed function oxidases) [70,71]. Employing a modified Ames test, the influence of antioxidant supplementation (containing BHT) was assessed for the activation of N-2-fluorenylacetamide, a potent hepatocarcinogen [72]. Mutation frequencies in antioxidant-supplemented animals increased twofold, creating a risk to the host it was intended to benefit. Indeed, BHT has been shown to induce the cell proliferation of lung epithelia [73] and to act as a tumor promoter in promotion-sensitive mice [74].
NAC (N-Acetylcysteine) is a stable form of l-cysteine that is a necessary precursor for glutathione synthesis. Glutathione (GSSG) is a major antioxidant, as depicted in Figure 1 NAC was reported to be effective in diminishing T2DM complications, inferring that glucose homeostasis was maintained and that ROS production was diminished [75]. However, a NAC supplementation of hyperglycemic T2DM patients exhibited no benefits in the markers of glucose metabolism, the ß-cell response, or oxidative status and it was concluded that NAC supplementation was unlikely to represent an effective therapeutic prospect [76]. Further, NAC supplementation has been shown to increase melanoma metastasis in experimental animals [47], a consequence of the differential role that ROS plays in the early and late stages of cancer [29]. However, when glycine was added to a NAC supplement, (GlyNAC) and supplemented to T2DM patients, it improved mitochondrial dysfunction, insulin resistance, increased glutathione synthesis, and lowered oxidative stress [76,77]. GlyNAC, if proven effective and safe, may circumvent the disappointing results that have resulted in the questioned use of antioxidant supplementation as a means to reduce cancer risk in the general public. It approaches the catalytic antioxidant that Brownlee [78] posited that would act continuously to reduce ROS. GlyNAC would act endogenously to regenerate glutathione, a major antioxidant, and restore and maintain the redox balance. This appears to be a promising avenue for future research.
A number of clinical trials, primarily observational studies of case-control designs, have reported lower antioxidant levels being inversely associated with cancer risk. These include skin, breast, prostate, colon, and lung cancers [44]. However, when intervention trials (the gold standard) reported adverse effects of antioxidant therapy with the same antioxidants, the safety and effectiveness of antioxidant supplementation was questioned. Several reports on the nominal or adverse effects of antioxidant supplementation appeared [42,43,47,63,76]. A review and meta-analysis reported a significant (16%) increase in the all-cause mortality of trial participants receiving ß-carotene and vitamins A and C treatments [79]. Antioxidant supplementation was reported to negate some of the health-promoting effects of physical exercise [80]. When vitamins C and E were supplemented to T2DM patients, blocking the exercise-dependent formation of ROS, the health-promoting effects of physical exercise, i.e., by promoting insulin sensitivity and the antioxidant defense, were negated. The disappointing and conflicting results of large doses of vitamins C and E and ß-carotene should not be too surprising [29]. Supplementing the highly complex and intricate natural antioxidant defense system with a high level of one antioxidant may alter the stoichiometry of the antioxidant pathways and push the pathway from an antioxidant to a pro-oxidant state or through other pro-oxidant paths [44,81] As a result of these reports on adverse effects of antioxidant supplementation on cancer risk and incidence, the World Cancer Fund/American Institute for Cancer Research and the IARC withdrew recommendations for dietary antioxidant supplementation as a means for cancer prevention [82,83]. This should not deter efforts to seek endogenously metabolized agents that maintain the redox balance and thus have the potential to prevent T2DM complications and cancer. It may require a reassessment of our current methods and the development of new algorithms for safety testing and overall risk/benefit analyses, weighing the risk of one form of cancer to other forms and adverse pathophysiological responses [84]. The application of new methods, e.g., liquid biopsy, might be employed for T2DM patients to determine when antioxidant therapy must cease to avoid antioxidant/ROS inhibition, leading to cancer development and metastasis [85]. The timing of antioxidant therapy is important to the oxidative stress/ROS link of T2DM and cancer. The reduction of ROS and oxidative stress, however, remains a fertile avenue of investigation to preventing and ameliorating both T2DM conditions and the cancers that are linked.
3. Mitochondria, the Electron Transport Chain (ETC), and Respiration Mitochondria and ROS
Mitochondria are the primary source of endogenous ROS. Usually, ROS are formed in the normal (euglycemic, insulin-regulated glucose metabolism) oxidation of foodstuffs, primarily glucose, accounting for about 1–2% of oxygen consumed, and are essential for regulating cell development and normal cellular processes, cellular signaling, and cell death. However, under a hyperglycemic, insulin-resistant glucose metabolism as manifested in T2DM, about 10% of the consumed oxygen may be lost as ROS [27]. Under these conditions, the respiratory chain is highly reduced with the formation of FADH2 and NADH that, in turn, yield ROS as electrons that are passed through the ETC (depicted in Figure 2. Mitochondrial ROS formation is involved in the uncoupling of phosphorylation/oxidation and cell signaling, which dictate a number of physiological and clinical outcomes [86,87,88,89,90,91].
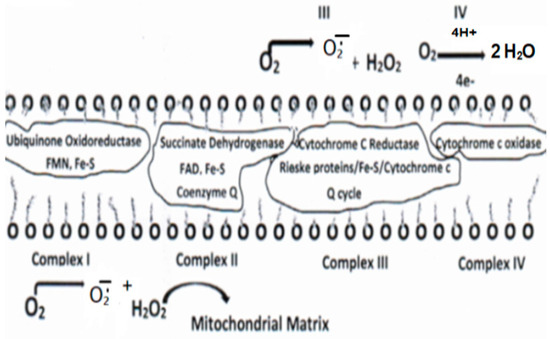
Figure 2.
Protein complexes I–IV depicting the electron transport chain and sites of ROS formation.
4. Diabetic versus Cancer Metabolism and Antioxidants
4.1. Diabetic Metabolism Linking Cancer
It should be noted that the pathophysiological responses in T2DM are complex and both cell- and tissue-specific. Likewise, the carcinogenic process is equally complex and also cell- and tissue-specific. Recognizing the complexity of these two diseases, the description of diabetic metabolism are synthesized herein, to present a general, comprehensive overview. It is imperative that the role of oxidative stress and ROS formation in diabetes be described, as the confluence of these diabetic responses to ROS sets the background linking this disorder to cancer. In this regard, persistent hyperglycemia leads to repeated, acute changes in cellular metabolism initiating four metabolic pathways induced through ROS production that, in turn, lead to higher levels of ROS and oxidative stress. These pathways are activated by increased ROS levels that elevate PARP ((poly (ADP-ribose) polymerase)) levels and downregulate GADPH (glyceraldehyde-3-phosphate dehydrogenase) levels. The latter activate the Polyol pathway (Scheme 1), the Hexosamine pathway (Scheme 2)] the Protein Kinase C pathway (Figure 3), and the formation of advanced glycation products [28,78,86,87,91].

Scheme 1.
Polyol Pathway. It is estimated that under persistent hyperglycemic conditions, the Polyol pathway is activated by elevated ROS levels that, in turn, elevate PARP levels and downregulate GADPH levels. The latter activate and upregulate the Polyol and Hexosamine pathways, accelerate the formation of advanced glycation products (AGE), and activate the Protein Kinase C signal transduction pathway (PKC)—all of which lead to further ROS production, tissue damage, and diabetic complications.

Scheme 2.
Hexosamine Pathway. GFAT, glutamine: fructose-6-phosphate-aminotransferase; UDP-GlucNAc, Uridine-5-diphosphate-N-acetylglucosamine; GlutN, glutamine; Glu, glutamic acid; UDP, Uridine 5′-diphosphate; Pi, inorganic phosphate.
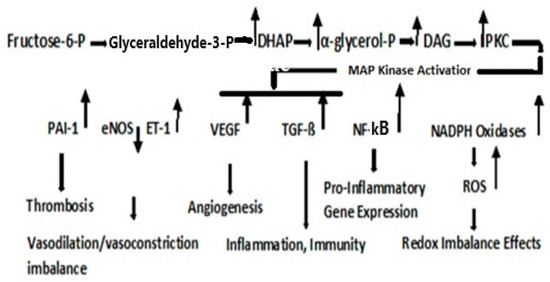
Figure 3.
Protein Kinase C Pathway. Metabolism of fructose-6-phosphate results in the upregulation in dihydroxyacetone phosphate (DHAP), α-glycerol-phosphate, and diacylglycerol (DAG). PKC activation, although cell type- and isoform-specific, is generally activated via DAG; although the flux through the Hexosamine pathway, regulated by GFAT, may also be involved in the activation of PKC [87].
As the high levels of intracellular glucose are reduced by aldose reductase to sorbitol, the reaction consumes NADPH, a cofactor critical for the regeneration of the natural antioxidant, reduced glutathione (refer to Figure 1). Decreasing the level of reduced glutathione increases intracellular oxidative stress [86]. Two NAD+ degradative reactions, a mitochondrial family of signaling proteins (Sirtuins), a histone deacetylase, and an ADP ribosyl transferase are NAD+ dependent, the latter consumed in the formation of nicotinamide, thus contributing to the NADH/NAD redox imbalance [92,93]. Thus, the Polyol pathway plays a critical part in pathophysiology, which contributes to diabetic complications and is initiated by the formation of ROS.
Fructose-6-phosphate is metabolized either through the Embden–Meyerhof–Parnas pathway, or under conditions when the rate of the reoxidation of NADPH is limited, and may be produced when the glucose metabolism is diverted through the pentose phosphate shunt. Nevertheless, fructose-6-phosphate is the initial point of both the Hexosamine and PKC pathways. The Hexosamine Pathway is depicted in Scheme 2 [28].
GFAT regulates the flux through the Hexosamine pathway and is involved in the etiology of diabetic nephropathy [94]. Uridine-5-diphosphate-N-acetylglucosamine (UDP-GlucNAc) is the precursor of amino sugars required for the synthesis of proteoglycans, glycolipids, and glycoproteins.
The PKC pathway is a signaling pathway that not only results in pathophysiological manifestations of T2DM but is also a direct link to cancer.
The activation of PKC leads to the activation of a cascade of kinases, including MAP kinases that regulate a number of factors that are not only important to T2DM but also to cancer. PAI-1, Plasminogen Activator Inhibitor-1 (serpin E1), is a risk factor for thrombosis and atherosclerosis [95]. A downregulation in eNOS, nitric oxide synthase, results in the vasodilation and upregulation of ET-1, Endothelin-1, a vaso-constrictor [96]. Vascular Endothelial Growth Factor (VEGF, a glycoprotein) mediates retinopathy and nephropathy in T2DM [97]. VEGF-D plays a role in lymph angiogenesis and promotes lung metastasis by regulating prostaglandins produced by collecting lymphatic endothelium [98,99]. Transforming Growth Factor-Beta (TGF-ß), a multifunctional cytokine, has a pro-inflammatory property that is important in host immunity [100]. TGF-ß signaling is known to play a role in a large number of human cancers [101,102,103]. Over 40 TGF-ß proteins have thus far been identified, and this family of ligands exert their activities as homodimers or heterodimers that are covalently linked by disulfide bonds, the latter a product of the oxidation (ROS?) [104] of two sulfhydryl groups and a vulnerable target for antioxidant action. TGF-ß manifests dual faces in a complex signaling system with respect to ROS. The signaling pathway exerts an anti-tumorigenic function during the early stages of cancer formation and a pro-tumorigenic effect at later stages, promoting metastasis [103,104]. This dual function for ROS has also been reported in pancreatic cancer [104]. A downregulation in the insulin receptor substance by ROS favors premalignant tumor formation, whereas elevated ROS levels enhance metastasis. The specific response to TGF-ß during tumor progression has been attributed to a range of definitive factors, among which are changes in receptor expression, downstream signaling, evasion of the immune response, stimulation of inflammation, and recruitment of cell types that favor tumor growth or promote angiogenesis [105]. NF-kß, nuclear factor-kappa B, is a DNA binding protein factor required for transcription of pro-inflammatory gene expression [106]. NF-ķß is a central mediator of the inflammatory response and a major participant in innate and adaptive immune responses [106,107]. Thus, it plays a major role in the link between inflammation and cancer [108]. Importantly, NF-ķß regulates the ability of preneoplastic and malignant cells to avert apoptosis-mediated tumor surveillance. The complexity of the NF-ķß signaling pathways is now becoming apparent and, as in T2DM, it is clear that various effects of NF-κB on cancer initiation, promotion, and progression are cell type-, tissue-, and context-specific [108]. NADPH Oxidase, nicotinamide adenine dinucleotide phosphate oxidase (a flavocytochrome B heterodimer), is a major source of ROS in T2DM [109,110]. Increased ROS results in a decrease in glyceraldehyde-3-phosphate dehydrogenase (GADPH) and an increase in methylglyoxal, a strong glycating agent that is a source of advanced glycation end-products (AGEs). The latter contributes to diabetic complications [78]. An additional burst of ROS is generated when AGEs are bound to their receptor sites (RAGEs). The overall effect of NAPDH Oxidases has been redox imbalance [111]. Nevertheless, the discovery of a family of NADPH oxidases (NOXs 1–5 and dual oxidases DUOX1/2 has provided a mechanism for ROS formation in tumor cells. Evidence suggests that these oxidases produce ROS in the G.I. tract under chronic inflammatory stress and may contribute to colorectal and pancreatic carcinomas in patients with inflammatory bowel disease and chronic pancreatitis [112]. The NOX5 isoform is highly expressed in melanoma, prostate cancer, and Barrett’s esophageal adenocarcinomas. The deregulation of these NADPH oxidases, leading to elevated ROS levels, has been recognized as a potential target for clinical therapy. An inhibitor, VAS2870 [3-Bezyl-7-(2-benzoaxozolyl) thio-1,2,3-triazolo(4,5-d) pyrimidine], has been shown to block ROS production, decrease cell proliferation and enhance the apoptotic response induced by TGF-beta in hepatic cell carcinoma [111].
4.2. Dyslipidemia and Cell Signaling
It is known that hyperglycemia, as it occurs in T2DM, results in an elevation of circulating triglycerides and free fatty acids (FFAs). This condition denotes serious dysfunction in lipid dynamics and leads to severe diabetic clinical complications [112]. Dyslipidemia is also a major risk factor for cardiovascular disease (CVD) [113]. Indeed, CVD and cancer share several similar risk factors. e.g., obesity, T2DM, dyslipidemia, chronic inflammation, oxidative stress, and cytokine production—all mediators that contribute to the connection of T2DM/CVD and cancer [113,114,115,116,117,118]. Although conflicting evidence of CVD and cancer lipidomic risk profiles have been observed [119], other studies have demonstrated that elevated levels of lipid biomarkers are independently associated with all-cause mortality as well as CVD risk [118]. Further evidence of a CVD and cancer link was found when study participants who met 6–7 of Life’s simple seven ideal health ASCV (atherosclerotic cardiovascular) metrics [119], exhibited a 51% lower risk for cancer incidence, and those at high CV risk exhibited a >3-fold increased risk of cancer, compared with low-CV risk subjects [119]. Thus, a link runs from T2DM to CVD to cancer, although the complexity of this link is multifaceted.
Type 2 Diabetes Mellitus-associated dyslipidemia may partially be a consequence of a systemic FFA flux secondary to insulin resistance [120,121]. At least 35% of gluconeogenesis in T2DM patients is FFA dependent. Efforts to explain the competitive oxidation between glucose and FFA resulted in what became known as the Glucose Fatty-Acid cycle, or the Randle cycle [122]. Fatty acids are first transported across the cell membrane by Fatty Acid Transport Protein 1 (FATP1), where an FA acyl-CoA synthetase yields acetyl- CoA in the cytosol or a carnitine palmitoyl transfers acyl-CoA across the mitochondrial membrane. A major convergence of signaling pathways involves Protein Kinase C (PKC) activation. Under hyperglycemic conditions in T2DM, incoming surplus energy from obesity is stored in adipocytes in the form of lipids or triacylglyceride (TAG). TAG is converted to diacylglyceride (DAG) by triglyceride lipase (TGL). An upregulation in DAG occurs through the metabolism of Fructose-6-P (Figure 3). The convergence of these pathways to produce DAG is depicted in Figure 4. PKC upregulation is generally activated by DAG [95]
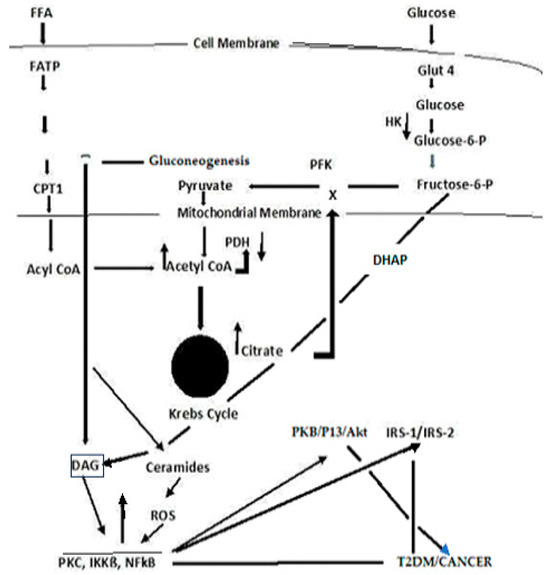
Figure 4.
A simplified schema depicting the confluence of glucose and free fatty acid ROS-induced signaling that links T2DM and cancer.
Although the Randle cycle has been discussed previously, with respect to T2DM [28], a recapitulation is necessary to place the importance of these reactions in perspective to the link between T2DM and cancer. Acyl-CoA formation, in both the cytosol and mitochondria, induces ß-oxidation of FAs [123,124,125]. In the mitochondria, this results in the formation of Acetyl-CoA, which feeds into the Krebs cycle, increasing citrate that, in turn, inhibits hexokinase and pyruvate dehydrogenase activities. The inhibition of hexokinase results in the downregulation of glucogenesis. Elevated Krebs cycle activity results in an increased formation of FADH2 and NADH that yield ROS as electrons that are passed down the ETC [125]. The Acyl-CoA formed in the cytosol stimulates ß-oxidation in the peroxisome and upregulates the glyoxylate cycle by which glucose is synthesized.. DAG activates PKC, IKKB, and NF-kß; the latter acts to suppress PKB, P13/Akt, and IRS-1 and 2. The inhibition of PKB function enhances T2DM, whereas the gain of function drives cancer. The deregulation of P13 plays an important role in proliferation and tumorigenesis. Akt protein plays a critical role in cell survival and the epithelial–mesenchymal transition for cancer formation.
A ROS attack on PUFA leads to increased lipid peroxidation that aggravates systemic inflammation [126]. The relationship between inflammation and cancer was recognized in the mid-1800s when Virchow theorized that cancers originated at sites of chronic inflammation. The causal relationship between inflammation, innate immunity, and cancer is now recognized [127,128,129,130]. Cytokines, and this includes “chemokines”, which are chemotactic cytokines, are the messengers for most of the biologic effects of the immune system, e.g., cell-mediated immunity and allergic responses [128]. The major source of cytokines/chemokines are T lymphocytes. Chemokines play a central role in the development and homeostasis of the immune system and are involved in all protective or destructive immune and inflammatory responses [129,130]. T lymphocytes are characterized by the presence of cell surface molecules, CD4 or CD8. Those lymphocytes expressing CD4 are known as helper T-cells and are prolific cytokine producers and are further subdivided into subsets Th1 and Th2. Th1 produces pro-inflammatory responses. The pro-inflammatory cytokine, Tumor Necrosis Factor-α, (TNF-α), a proinflammatory cytokine—regulates inflammatory cell populations, but once homoeostasis is imbalanced and both Th1 and Th2 arms produce an overabundance of proinflammatory cytokines, rapid tumor growth and proliferation occur [128]. (Figure 5). Indeed, in studies on particulate lung carcinogenesis, it has been shown that chronic inflammation alone can initiate tumor growth without direct interaction with DNA [131].
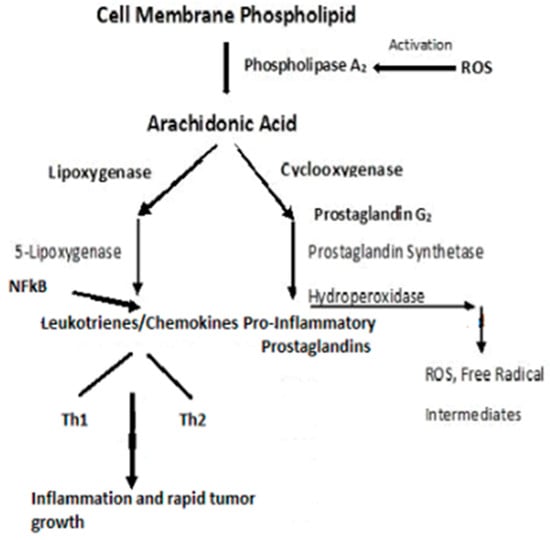
Figure 5.
Pro-inflammatory schema leading to inflammation and rapid tumor growth.
PKC isozymes, phospholipid-dependent serine/threonine kinases, signal through multiple transduction pathways and, in cancer cells, are implicated in angiogenesis, cell proliferation, tumor promotion, invasion, migration, metastasis, and apoptosis (cell survival) [132,133,134]. The PKCs’ role in normal cell function and that in cancer are complicated in that, based upon their structural and activation properties, three subfamilies are classified as classic PKC isozymes that require DAG as an activator and Ca2+ as a cofactor; non-classic, regulated by DAG with no Ca2+ required; and atypical PKC that is not activated by DAG [135]. Thus, the various isoforms may exhibit different responses dependent upon their target proteins and their subsequent signaling responses, with some isoforms even acting as tumor suppressors [134]. Generally, however, it is recognized that PKCs are associated with a number of types of cancer, including breast [11,136], bladder [137], colon [138], gastric [139], glioma [140,141], head and neck [142,143], lung [144,145], melanoma [146,147], and some types of leukemia [148,149,150,151]. The most widely studied isozymes, in relation to cancer, have been PKC α, β, ε, and δ [134]. The theta isozyme, PKCѲ, is classified in a novel PKC subfamily, and its expression is limited to only a few cell types. However, it controls T-cell activation, survival, and differentiation [152]. PKCѲ is highly expressed in T-cell immune responses, playing a critical role for the T-helper (Th2 and Th17)-mediated responses, while the cytotoxic T-cell-driven responses remain relatively intact. Although the function and mode of action of this isoform are different, depending on the type of cancer, in most cancers, the presence of an elevated PKCѲ leads to an abnormal proliferation, migration, and invasion of cancer cells and, thus, promotes tumor aggressiveness [152].
IKKß (inhibitory kß phosphorylase) is a serine/threonine kinase that is upregulated either through the conversion of triacylglyceride (TAG) to diacylglyceride (DAG) in the FFA arm of the Randle cycle or upregulation in DAG through the metabolism of Fructose-6-P in the glucose arm of the cycle [153,154] (Figure 4). IKKß phosphorylates and deregulates NF-ķß, a serine/threonine kinase nuclear transcription factor that has critical roles in inflammation, immunity, cell proliferation, and apoptosis [153,154,155,156]. NF-ķß may also be activated by proinflammatory cytokines, e.g., Tumor Necrosis Factor (TNF)-α [156]. Most known hallmarks of cancer are affected by NF-ķß activation [156]. Targeting IKKß, via inhibitors, may provide therapeutic opportunities [154].
PKC activation induces a cascade of kinases, including mitogen-activated serine/threonine proteins (MAP Kinases) that regulate cell differentiation, proliferation, and apoptosis [156]. Although the downstream secondary signaling cascades are numerous and complex, one, depicted in Figure 4, has particular importance with respect to T2DM and cancer, i.e., the NF-ķß suppression of IRS, PKB/Akt, Serine/threonine-based proteins, protein kinase B (PKB), are also known as Ant, as the widely expressed isoforms of PKB; PKB α, ß, and γ are also known as Ant1, Ant2, and Ant3 [157,158,159,160].
IRS-1, Insulin Receptor Substrate, phosphorylation and dysregulation reduce GLUT4 translocation to the cell surface, decreasing insulin-stimulated glucose transport and glucose uptake. IRS-1 also disrupts subsequent cell signaling pathways, contributing to the development of T2DM [161,162]. NF-ķß targets P13 (Phosphatidylinositol 3-kinase), which plays a central role in a complex, multi-armed signaling network that orchestrates cell responses, including cell survival, growth, proliferation, angiogenesis, migration, and glucose metabolism [157,158]. PI3K is presumed to activate most of its downstream targets via Akt, a serine/threonine kinase, that affects the aforementioned cellular responses and may provide a therapeutic target [163,164,165].
Contributing to an already intricate and complex picture, is the formation of ceramides [164,165] (Figure 4). Ceramide is a core sphingolipid and generally produces antiproliferative responses, e.g., cell growth inhibition, apoptosis induction, and cell invasiveness—thus acting as a tumor suppressor. However, the ceramide metabolism involves glycosylation to produce an AGE that reacts with receptor sites to form a RAGE that, in turn, releases ROS. The tumor suppressor function is lost, and ROS activate the PKC signal transduction pathway. Ceramide glycosylation is closely linked to drug resistance, and this has become a high interest area of investigation for therapeutic targets for cancer [165].
In summary, dyslipidemia is a hallmark of T2DM and cancer [28,112,123]. Dyslipidemia has also been associated with a range of cancers, including secondary cancers, e.g., colon, liver, pancreas, ovary, prostate, kidney, bladder, and brain cancers [166,167].
4.3. Cancer Cell Metabolism, ROS, and Antioxidant Therapy
Otto Warburg first observed, in 1922, that cancer cells exhibit a specific metabolic pattern—one characterized by a shift from aerobic respiration to anaerobic fermentation (the Warburg Effect) [168]. The aerobic respiratory metabolic pattern of normal cells and anaerobic fermentation of cancer cells are depicted in Figure 6.
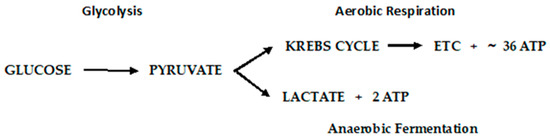
Figure 6.
Metabolic pathways of glucose metabolism of normal (aerobic respiration) and cancer cells lactic acid fermentation).
The Warburg Effect raises a number of questions that have been systematically addressed [169]. First, how does a switch to a much lower yielding ATP pathway as fermentation sustain tumor growth? Using mouse ascites (cancer) cells that obtain ~100% of their energy from fermentation, it was determined that normal mouse cells consumed an average of seven mm3 of oxygen.mg/h., whereas fermentation produced 60 mm3 of lactic acid/mg/h. Converted to energy equivalents, cancer cells obtain approximately the same amount of energy from fermentation as normal cells do through aerobic respiration. A second question, recognizing that the respiration of all cancer cells is irreversibly damaged (irreversible damage occurs as the restoration of oxygen does not restore cells’ normal respiration), is how this damage is induced without killing the cells? Warburg postulated that damage to the respiratory system could be induced by decreasing the oxygen consumption, consequently decreasing the yield of ATP, or the uncoupling of respiration and ATP production with undiminished oxygen consumption. Injury to respiration will be irreversible, and this is common to all cancer incitants. The calculation of metabolic quotients demonstrated that the first phase of carcinogenesis (the irreversible damaging of respiration) need not involve a decrease in the respiratory quotient but entail the uncoupling of oxidative phosphorylation without undiminished oxygen consumption. Warburg provided striking confirmation of his main conclusions from metabolic studies on the C3H/He mouse cell lines developed at the NCI. Two cell lines, developed from a single cell, demonstrate a high and low malignancy rate when injected into C3H/He mice. The anaerobic glycolysis quotient for the high malignancy line was QMN2 = 60–80; that of the low malignancy rate was 20–30. The aerobic glycolysis values, QMO2, was 30 vs. 10 for the high and low malignancy lines, respectively. They were of lower magnitude because of the Pasteur Effect, which was greater in the high malignancy cell line. The Pasteur Effect is the inhibiting effect of oxygen upon fermentation. As oxygen is increased, the accumulation of fermentation products is repressed, and there is a decline in the rate of carbohydrate dissimilation. Conversely, the rate of oxygen consumption (QO2 = 5–10) in the high malignancy line was less than that of the low malignancy line (QO2 = 10–15), corresponding to a greater level of respiratory damage in the high malignancy line. In toto, there is strong evidence consistent with the Warburg Effect, but the question of how this irreversible damage is induced remains open and a very active area of investigation.
Some critics of Warburg’s hypothesis, i.e., that the “driver of tumorigenesis is defective cellular respiration” have proposed alternative possibilities, particularly with the activation of oncogenes and inactivation of tumor suppressor genes [168,169,170]. It is posited that damaged mitochondria are not the root cause of the aerobic glycolytic lesion exhibited by most tumor cells but result from oncogene-directed metabolic reprogramming required to support anabolic growth [171]. It is argued that most tumor mitochondria are not defective in their ability to conduct oxidative phosphorylation and that anabolic growth is the result of oncogene-directed metabolic programming and that the metabolites can be oncogenic by altering cell signaling and blocking cellular differentiation. In this scenario, the activation of the P13/Akt pathway leads to enhanced glucose uptake, glycolysis, increased glucose transporter expression, and the activation of hexokinase. Increased nutrient intake, glucose and glutamine, supports the anabolic requirements of cell growth, whereas proliferating cells use strategies to decrease their ATP production. The overall hypothesis is that the reprogramming of the cell’s metabolism toward macromolecular synthesis is critical for maintaining cell mass and reaching G2 phase in preparation for cell division. In this reprogrammed metabolism, the need is greater for reduced carbon and nitrogen and NADPH for reductive biosynthetic reactions. However, with respect to a link between T2DM and cancer, it should be noted that hyperglycemia leads to the activation of the Hexosamine pathway, which would limit glutamine availability [28]. Further, in insulin-resistant cells, mitochondrial respiration, glycolysis, and ATP levels decrease (in part due to changes in the glucose transporter (GLUT4)—all conditions associated with cancer cells.
Seeming contradictory to the argument that most tumor mitochondria are not defective in their ability to carry-out oxidative phosphorylation, control of the latter’s metabolic machinery resides in the mitochondrial DNA (mtDNA), and there have been extensive studies conducted to examine the mitochondrial genome [172,173]. It is posited that long-term consequences induced by ROS are the results of alterations in mtDNA and indeed, mutations in Complex 1, ubiquinone oxidoreductase, of the ETC are derived from mutations in mtDNA [172]. Although mutations in mtDNA occur at high frequency, the question of whether these mutations alter tumor behavior has been difficult to discern. Using the mtDNA from two tumor cell lines, one highly metastatic, the other of low metastatic potential, the transfer of the mtDNA into recipient tumor cells conveyed the metastatic potential of the transferred mtDNA. The mutations produced a deficiency in respiratory Complex I and produced an overabundance of ROS. Experimental results indicated that mtDNA mutations contributed to tumor progression by enhancing the metastatic potential of tumor cells [172]. A commentary to this study suggested that, using the methodology employed, the researchers failed to show evidence for the formation of superoxide and hydrogen peroxide that was presumed to be generated from the Complex I deficiency associated with mtDNA mutations [173]. Nevertheless, all of the mutations that affect Complex I have similar consequences, i.e., they promote an increase in ROS, increase succinate, inhibit mitochondrial pyruvate dehydrogenase (reducing the flux of pyruvate into the Krebs cycle), and stabilize Hypoxia-Inducing Factor 1-α (HIF-1α) [174].
Hypoxia (low oxygen tension) is thought to be one of the main elements in the switch between glycolysis and respiration [174,175]. Hypoxia induces a complex of intracellular signaling pathways including P13/Akt, MAPK, NFkß, and HIF—all of which are involved in cell proliferation, apoptosis, glucose metabolism, metastasis, and inflammation [175]. Low-oxygen availability inhibits oxidative phosphorylation. The adaption of a cell to hypoxia is partially dependent on the expression and stabilization of Hypoxia-Inducing Factor 1-α (HIF-1α), a transcription protein that when overexpressed, is implicated in promoting tumor growth and metastasis. The overexpression of HIF-1α in tumor cells and rapidly growing normal cells stimulates glycolysis and restricts mitochondrial respiration. The inadequate regulation of hypoxia is an important contributor to the malignant phenotype. Hypoxia also leads to immune resistance and immune suppression that aid tumor cells to escape immune surveillance [176].
Considering the second possibility for the transition to the Warburg phenotype, i.e., the uncoupling of respiration and oxidative phosphorylation, mitochondrial uncoupling proteins (UCPs) catalyze a regulated proton leak across the inner mitochondrial membrane without the generation of ATP [177]. ROS (superoxide) and long-chain fatty acids activate UCP-1, which can be inhibited by purine nucleotides, e.g., ATP [178,179]. There are five isoforms of UCP that have been identified thus far, each with specific functions [180]. An increased expression of UCP-1 has been shown to play a relevant role in immune infiltration by regulating oncogene levels in ovarian cancer [180]. Based on cancer single-cell sequencing data, tumor functional status analyses suggest that UCP-1 may down regulate invasion, epithelial-mesenchymal transition, metastasis, DNA repair, and angiogenesis. UCP-2 inhibits ROS production, which results in a reduced ADP yield and reduced insulin secretion. UCP-2 also catalyzes the exchange and transport of intramitochondrial C4 intermediates (e.g., oxaloacetate), which negatively controls the oxidation of acetyl-CoA-producing substrates through the Krebs cycle. This lowers the redox pressure on the mitochondrial respiratory chain, the ATP–ADP ratio, and ROS production [181]. Employing a UCP-2 knockout mouse, the first in vivo evidence reported that UCP-2 significantly reduced the chemically induced formation of papilloma and malignant squamous cell carcinomas of the skin while not affecting apoptosis [182]. Lactate generation was significantly increased in the carcinogen-treated wild-type mice, but there was no difference between carcinogen-treated and vehicle-treated UCP-2 knockout mice. An upregulation in UCP-2 is known to promote aerobic glycolysis and increase lactate levels. Mitochondrial phospholipase A2, activated by ROS, induces uncoupling in heart mitochondria [183].
Seeking the “switch” that turns normal cells into the cancer phenotype seems inexpedient considering the cacophony of biological responses that may occur simultaneously within an indeterminate time span. As an example, consider cell signaling pathways, including P13/Akt, MAPK, NFkß, and HIF, and their myriad of secondary responses; oxygen tension and the overexpression and stabilization of HIF-1α; mutations in mtDNA, especially those affecting complexes in the respiratory chain; and the uncoupling of respiration from oxidative phosphorylation, are all involved in the reprogramming of the metabolism of the cell [171,184]. If there is a single “switch”, ROS must be considered the prime candidate. Indeed, while recognizing that cancer initiation may be due to multifactorial stressors, evidence suggests a close relationship between oxidative stress and carcinogenesis [185,186]. In addition, ROS not only leads to redox imbalance but functions as signaling molecules that activate signal transduction pathways that promote many aspects of tumor development and progression, expressed in the cancer phenotype. This is particularly relevant in the case of obesity-related hyperglycemia in T2DM, where elevated ROS are produced, and these act as signal molecules to initiate signal transduction pathways (Scheme 3).
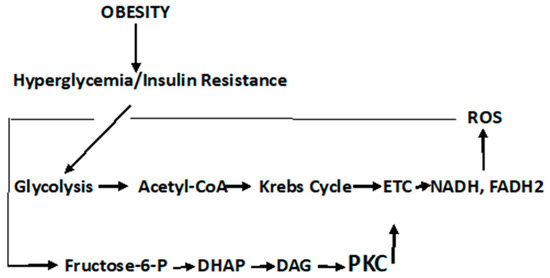
Scheme 3.
Hyperglycemic-induced ROS that activates signal transduction pathways.
Through a somewhat tortuous journey of possibilities, the conclusion remains the same for the initiation of the cancer phenotype, i.e., “reprogrammed metabolism should now be considered as a core hallmark of cancer” [113,171], and cancer joins T2DM, Metabolic Syndrome, and CVD, as a metabolic disease.
4.4. ROS’ Role in the Early and Late Stages of Cancer
Mitochondrial damage and the resulting dysfunction of respiratory components that are induced by ROS, lead to malignant transformations [35]. These organelles are essential for energy metabolism, apoptosis regulation, and cell signaling [186]. The overproduction of ROS, as it occurs in hyperglycemia, induces cancer development by causing genomic instability (mtDNA), inducing signal transduction pathways that modify gene expression (tumor proto-oncogenes and tumor suppressor genes), and impairs oxidative phosphorylation. The latter results in the production of more ROS, which aggravates physiological and metabolic dysfunctions. These elevated levels of ROS can cause cell apoptosis. ROS levels critically determine whether ROS augment tumorigenesis or terminate tumorigenesis via apoptosis [29]. Thus, a homeostatic balance of ROS must be maintained for normal cell survival [29,35,186].
The role of ROS in initiating the early stages of carcinogenesis is depicted in Figure 7. ROS are elevated from the Hyperglycemic metabolism. Elevated ROS results in malignant cell transformations. High ROS levels can lead to apoptosis; therefore, any effective antioxidant therapy should be administered prior to cancer cell proliferation. When antioxidants are administered late in the carcinogenic continuum, ROS levels are suppressed, and apoptosis is lowered. The cancer cells lose their polarity, cell–cell adhesion, and gain mobility. This epithelial to mesenchymal transition is a major cause of tumor metastasis [187,188]. As seen in Figure 7, antioxidant therapy at this late stage of carcinogenesis results in a boost in metastasis. It has been demonstrated that increased activity (decreased stress) of the GPX4 (a selenocysteine-containing protein pathway), known to be regulated via redox homeostasis, is causally linked to the enhanced metastatic activity of cancer cells [187]. Dysregulated cholesterol homeostasis also results in resistance to ferroptosis, which results in increased tumorigenesis and metastasis [188]. A dietary supplementation of antioxidants or induced glutathione synthesis can increase GPX4 activity and promote distant metastasis in animal models of lung cancer and melanoma [189,190]. Thus, the timing of antioxidant therapy is obviously important and should be initiated as early as possible. If dietary interventions are not successful in addressing dietary risks leading to hyperglycemia, the antioxidant therapy should begin in the pre-diabetic phase of T2DM. The discontinuation of antioxidant therapy should take place before the later stages of carcinogenesis and metastasis. Unfortunately, up to this point, antioxidant supplementation, in clinical trials, in order to maintain or restore redox balance and prevent cancer, have largely either failed or been controversial for reasons given previously [42,43,44]. GlyNAC has shown some promise when it is shown to avoid the detrimental effects of NAC supplementation [76,77,78,79]. A recent report describes a mitochondrion-targeting nanoparticle that delivers an antioxidant directly to mitochondria, the major source of endogenous ROS [191]. The perfection of these targeting systems hold promise for targeted antioxidant therapy.
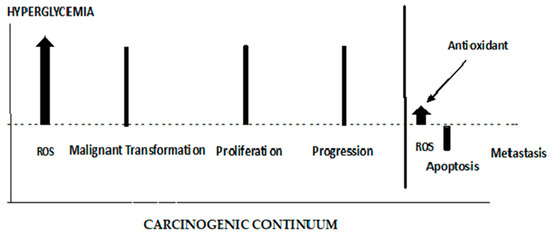
Figure 7.
The different roles of ROS and antioxidants in the early and late stages of cancer.
5. Discussion and Conclusions
Globally, the combined incidence of both T2DM and cancer accounts for a significantly high mortality rate and exacts enormous financial and societal burdens. T2DM is a known risk factor for many forms of cancer, and both manifest a reprogrammed glucose metabolism, the latter leading to the “Warburg cancer phenotype”.
An altered glucose metabolism provides the link of T2DM to cancer. This is the first point at which this link could be addressed—through dietary interventions to reduce the major risk factors for T2DM. Once insulin resistance and hyperglycemia occur, the altered metabolism associated with these conditions results in elevated ROS. ROS not only impact the cell’s redox balance (oxidative stress), but also can attack DNA, proteins, and lipids. ROS may have direct effects on mtDNA, resulting in mutations in respiratory complexes I–IV with further elevation of ROS levels. ROS may create hypoxic conditions through the overexpression of HIF-1α, which results in the stimulation of glycolysis; restricts mitochondrial respiration; and promotes tumor growth and metastasis. A further complecting response to ROS is the activation of mitochondrial uncoupling proteins (UCPs). These proteins catalyze a regulated proton leak across the inner mitochondrial membrane without the formation of ATP. There are five known isoforms of UCP, some exerting counter responses. An upregulation in the isoform, UCP-2, is known to promote aerobic glycolysis and increase lactate levels. Overall, the ROS regulation of HIF-1a and UCP must be important contributing factors that drive cells to anaerobic fermentation and the Warburg cancer phenotype.
The hyperglycemia-mediated elevation of ROS levels lead to the activation of the Polyol and Hexosamine pathways; advanced glycation end products; and the PKC-NF-kß pathways. The PKC signal transduction pathway leads to MAPK activation, and a downstream cascade of kinases that regulate a number of factors, e.g., VEGF, TGF-ß, NF-kß, and NADPH oxidase, which are important to the clinical symptoms and pathophysiologic manifestations of both T2DM and cancer. As an example, VEGF mediates retinopathy and nephropathy in T2DM and promotes lung metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium; TGF-ß is a pro-inflammatory cytokine that is important in host immunity, and TGF-ß signaling is known to play a role in a number of human cancers; NF-kß plays a major role in the link between inflammation and innate and adaptive immune responses, and it regulates the ability of preneoplastic and malignant cells to avert apoptosis-mediated tumor surveillance; NADPH Oxidase deregulation leads to redux imbalance (oxidative stress) and to elevated ROS levels with its inherit consequences in T2DM and cancer. It should be emphasized that many of these effects are cell type-, tissue-, and context-specific and that we are addressing very complex pathologies in T2DM and cancer.
PKC activation can also result from dyslipidemia—a major hallmark of both T2DM and cancer. Secondary to insulin resistance, elevated systemic FFA results in a competitive oxidation between glucose and FA in adipose cells—known as the Randle cycle. FFA flux yields acetyl-CoA that is oxidized in the mitochondria and results in the formation of FADH2 and NADH (also products of the ROS-induced ß-oxidation of FAs). The electron carriers yield ROS as they are carried through the ETC. The ROS attacks on PUFA lead to increased lipid peroxidation that results in systemic inflammation, producing pro-inflammatory prostaglandins, cytokines, and chemokines, particularly IL-6, IL-1, and TNF-α, which results in the modulation of immunity, tumor proliferation, and rapid tumor growth. ROS also activate phospholipase A2, which cleaves arachidonic acid that is oxidized through the lipoxygenase and cyclooxygenase pathways through which these pro-inflammatory products are produced. It is evident that ROS-induced effects on cell signaling, lipid oxidation/peroxidation, chronic inflammation, and immunity are etiologic factors in the manifestation of both T2DM and cancer.
Although the rational approach to controlling redox balance and ROS levels would be antioxidant therapy, the differential role of ROS in the early and late stages of cancer make such therapies precarious. Indeed, the safety and effectiveness of antioxidant supplementation was questioned after several reports on the nominal or adverse effects of antioxidant supplement appeared. Thus, the World Cancer Fund/American Institute for Cancer Research and the ARC withdrew recommendations for dietary antioxidant supplementation as a means of cancer prevention. The antioxidant supplementation of T2DM patients has also not been encouraging. It was proposed that to reduce direct oxidative damage, the amount of superoxide anion must be reduced and that conventional antioxidants were unlikely to do so effectively. Nevertheless, a recent report on a system that delivers an antioxidant directly to the mitochondria may be useful in inhibiting the formation of ROS (particularly to inhibit superoxide anion) early in the pre-diabetic stage that would then break the link to cancer. Currently, antioxidant therapy should be halted in later stages to retard metastasis until efficient delivery systems of effective antioxidants are developed. The timing of antioxidant therapy is important to the oxidative stress/ROS link of T2DM and cancer and may be responsible for some of the controversial results obtained earlier. Nonetheless, a reduction in ROS and the maintenance of redux homeostasis remains a fertile investigative approach to preventing and ameliorating T2DM and the cancers that are linked.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The author declares no conflicts of interest.
References
- International Diabetes Federation. IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021; Available online: https://www.diabetesatlas.org (accessed on 2 November 2022).
- Abudawood, M. Diabetes and Cancer: A comprehensive review. J. Res. Med. Sci. 2019, 24, 94. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. National Diabetes Statistics Report; Centers for Disease Control and Prevention, U.S. Dept of Health and Human Services: Atlanta, GA, USA, 2020. Available online: https://cdc.gov/diabetes/data/statistics-report/index.html (accessed on 25 January 2022).
- American Diabetes Association. Diabetes Care. J. Clin. Appl. Res. Educ. 2022, 45 (Suppl. S1), S17–S36. Available online: https://www.Diabetes.org/DibetesCare (accessed on 2 November 2022).
- Polonsky, K.S. The Past 200 Years in Diabetes. N. Engl. J. Med. 2012, 367, 1332–1340. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- American Cancer Society. Global Cancer Facts & Figures, 4th ed.; American Cancer Society: Atlanta, GA, USA, 2018. [Google Scholar]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jerma, A. Global Cancer Incidence and Mortality Rates and Trends—An Update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Bray, F. The Evolving Scale and Profile of Cancer Worldwide: Much Ado About Everything; Cancer Surveillance Section, International Agency for Research on Cancer: Lyon, France, 2016. [Google Scholar] [CrossRef]
- World Health Organization. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detai;/cancer (accessed on 27 November 2022).
- Jemal, A.; Torre, L.; Soerjomataram, I.; Bray, F. (Eds.) The Cancer Atlas, 3rd ed.; American Cancer Society: Atlanta, GA, USA, 2019; Available online: http://www.cancer.org/canceratlas (accessed on 2 November 2022).
- Collins, K.K. The Diabetes-Cancer Link. Diabetes Spectr. 2014, 27, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Ben, Q.; Xu, M.; Ning, X.; Liu, J.; Hong, S.; Huang, W.; Zhang, H.; Li, Z. Diabetes mellitus and risk of pancreatic cancer: A meta-analysis of cohort studies. Eur. J. Cancer 2011, 47, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Ben, Q.; Shen, H.; Lu, W.; Zhang, Y.; Zhu, J. Diabetes mellitus and incidence and mortality of colorectal cancer: A systematic review and meta-analysis of cohort studies. Eur. J. Epidemiol. 2011, 26, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, X.; Gong, G.; Ben, Q.; Qiu, W.; Chen, Y.; Li, G.; Wang, L. Increased risk of hepatocellular carcinoma in patients with diabetes mellitus: A systematic review and meta-analysis of cohort studies. Int. J. Cancer 2012, 130, 1639–1648. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Orsini, N.; Brismar, K.; Wolk, A. Diabetes mellitus and risk of bladder cancer: A meta-analysis. Diabetologia 2006, 49, 2819–2823. Available online: https://link.springer.com/article/10.1007/s00125-006-0468-0 (accessed on 12 March 2023). [CrossRef] [PubMed]
- Larsson, S.C.; Mantzoros, C.S.; Wolk, A. Diabetes mellitus and risk of breast cancer: A meta-analysis. Int. J. Cancer 2007, 121, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Boyle, P.; Boniol, M.; Koechlin, A.; Robertson, C.; Valentini, F.; Coppens, K.; Fairley, L.-L.; Zheng, T.; Zhang, Y.; Pasterk, M.; et al. Diabetes and breast cancer risk: A meta-analysis. Br. J. Cancer 2012, 107, 1608–1617. [Google Scholar] [CrossRef] [PubMed]
- Friberg, E.; Orsini, N.; Mantzoros, C.S.; Wolk, A. Diabetes mellitus and risk of endometrial cancer: A meta-analysis. Diabetologia 2007, 50, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Mitri, J.; Castillo, J.; Pittas, A.G. Diabetes and risk of non-Hodgkin’s lymphoma: A meta-analysis of observational studies. Diabetes Care 2008, 31, 2391–2397. [Google Scholar] [CrossRef] [PubMed]
- Kasper, J.S.; Giovannucci, E. A meta-analysis of diabetes mellitus and the risk of prostate cancer. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2056–2062. Available online: https://d.docs.live.net/d89a4d69f41936fc/Documents/Cancer%20MS2.docx (accessed on 12 March 2023). [CrossRef]
- Snyder, C.F.; Stein, K.B.; Barone, B.B.; Peairs, K.S.; Yah, H.C.; Derr, R.L.; Wolff, A.C.; Carducci, M.A.; Brancati, F.L. Does pre-existing diabetes affect prostate cancer prognosis? A systematic review. Prostate Cancer Prostatic Dis. 2010, 13, 58–64. Available online: https://link.springer.com/article/10.1007/s10552-013-0334-6 (accessed on 12 March 2023). [CrossRef] [PubMed]
- Bensimon, L.; Yin, H.; Suissa, S.; Pollak, M.N.; Azoulay, L. Type 2 diabetes and the risk of mortality among patients with prostate cancer. Cancer Causes Control. 2014, 25, 329–338. Available online: https://link.springer.com/article/10.1007/s10552-013-0334-6 (accessed on 12 March 2023). [CrossRef]
- Body Weight, Physical Activity, Diet & Alcohol. The Cancer Atlas. Available online: https://canceratlas.cancer.org/risk-factors/nutrition-and-physical-activity (accessed on 12 March 2023).
- Cloana, M.; Deng, J.; Nadarajah, A.; Hou, M.; Qiu, Y.; Chen, S.S.J.; Rivas, A.; Banfield, L.; Toor, P.P.; Zhou, F.; et al. The Prevalence of Obesity among Children with Type 2 Diabetes A Systematic Review and Meta-analysis. JAMA Netw. Open 2002, 5, e2247186. [Google Scholar]
- Stattin, P.; Bjor, O.; Lukanova, A.; Lenner, P.; Lindahl, B.; Hallmans, G.; Kaaks, R. Prospective study of hyperglycemia and cancer risk. Diabetes Care 2007, 30, 561–567. [Google Scholar] [CrossRef]
- Turrens, J.F.; Freeman, B.A.; Crapo, J.D. Hyperoxia increases H2O2 release by lung mitochondria and microsomes. Biochem. Biophys. 1982, 217, 411–421. [Google Scholar] [CrossRef]
- Black, H.S. A Synopsis of the Associations of Oxidative Stress, ROS, and Antioxidants with Diabetes Mellitus. Antioxidants 2022, 11, 2003. [Google Scholar] [CrossRef]
- Assi, M. The differential role of reactive oxygen species in early and late stages of cancer. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2017, 313, R646–R653. [Google Scholar] [CrossRef]
- Anbar, A.D. Elements and Evolution. Science 2008, 322, 1481–1483. [Google Scholar] [CrossRef]
- Mittler, R. ROS are good. Trends Plant Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef]
- Alper, T.; Howard-Flanders, P. Role of oxygen in modifying the radiosensitivity of E. coli B. Nature 1956, 178, 978–979. [Google Scholar] [CrossRef]
- Gerschman, R.; Gilbert, D.L.; Mye, S.W.; Dwyer, P.; Fenn, W.O. Oxygen poisoning and X-irradiation: A mechanism in common. Science 1954, 119, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Wright, E., Jr.; Scism-Bacon, J.L.; Glass, L.C. Oxidative stress in type 2 diabetes: The role of fasting and postprandial glycaemia. Int. J. Clin. Pract. 2006, 60, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Xia, T.; Chen, D.; Piao, H.-l.; Liu, H.-X. The double-edged role of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef] [PubMed]
- Black, H.S. Potential involvement of free radical reactions in ultraviolet light-mediated cutaneous damage. Photochem. Photobiol. 1987, 46, 213–221. [Google Scholar] [CrossRef]
- Saikolappan, S.; Kumar, B.; Shishodia, G.; Koul, S.; Koul, H.K. Reactive Oxygen species and cancer: A complex interaction. Cancer Letters. 2019, 452, 132–143. [Google Scholar] [CrossRef]
- Cerutti, P.A. Prooxidant states and tumor promotion. Science 1985, 227, 375–381. [Google Scholar] [CrossRef]
- Proctor, P.H.; Reynolds, E.S. Free radicals and disease in man. Physiol. Chem. Phys. Med. NMR 1984, 16, 175–195. [Google Scholar]
- Black, H.S. The defensive role of antioxidants in skin carcinogenesis. In Oxidative Stress in Dermatology; Fuchs, J., Packer, L., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1993; pp. 243–269. [Google Scholar]
- Cadenaas, E.; Sies, H. Singlet oxygen formation detected by low-level chemiluminescence during enzymatic reduction of prostaglandin G2 to H2. Hoppe-Seylers’ Z. Physiol. Chem. 1983, 364, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of gastrointestinal cancers: A systematic review and meta-analysis. Lancet 2004, 364, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- The α-Tocopherol, ß-Carotene Cancer Prevention Study Group. The effect of vitamin E and ß-carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1996, 330, 1029–1035. [Google Scholar] [CrossRef]
- Black, H.S.; Boehm, F.; Edge, R.; Truscott, T.G. The benefits and risks of certain dietary carotenoids that exhibit both anti- and pro-oxidative mechanisms—A comprehensive review. Antioxidants 2020, 9, 264. [Google Scholar] [CrossRef] [PubMed]
- Udenfriend, S.; Clark, C.T.; Axelrod, J.; Brodie, B.B. Ascorbic acid in aromatic hydroxylation.I. A model system for aromatic hydroxylation. J. Biol Chem 1954, 208, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.; Folsom, A.R.; Harnack, L.; Halliwell, B.; Jacobs, D.R., Jr. Does supplemental vitamin C increase cardiovascular disease risk in women with diabetes? Am. J. Clin. Nutr. 2004, 80, 1194–2000. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyürek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8. Available online: www.ScienceTranslationalMedicine.org (accessed on 15 March 2023). [CrossRef] [PubMed]
- McArdle, F.; Rhodes, L.E.; Parslew, R.A.G.; Close, G.L.; Jack, C.I.A.; Friedmann, P.S.; Jackson, M.J. Effects of oral vitamin E and beta-carotene supplementation on ultraviolet radiation-induced oxidative stress in human skin. Am. J. Clin. Nutr. 2004, 80, 1270–1275. [Google Scholar] [CrossRef]
- Berman, A.Y.; Motechin, R.A.; Wiesenfeld, M.Y.; Holz, M.K. The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis Oncol. 2017, 1, 35. [Google Scholar] [CrossRef] [PubMed]
- AL-Ishag, R.K.; Abotaleb, M.; Kubatka, P.; Kajo, K.; Busselberg, D. Flavonoids and their anti-diabetic effects: Cellular mechanisms and effects to improve blood sugar levels. Biomolecules 2019, 9, 430. [Google Scholar] [CrossRef]
- Buttuzzi, S.; Brausi, M.; Rizzi, F.; Castagnetti, G.; Peeracchia, G.; Corti, A. Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high-grade prostateintraepithelial neoplasia: A preliminary report from a one-year proof-of-principle study. Cancer Res. 2006, 66, 1234–1240. Available online: https://d.docs.live.net/d89a4d69f41936fc/Documents/Cancer%20MS2.docx (accessed on 15 March 2023). [CrossRef]
- Bushman, J.L. Green tea and cancer in humans: A review of the literature. Nutr. Cancer 1998, 31, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.S.; Acharya, A.; Ray, R.S.; Agrawal, R.; Raghuwanshi, R.; Jain, P. Cellular and molecular mechanisms of curcumin in prevention and treatment of disease. Crit. Rev. Food Sci. Nutr. 2020, 60, 887–939. Available online: https://d.docs.live.net/d89a4d69f41936fc/Documents/Cancer%20MS2.docx (accessed on 15 March 2023). [CrossRef] [PubMed]
- Yuan, J.H.; Li, Y.Q.; Yang, X.Y. Protective effects of epigallocatechin gallate on colon preneoplastic lesions induced by 2-amino-3-methylimidazole [4-5 f] quinoline in mice. Mol. Med. 2018, 31, 88–96. Available online: https://d.docs.live.net/d89a4d69f41936fc/Documents/Cancer%20MS2.docx (accessed on 15 March 2023).
- Sluijs, I.; Cadier, E.; Beulens, J.W.J.; vad der A, D.L.; Spijkerman, A.M.W.; van der Schouw, Y.T. Dietary intake of carotenoids and risk of type 2 diabetes. Nutr. Metab. Cardiovasc. Dis. 2014, 25, P376–P381. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, Y.J.; Lim, Y.; Oh, B.; Kim, J.Y.; Bouwman, J.; Kwon, O. Combination of Diet Quality Score, Plasma Carotenoids, and Lipid Peroxidation to Monitor Oxidative Stress. Oxidative Med. Cell. Longev. 2018, 2018, 8601028. [Google Scholar] [CrossRef] [PubMed]
- Marcelino, G.; Machate, D.J.; de Cássia Freitas, K.; Hiane, P.A.; Maldonade, I.R.; Pott, A.; Asato, M.A.; de Cássia Avellaneda Guimarães, R. β-carotene: Preventive role for type 2 diabetes mellitus and obesity: A review. Molecules 2020, 25, 5803. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, L.; Facey, A.; Omoruyi, F. Diabetes Mellitus and Its Metabolic Complications: The Role of Adipose Tissues. Int. J. Mol. Sci. 2021, 22, 7644. [Google Scholar] [CrossRef]
- Raut, S.K.; Khullar, M. Oxidative stress in metabolic diseases: Current scenario and therapeutic relevance. Mol. Cell Biochem. 2023, 478, 185–196. [Google Scholar] [CrossRef]
- Peto, R.; Doll, R.; Buckley, J.D.; Sporn, M.B. Can dietary β-carotene materially reduce human cancer rates? Nature 1981, 290, 201–208. [Google Scholar] [CrossRef]
- Mathews-Roth, M.M.; Krinsky, N.I. Carotenoid dose level and protection against UV-B- induced skin tumors. Photochem. Photobiol. 1985, 42, 35–38. [Google Scholar] [CrossRef]
- Black, H.S. Radical interception by carotenoids and effects on UV carcinogenesis. Nutr. Cancer 1998, 31, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Black, H.S.; Okotie-Eboh, G.; Gerguis, J. Diet potentiates the UV-carcinogenic response to β-carotene. Nutr. Cancer 2000, 37, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Black, H.S.; Gerguis, J. Modulation of dietary vitamins E and C fails to ameliorate β-carotene exacerbation of UV Carcinogenesis in mice. Nutr. Cancer 2003, 45, 36–45. [Google Scholar] [CrossRef]
- Burton, G.W.; Ingold, K.U. β-carotene: An unusual type of lipid antioxidant. Science 1984, 224, 569–573. [Google Scholar] [CrossRef]
- Black, H.S.; Chan, J.T. Suppression of ultraviolet light-induced tumor formation by dietary antioxidants. J. Investig. Dermatol. 1975, 65, 412–414. [Google Scholar] [CrossRef] [PubMed]
- Black, H.S.; Chan, J.T.; Brown, G.E. Effects of dietary constituents on ultraviolet light-mediated carcinogenesis. Cancer Res. 1978, 38, 1384–1387. [Google Scholar]
- Koone, M.D.; Black, H.S. A mode of action for butylated hydroxytoluene-mediated photocarcinogenesis. J. Investig. Dermatol. 1986, 87, 343–347. [Google Scholar] [CrossRef]
- Black, H.S. Nutritional lipid and antioxidant supplements: Risks versus benefits. Expert Rev. Dermatol. 2012, 7, 483–492. [Google Scholar] [CrossRef]
- Chan, J.T.; Ford, J.O.; Rudolph, A.H.; Black, H.S. Physiological changes in hairless mice maintained on an antioxidant supplemented diet. Experientia 1977, 33, 41–42. [Google Scholar] [CrossRef]
- Malkinson, A.M. Review: Putative mutagens and carcinogens in foods. III. Butylated hydroxytoluene (BHT). Environ Mutat. 1983, 5, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Black, H.S.; Gerguis, J. Use of the Ames test in assessing the relation of dietary lipid and antioxidants to N-2-fluorenylacetamide activation. J. Environ. Pathol. Toxicol. 1980, 4, 131–138. [Google Scholar] [PubMed]
- Witschi, H.; Lock, S. Mechanisms of tumor promotion and carcinogenesis. In Carcinogenesis; Slaga, T.J., Sivak, A., Boutwell, K., Eds.; Raven Press: New York, NY, USA, 1978; Volume 2, pp. 465–474. [Google Scholar]
- Bauer, A.K.; Dwyer-Nield, L.D.; Hankin, J.A.; Murphy, R.C.; Malkinson, A.M. The lung tumor promoter, butylated hydroxytoluene (BHT) causes chronic inflammation in promotion-sensitive BALB/cByJ mice but not in promotion-resistant CXB4 mice. Toxicology 2001, 169, 1–15. [Google Scholar] [CrossRef]
- Bajaj, S.; Khan, A. Antioxidants and diabetes. Indian J. Endocrinol. Metab. 2012, 16 (Suppl. S2), S267–S271. [Google Scholar] [CrossRef]
- Szkudlinska, M.A.; von Frankenberg, A.D.; Utzschneider, K.M. The antioxidant N-Acetylcysteine does not improve glucose tolerance or ß-cell function in type 2 diabetes. J. Diabetes Its Complicat. 2016, 30, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, R.V. GlyNAC (Glycine and N-Acetylcysteine) supplementation improves impaired mitochondrial fuel oxidation and lowers insulin resistance in patients with type 2 diabetes: Results of a pilot study. Antioxidants 2022, 11, 154. [Google Scholar] [CrossRef]
- Brownlee, M. The Pathology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA 2007, 297, 842–857. [Google Scholar] [CrossRef]
- Ristow, M.; Zarse, K.; Oberbach, A.; Kloting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Bluher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Black, H.S. The role of nutritional lipids and antioxidants in UV-induced skin cancer. Front. Biosci. Sch. 2015, 7, 30–39. [Google Scholar] [CrossRef] [PubMed]
- World Cancer Research Fund/American Institute for Cancer Research. Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective; AICR: Washington, DC, USA, 2007. [Google Scholar]
- IARC Working Group on the Evaluation of Cancer-preventive Agents. IARC Handbooks of Cancer Prevention: Carotenoids; International Agency for Research on Cancer: Lyon, France, 1998; Volume 2. [Google Scholar]
- Black, H.S. Reassessment of a free radical theory of cancer with emphasis on ultraviolet carcinogenesis. Integr. Cancer Therapies. 2004, 3, 279–293. [Google Scholar] [CrossRef]
- Black, H.S. Liquid biopsy: A minimally invasive diagnostic tool to identify and characterize cancer cells. J. Integr. Oncol. 2019, 8, 2. [Google Scholar]
- Yan, L.J. Redox imbalance stress in diabetes mellitus: Role of the polyol pathway. Anim. Models Exp. Med. 2018, 1, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, E.D.; Weigert, C. Role of hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int. 2000, 58 (Suppl. S77), S13–S18. [Google Scholar] [CrossRef]
- Ahmad, M.; Wolberg, A.; Kahwaji, C.I. Biochemistry, Electron Transport Chain; StatPearls Publishing: Treasure Island, FL, USA, 2022; Available online: https://d.docs.live.net/d89a4d69f41936fc/Documents/CancersMs.docx (accessed on 15 March 2023).
- Hirst, J. Energy transduction by respiratory complex I—An evaluation of current knowledge. Biochem. Soc. Trans. 2005, 33, 525–529. [Google Scholar] [CrossRef]
- Cardenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Bioenergetics 2018, 1859, 940–950. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef]
- Turkmen, K.; Karagoz, A.; Kucuk, A. Sirtuins as novel players in the pathogenesis of diabetes mellitus. World J. Diabetes 2014, 5, 894–900. [Google Scholar] [CrossRef]
- Wu, J.; Jin, Z.; Zheng, H.; Yan, L.J. Sources and implications of NADH/NAD+ redox imbalance in diabetes and its complications. Diabetes Metab. Syndr. Obes. 2016, 10, 145–153. [Google Scholar] [CrossRef]
- Szabo, C.; Biser, A.; Benko, R.; Bottinger, E.; Susztak, K. Poly (ADP-Ribose) Polymerase Inhibitors Ameliorate Nephropathy of Type 2 Diabetic Leprdb/db Mice. Diabetes 2006, 55, 3004–3012. [Google Scholar] [CrossRef]
- Nishizuka, Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995, 9, 484–496. [Google Scholar] [CrossRef]
- Marasclulo, F.L.; Montagmani, M.; Potenza, M.A. Endotheliiiiin-1: The yin and yang of vascular function. Curr. Med. Chem. 2006, 13, 1655–1665. [Google Scholar] [CrossRef]
- Aiello, L.P.; Wong, J. VEGF—Vascular endothelial growth factor in diabetic vascular complications. Kidney Int. Suppl. 2000, 77, S113–S119. [Google Scholar] [CrossRef]
- Kopfstein, L.; Veikkola, T.; Djonov, V.G.; Baeriswyl, V.; Schomber, T.; Strittmatter, K.; Stacker, S.A.; Achen, M.G.; Alitalo, K.; Christofori, G. Distinct roles of vascular endothelial growth factor-D in lymphangiogenesis and metastasis. Am. J. Pathol. 2007, 170, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Karnezis, T.; Shayan, R.; Caesar, C.; Roufail, S.; Harris, N.C.; Ardipradja, K.; Zhang, Y.F.; Williams, S.P.; Farnsworth, R.H.; Chai, M.G.; et al. VEGF-D promotes tumor metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium. Cancer Cell 2012, 21, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Gomes, K.B.; Rodrigues, K.F.; Fernandes, A.P. The role of transforming growth factor-beta in Diabetic Nephropathy. Int. J. Med. Genet. 2014, 2014, 180270. [Google Scholar] [CrossRef]
- Hachana, S.; Larrivée, B. TGF-β superfamily signaling in the eye: Implications for ocular pathologies. Cells 2022, 11, 2336. [Google Scholar] [CrossRef] [PubMed]
- Stacker, S.A.; Achen, M.G. The VEGF signaling pathway in cancer: The road ahead. Chin. J. Cancer 2013, 32, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. TGF-ß and cancer. Cytokine Growth Factor Rev. 2006, 17, 29–40. [Google Scholar] [CrossRef]
- Chang, C.H.; Pauklin, S. ROS and TGFβ: From pancreatic tumour growth to metastasis. J. Exp. Clin. Cancer Res. 2021, 40, 152. [Google Scholar] [CrossRef]
- Madambath, I.; Appu, A.P. Role of NF-kapa B (NF-kB) in diabetes. Forum Immunopathol. Dis. Ther. 2013, 4, 111–132. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-ķß and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. NF-ķß and cancer: Mechanisms and targets. Mol. Carcinog. 2006, 45, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Karin, M. NF-kappaB and cancer-identifying targets and mechanisms. Curr. Opin. Genet. Dev. 2008, 18, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Mann, G.E. Vascular NAD(P)H oxidase activation in diabetes: A double-edged sword in redox signaling. Cardiovasc. Res. 2009, 82, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Wu, Y.; Meitzler, J.L.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Antony, S.; Doroshow, J.H. NADPH Oxidases and Cancer. Clin. Sci. 2015, 128, 863–875. [Google Scholar] [CrossRef]
- Weyemi, U.; Redon, C.E.; Parekh, P.R.; Dupuy, C.; Bonner, W.M. NADPH Oxidases NOXs and DUOXs as putative targets for cancer therapy. Anti-Cancer Agents Med. Chem. 2013, 13, 502–514. [Google Scholar]
- McGarry, J.D. Banting Lecture 2001: Dysregulation of fatty acid metabolism in the etiology of Type 2 diabetes. Diabetes 2002, 51, 7–18. [Google Scholar] [CrossRef]
- Reaven, G.M. Role of Insulin Resistance in Human Disease (Syndrome X): An Expanded Definition. Annu. Rev. Med. 1993, 44, 121–131. [Google Scholar] [CrossRef]
- Lau, E.S.; Paniagua, S.M.; Liu, E.; Jovani, M.; Li, S.X.; Takvorian, K.; Suthahar, N.; Cheng, S.; Splansky, G.L.; Januzzi, J.L.; et al. Shared risk factors in cardiovascular disease and cancer. J. Am. Coll. Cardiol. 2021, 3, 48–58. [Google Scholar] [CrossRef]
- Koene, R.J.; Prizment, A.E.; Blaes, A.; Konety, S.H. Shared risk factors in cardiovascular disease and cancer. Circulation. 2016, 133, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Duarte, C.W.; Lindner, V.; Francis, S.A.; Schoormans, D. Visualization of cancer and cardiovascular disease co-occurrence with network methods. JCO Clin. Cancer Inform. 2017, 1, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Katzie, V.A.; Sookthai, D.; Johnson, T.; Kühn, T.; Kaaks, R. Blood lipids and lipoproteins in relation to incidence and mortality risks for CVD and cancer in the prospective epic-Heidelberg cohort. BMC Med. 2017, 15, 218. [Google Scholar] [CrossRef]
- Nomikos, T.; Panagiotakos, D.; Georgousopoulou, E.; Metaxa, V.; Chrysohoou, C.; Skoumas, I.; Antonopoulou, S.; Tousoulis, D.; Stefanadis, C.; Pitsavos, C.; et al. Hierarchical modelling of blood lipids’ profile and 10-year (2002–2012) all-cause mortality and incidence of cardiovascular disease: The Attica study. Lipids Health Dis. 2015, 14, 108. [Google Scholar] [CrossRef] [PubMed]
- Hasbani, N.R.; Ligthart, S.; Brown, M.R.; Heath, A.S.; Bebo, A.; Ashley, K.E.; Boerwinkle, E.; Morrison, A.C.; Folsom, A.R.; Aguilar, D.; et al. American Heart Association’s life’s simple 7: Lifestyle recommendations, polygenic risk, and lifetime risk of coronary heart disease. Circulation 2022, 145, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D. Dyslipidemia in type 2 diabetes mellitus. Nat. Clin. Prac. Endocrinol. Metab. 2009, 5, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Korac, B.; Kalezic, A.; Pekovic-Vaughan, V.; Koras, A. Redox changes in obesity, metabolic syndrome, and diabetes. Redox Biol. 2021, 42, 101887. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Delarue, J.; Magnan, C. Free fatty acids and insulin resistance. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 142–148. [Google Scholar] [CrossRef]
- Houten, S.M.; Wanders, R.J.A. General introduction to the biochemistry of mitochondrial fatty acid ß-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef]
- Kaur, S.; Auger, C.; Jeschke, M.G. Adipose tissue metabolic function and dysfunction: Impact of burn injury. Front. Cell Dev. Biol. 2020, 8, 599576. [Google Scholar] [CrossRef]
- De Souza Bastros, A.; Graves, D.T.; de Melo Loureiro, A.P.; Junior, C.R.; Corbi, S.C.T.; Frizzera, F.; Orrico, S.R.P. Diabetes and increased lipid peroxidation are associated with systemic inflammation even in well-controlled patients. J. Diabetes Complicat. 2016, 30, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Kim, D.; Chung, H.; Lee, J.E.; Kim, J.; Hwang, J.; Chung, Y. Immunologic Aspects of Dyslipidemia: A Critical Regulator of Adaptive Immunity and Immune Disorders. J. Lipid Atheroscler. 2021, 10, 184–201. [Google Scholar] [CrossRef]
- Berger, A. Th1 and Th2 responses: What are they? Brit. Med. J. 2000, 321, 424. [Google Scholar] [CrossRef]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Ledford, H. How air pollution causes lung cancer—Without harming DNA. Nature 2023, 616, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-H. Protein kinase C (PKC) isozymes and cancer. New J. Sci. 2014, 2014, 231418. [Google Scholar] [CrossRef]
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.B.; Abba, M.; Kazanietz, M.G. Protein Kinase C and cancer: What we know and what we do not. Oncogene 2014, 33, 5225–5237. [Google Scholar] [CrossRef] [PubMed]
- Isakov, N. Protein kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression. Semin. Cancer Biol. 2018, 48, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Griner, E.; Kazanietz, M. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 2007, 7, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Galoforo, S.S.; Berns, C.M.; Martinez, A.A.; Guan, K.L.; Lee, Y.J. Elevated levels of ERK2 in human breast carcinoma MCF-7 cells transfected with protein kinase Cα. Cell Prolif. 1996, 29, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Varga, A.; Czifra, A.; Tállai, B.; Németh, T.; Kovács, I.; Kovács, L.; Bıró, T. Tumor grade-dependent alterations in the protein kinase C isoform pattern in urinary bladder carcinomas. Eur. Urol. 2004, 46, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Kho, D.H.; Bae, J.A.; Lee, J.H.; Cho, H.J.; Cho, S.H.; Seo, Y.-W.; Ahn, K.Y.; Chung, I.J.; Kim, K.K. KITENIN recruits Dishevelled/PKCd to form a functional complex and controls the migration and invasiveness of colorectal cancer cells. Gut 2009, 58, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-S.; Kim, T.Y.; Kim, Y.-B.; Lee, S.-Y.; Ko, S.-G.; Jong, H.-S.; Bang, Y.-J.; Lee, J.W. The Signaling Network of Transforming Growth Factor β1, Protein Kinase Cδ, and Integrin Underlies the Spreading and Invasiveness of Gastric Carcinoma Cells. Mol. Cell. Biol. 2005, 25, 6921–6936. [Google Scholar] [CrossRef] [PubMed]
- Mandil, R.; Ashkenazi, E.; Blass, M.; Kronfeld, I.; Kazimirsky, G.; Rosenthal, G.; Umansky, F.; Lorenzo, P.S.; Blumberg, P.M.; Brodie, C. Protein kinase Cα and protein kinase Cδ play opposite roles in the proliferation and apoptosis of glioma cells. Cancer Res. 2001, 61, 4612–4619. [Google Scholar]
- Kim, M.; Kim, R.; Yoon, C.; An, S.; Hwang, S.-G.; Suh, Y.; Park, M.-J.; Chung, H.Y.; Kim, I.G.; Lee, S.-J. Importance of PKCδ signaling in fractionated radiation-induced expansion of glioma-initiating cells and resistance to cancer treatment. J. Cell Sci. 2011, 124, 3084–3094. [Google Scholar] [CrossRef]
- Yang, Y.-L.; Chu, J.-Y.; Luo, M.-L.; Wu, Y.-P.; Zhang, Y.; Feng, Y.-B.; Shi, Z.-Z.; Xu, X.; Han, Y.-L.; Cai, Y.; et al. Amplification of PRKCI, located in 3q26, is associated with lymph node metastasis in esophageal squamous cell carcinoma. Genes Chromosomes Cancer 2008, 47, 127–136. [Google Scholar] [CrossRef]
- Liu, S.-G.; Wang, B.-S.; Jiang, Y.-Y.; Zhang, T.-T.; Shi, Z.-Z.; Yang, Y.; Yang, Y.-L.; Wang, X.-C.; Lin, D.-C.; Zhang, Y.; et al. Atypical protein kinase Cι (PKCι) promotes metastasis of esophageal squamous cell carcinoma by enhancing resistance to anoikis via PKCι-SKP2-AKT Pathway. Mol. Cancer Res. 2011, 9, 390–402. [Google Scholar] [CrossRef]
- Lahn, M.; Su, C.; Li, S.; Chedid, M.; Hanna, K.R.; Graff, J.R.; Sandusky, G.E.; Ma, D.; Niyikiza, C.; Sundell, K.L.; et al. Expression levels of protein kinase C-α in non-small-cell lung cancer. Clin. Lung Cancer 2004, 6, 184–189. [Google Scholar] [CrossRef]
- Bae, K.; Wang, H.; Jiang, G.; Chen, M.G.; Lu, L.; Xiao, L. Protein kinase Cε is overexpressed in primary human non-small cell lung cancers and functionally required for proliferation of non-small cell lung cancer cells in a p21/Cip1-dependent manner. Cancer Res. 2007, 67, 6053–6063. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Kikkawa, U. Protein kinase C in melanoma. Cancer Metastasis Rev. 2005, 24, 287–300. [Google Scholar] [CrossRef] [PubMed]
- la Porta, C.A.M.; di Dio, A.; Porro, D.; Comolli, R. Overexpression of novel protein kinase C δ in BL6 murine melanoma cells inhibits the proliferative capacity in vitro but enhances the metastatic potential in vivo. Melanoma Res. 2000, 10, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.T.; Lakum, T.; Lin, K.; Jones, G.M.; Treweeke, A.T.; Farahani, M.; Hughes, M.; Zuzel, M.; Slupsky, J.R. B-cell receptor signaling in chronic lymphocytic leukemia cells is regulated by overexpressed active protein kinase CβII. Blood 2007, 109, 1193–1201. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Holler, C.; Piñón, J.D.; Denk, U.; Heyder, C.; Hofbauer, S.; Greil, R.; Egle, A. PKCβ is essential for the development of chronic lymphocytic leukemia in the TCL1 transgenic mouse model: Validation of PKCβ as a therapeutic target in chronic lymphocytic leukemia. Blood 2009, 113, 2791–2794. [Google Scholar] [CrossRef] [PubMed]
- Kabir, N.N.; Rönnstrand, L.; Kazi, J.U. Protein kinase C expression is deregulated in chronic lymphocytic leukemia. Leuk. Lymphoma 2013, 54, 2288–2290. [Google Scholar] [CrossRef][Green Version]
- Espinosa, I.; Briones, J.; Bordes, R.; Brunet, S.; Martino, R.; Sureda, A.; Prat, J.; Sierra, J. Membrane PKC-beta 2 protein expression predicts for poor response to chemotherapy and survival in patients with diffuse large B-cell lymphoma. Ann. Hematol. 2006, 85, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Nicolle, A.; Zhang, Y.; Belguise, K. The emerging function of PKC theta in cancer. Biomolecules 2021, 11, 221. [Google Scholar] [CrossRef]
- Page, A.; Navarro, M.; Suarez-Cabrera, C.; Bravo, A.; Ramirez, A. Context-dependent role of IKKß in cancer. Genes 2017, 8, 376. [Google Scholar] [CrossRef]
- Israël, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef]
- Viatour, P.; Merville, M.-P.; Bours, V.; Chariot, A. Phosphorylation of NF-ķß and Ikß proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef]
- Lawlor, M.; Alessi, D.R. PKB/AKT: A key mediator of cell proliferation, survival and insulin responses? J. Cell Sci. 2001, 114, 2903–2910. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Revathidevi, S.; Munirajan, A. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019, 59, 80–91. [Google Scholar] [CrossRef]
- Li, W.; Liang, X.; Zeng, Z.; Yu, K.; Zhan, S.; Su, Q.; Yan, Y.; Mansai, H.; Qiao, W.; Yang, Q.; et al. Simvastatin inhibits glucose uptake activity and GLUT4 translocation through suppression of the IR/IRS-1/Akt signaling in C2C12 myotubes. Biomed. Pharmacother. 2016, 83, 194–200. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, N.; Yang, C.H.; Ding, H.S.; Luo, C.; Zhang, Y.; Wu, M.-J.; Zhang, X.-W.; Shen, X.; Jiang, H.-L.; et al. A novel anticancer agent, exerts its anti-proliferative activity by interfering with both PI3K-Akt-mTOR signaling and microtubule cytoskeleton. PLoS ONE 2009, 4, e4881. [Google Scholar] [CrossRef]
- Ponnusamy, S.; Meyers-Needham, M.; Senkal, C.E.; Saddoughi, S.A.; Sentelle, D.; Selvam, S.P.; Salas, A.; Ogretmen, B.; Martinez-Lostao, L.; de Miguel, D.; et al. Sphingolipids and cancer: Ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol. 2010, 10, 1603–1624. [Google Scholar] [CrossRef]
- Sheridan, M.; Ogretmen, B. The Role of Ceramide Metabolism and Signaling in the Regulation of Mitophagy and Cancer Therapy. Cancers 2021, 13, 2475. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, L.; Liu, D.; Wang, C. Ceramide glycosylation and related enzymes in cancer signaling and therapy. Biomed. Pharmacother. 2021, 139, 111565. [Google Scholar] [CrossRef]
- Neshat, S.; Rezaei, A.; Farid, A.; Sarallah, R.; Javanshir, S.; Ahmadian, S.; Chatrnour, G.; Daneii, P. The tangled web of dyslipidemia and cancer: Is there any association? J. Res. Med. Sci. 2022, 27, 93. [Google Scholar] [CrossRef]
- Ho, J.; Kim, E.; Han, M.; Jung, I.; Lee, J.; Suk Jo, Y. Impact of Dyslipidemia on the Risk of Second Cancer in Thyroid Cancer Patients: A Korean National Cohort Study. Ann. Surg. Oncol. 2021, 28, 4373–4384. [Google Scholar] [CrossRef]
- Ferreira, L.M. Cancer metabolism: The Warburg effect today. Exp. Mol. Pathol. 2010, 89, 372–380. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Garcia-Heredia, J.M.; Carnero, A. Decoding Warburg’s hypothesis: Tumor-related mutations in the mitochondrial respiratory chain. Oncotarget 2015, 6, 41582–41599. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even Warburg did not anticipate. Cancer Cell 2012, 20, 297–308. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.-I. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef]
- Zielonka, J.; Kalyanaraman, B. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis—A critical commentary. Free Rad. Biol. Med. 2008, 45, 1217–1719. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol.-Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef] [PubMed]
- Hagen, T.; Vidal-Puig, A. Mitochondrial uncoupling proteins in human physiology and disease. Minerva Medica 2002, 93, 41–57. [Google Scholar]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cardenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Huang, J.; Wang, G.; Liao, K.; Xie, N.; Deng, K. UCP1 modulates immune infiltration level and survival outcome in ovarian cancer patients. J. Ovarian Res. 2022, 15, 16. [Google Scholar] [CrossRef]
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 21, 960–965. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Jackson, K.; Shen, X.; Jin, R.; Li, G.; Kevil, C.G.; Gu, X.; Shi, R.; Zhao, Y. UCP2 knockout suppresses mouse skin carcinogenesis. Cancer Prev. Res. 2015, 8, 487–491. [Google Scholar] [CrossRef]
- Jezek, J.; Jaburek, M.; Zelenka, J.; Jezek, P. Mitochondrial phospholipase A2 activated by reactive oxygen species in heart mitochondria induces mild uncoupling. Physiol. Res. 2010, 59, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell. Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef]
- Drochiolu, G. Multifactorial distress, the Warburg Effect, and respiratory and pH imbalance in cancer development. Stresses 2023, 3, 500–528. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 13, 735. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Roh, J.-L. Targeting GPX4 in human cancer: Implications of ferroptosis induction for tackling cancer resilience. Cancer Lett. 2023, 559, 216119. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chakraborty, B.; Safi, R.; Kazmin, D.; Chang, C.-y.; McDonnell, D.P. Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat. Commun. 2021, 12, 5103. [Google Scholar] [CrossRef]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345.e22. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef]
- Hibino, M.; Maeki, M.; Tokeshi, M.; Ishitsuka, Y.; Harashima, H.; Yamada, Y. A system that delivers an antioxidant to mitochondria for the treatment of drug-induced liver injury. Nat. Sci. Rep. 2023, 13, 6961. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).