
Different Subtypes of Osteosarcoma: Histopathological Patterns and Clinical Behaviour



{kind=link}
{kind=link}
Abstract
1. Introduction to Sarcoma
2. Osteosarcoma
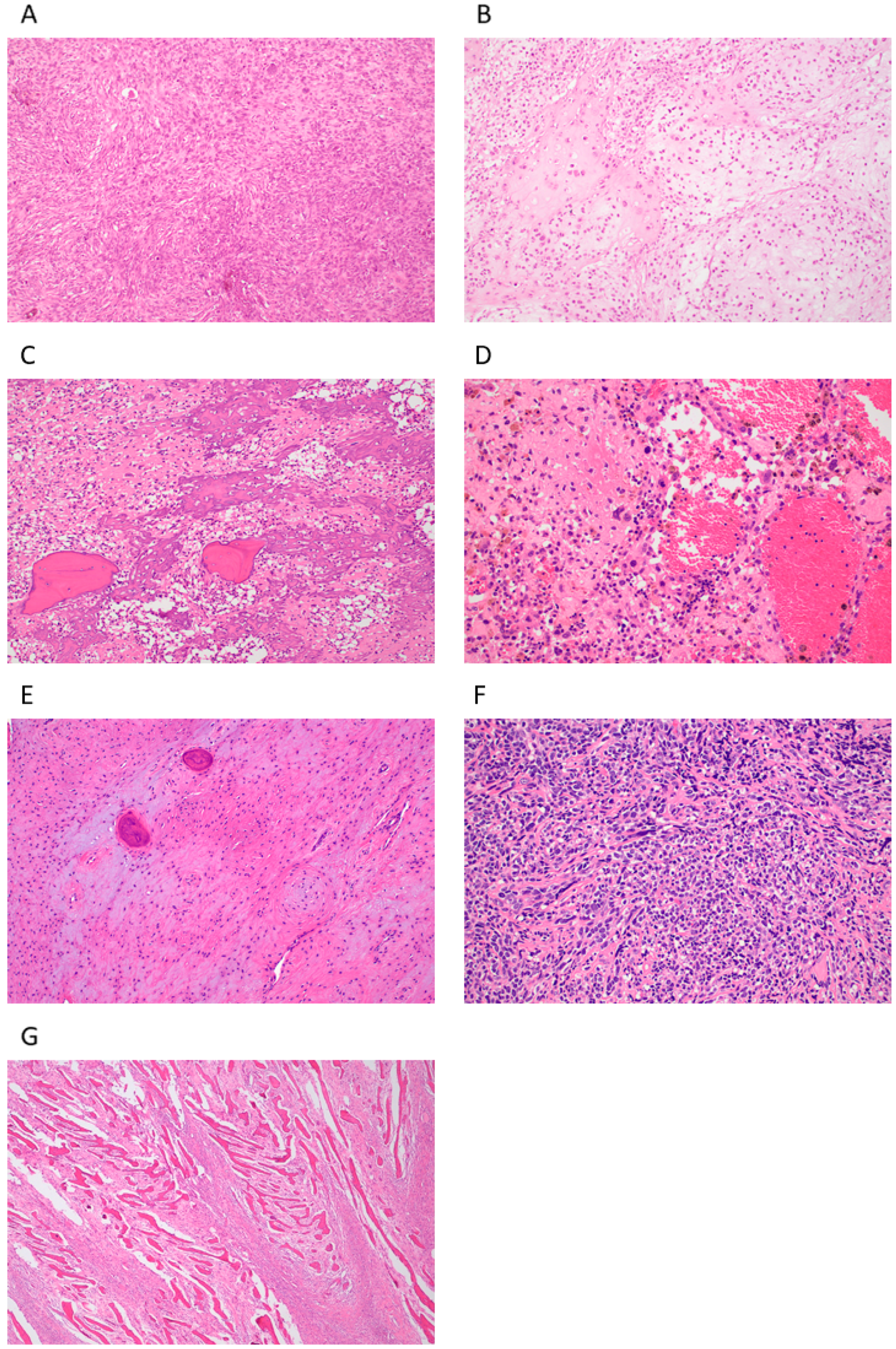
2.1. Central (Medullary) Osteosarcoma
2.1.1. Conventional Central Osteosarcoma (COS)
2.1.2. Telangiectatic Osteosarcoma (TOS)
2.1.3. Low-Grade Osteosarcoma (LGOS, Intraosseous, Well-Differentiated)
2.1.4. Small-Cell Osteosarcoma (SCOS)
2.2. Surface (Peripheral) Osteosarcoma
2.2.1. Parosteal Osteosarcoma (POS)
2.2.2. Periosteal Osteosarcoma (PeOS)
2.2.3. High-Grade Surface Osteosarcoma (HGSO)
3. Epidemiology and Aetiology
4. Symptoms, Diagnosis, and Treatment
5. Conclusions Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Skubitz, K.M.; D’Adamo, D.R. Sarcoma. Mayo Clin. Proc. 2007, 82, 1409–1432. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Staddon, A.P.; Shabason, J.E.; Sebro, R. Phase I and phase II clinical trials in sarcoma: Implications for drug discovery and development. Cancer Med. 2019, 8, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Hatina, J.; Kripnerova, M.; Houfkova, K.; Pesta, M.; Kuncova, J.; Sana, J.; Slaby, O.; Rodríguez, R. Sarcoma Stem Cell Heterogeneity. Stem Cells Heterog.-Nov. Concepts 2019, 1123, 95–118. [Google Scholar]
- Schöffski, P.; Blay, J.-Y.; Ray-Coquard, I. Cabozantinib as an emerging treatment for sarcoma. Curr. Opin. Oncol. 2020, 32, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.M.; Shenasa, E.; Nielsen, T.O. Sarcomas: Immune biomarker expression and checkpoint inhibitor trials. Cancer Treat. Rev. 2020, 91, 102115. [Google Scholar] [CrossRef]
- Brennan, M.F.; Antonescu, C.R.; Moraco, N.; Singer, S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann. Surg. 2014, 260, 416–421, discussion 421–422. [Google Scholar] [CrossRef]
- Tang, F.; Tie, Y.; Wei, Y.-Q.; Tu, C.-Q.; Wei, X.-W. Targeted and immuno-based therapies in sarcoma: Mechanisms and advances in clinical trials. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188606. [Google Scholar] [CrossRef]
- Poveda, A.; del Muro, X.G.; López-Guerrero, J.A.; Cubedo, R.; Martínez, V.; Romero, I.; Serrano, C.; Valverde, C.; Martín-Broto, J. GEIS guidelines for gastrointestinal sarcomas (GIST). Cancer Treat. Rev. 2017, 55, 107–119. [Google Scholar] [CrossRef]
- Young, R.; Natukunda, A.; Litière, S.; Woll, P.; Wardelmann, E.; van der Graaf, W. First-line anthracycline-based chemotherapy for angiosarcoma and other soft tissue sarcoma subtypes: Pooled analysis of eleven European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group trials. Eur. J. Cancer 2014, 50, 3178–3186. [Google Scholar] [CrossRef]
- Ferrari, S.; Serra, M. An update on chemotherapy for osteosarcoma. Expert Opin. Pharmacother. 2015, 16, 2727–2736. [Google Scholar] [CrossRef]
- Rothzerg, E.; Ho, X.D.; Xu, J.; Wood, D.; Märtson, A.; Kõks, S. Upregulation of 15 Antisense Long Non-Coding RNAs in Osteosarcoma. Genes 2021, 12, 1132. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Stitzlein, R.N.; Wojcik, J.; Sebro, R.A.; Balamuth, N.J.; Weber, K.L. Team Approach: Osteosarcoma of the Distal Part of the Femur in Adolescents. JBJS Rev. 2017, 5, e5. [Google Scholar] [CrossRef] [PubMed]
- Misaghi, A.; Goldin, A.; Awad, M.; Kulidjian, A.A. Osteosarcoma: A comprehensive review. SICOT-J 2018, 4, 12. [Google Scholar] [CrossRef]
- Sadeghi, H.M.; Karimi, A.; Derakhshan, S.; Aminishakib, P.; Parchami, K. Conventional osteosarcoma of the mandible: Report of a rare case. Clin. Case Rep. 2021, 9, e04843. [Google Scholar]
- Kimura, Y.; Tomihara, K.; Tachinami, H.; Imaue, S.; Nakamori, K.; Fujiwara, K.; Suzuki, K.; Yasuda, T.; Miwa, S.; Nakayama, E.; et al. Conventional osteosarcoma of the mandible successfully treated with radical surgery and adjuvant chemotherapy after responding poorly to neoadjuvant chemotherapy: A case report. J. Med. Case Rep. 2017, 11, 210. [Google Scholar] [CrossRef]
- McDonald, J.; DenOtter, T.D. Codman Triangle. In StatPearls; Statpearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Limaiem, F.; Kuhn, J.; Khaddour, K. Telangiectatic Osteosarcoma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Malhas, A.M.; Sumathi, V.P.; James, S.L.; Menna, C.; Carter, S.R.; Tillman, R.M.; Jeys, L.; Grimer, R.J. Low-Grade Central Osteosarcoma: A Difficult Condition to Diagnose. Sarcoma 2012, 2012, 764796. [Google Scholar] [CrossRef]
- Sinha, R.; Chowdhury, S.K.R.; Chattopadhyay, P.K.; Rajkumar, K. Low-Grade Osteosarcoma of the Mandible. J. Maxillofac. Oral Surg. 2010, 9, 186–190. [Google Scholar] [CrossRef]
- Vasiliadis, H.S.; Arnaoutoglou, C.; Plakoutsis, S.; Doukas, M.; Batistatou, A.; Xenakis, T.A. Low-grade central osteosarcoma of distal femur, resembling fibrous dysplasia. World J. Orthop. 2013, 4, 327–332. [Google Scholar] [CrossRef]
- Edeiken, J.; Raymond, A.K.; Ayala, A.G.; Benjamin, R.S.; Murray, J.A.; Carrasco, H.C. Small-cell osteosarcoma. Skelet. Radiol. 1987, 16, 621–628. [Google Scholar] [CrossRef]
- Nakajima, H.; Sim, F.H.; Bond, J.R.; Unni, K.K. Small cell osteosarcoma of bone. Review of 72 cases. Cancer 1997, 79, 2095–2106. [Google Scholar] [CrossRef]
- Righi, A.; Gambarotti, M.; Longo, S.; Benini, S.; Gamberi, G.; Cocchi, S.; Vanel, D.; Picci, P.; Bertoni, F.; Simoni, A.; et al. Small cell osteosarcoma: Clinicopathologic, immunohistochemical, and molecular analysis of 36 cases. Am. J. Surg. Pathol. 2015, 39, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Hang, J.F.; Chen, P.C. Parosteal osteosarcoma. Arch. Pathol. Lab. Med. 2014, 138, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Prabowo, Y.; Kamal, A.F.; Kodrat, E.; Prasetyo, M.; Maruanaya, S.; Efar, T.S. Parosteal Osteosarcoma: A Benign-Looking Tumour, Amenable to a Variety of Surgical Reconstruction. Int. J. Surg. Oncol. 2020, 2020, 4807612. [Google Scholar] [CrossRef]
- Sciot, R. MDM2 Amplified Sarcomas: A Literature Review. Diagnostics 2021, 11, 496. [Google Scholar] [CrossRef]
- Chen, P.C.; Yen, C.C.; Hung, G.Y.; Pan, C.C.; Chen, W.M. Gene amplification and tumor grading in parosteal osteosarcoma. J. Chin. Med. Assoc. 2019, 82, 889–894. [Google Scholar] [CrossRef]
- Liu, X.W.; Zi, Y.; Xiang, L.B.; Han, T.Y. Periosteal osteosarcoma: A review of clinical evidence. Int. J. Clin. Exp. Med. 2015, 8, 37–44. [Google Scholar]
- Cesari, M.; Alberghini, M.; Vanel, D.; Palmerini, E.; Staals, E.L.; Longhi, A.; Abate, M.; Ferrari, C.; Balladelli, A.; Ferrari, S. Periosteal osteosarcoma: A single-institution experience. Cancer 2011, 117, 1731–1735. [Google Scholar] [CrossRef]
- Deng, Z.; Huang, Z.; Ding, Y.; Su, Y.; Chan, C.M.; Niu, X. High-Grade Surface Osteosarcoma: Clinical Features and Oncologic Outcome. J. Bone Oncol. 2020, 23, 100288. [Google Scholar] [CrossRef]
- Staals, E.L.; Bacchini, P.; Bertoni, F. High-grade surface osteosarcoma: A review of 25 cases from the Rizzoli Institute. Cancer 2008, 112, 1592–1599. [Google Scholar] [CrossRef]
- Jafari, F.; Javdansirat, S.; Sanaie, S.; Naseri, A.; Shamekh, A.; Rostamzadeh, D.; Dolati, S. Osteosarcoma: A comprehensive review of management and treatment strategies. Ann. Diagn. Pathol. 2020, 49, 151654. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int. J. Cancer 2009, 125, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Linabery, A.; Ross, J.A. Trends in childhood cancer incidence in the U.S. (1992–2004). Cancer 2007, 112, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Savage, S.A.; Mirabello, L. Using Epidemiology and Genomics to Understand Osteosarcoma Etiology. Sarcoma 2011, 2011, 548151. [Google Scholar] [CrossRef]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Savage, Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, Epidemiology, and End Results Program. Cancer 2009, 115, 1531–1543. [Google Scholar] [CrossRef]
- Parkin, D.M.; Stiller, C.A.; Draper, G.J.; Bieber, C.A. The international incidence of childhood cancer. Int. J. Cancer 1988, 42, 511–520. [Google Scholar] [CrossRef]
- Blackwell, J.B.; Threlfall, T.J.; McCaul, K.A. Primary malignant bone tumours in Western Australia, 1972–1996. Pathology 2005, 37, 278–283. [Google Scholar] [CrossRef]
- Prater, S.; McKeon, B. Osteosarcoma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Beird, H.C.; Bielack, S.S.; Flanagan, A.M.; Gill, J.; Heymann, D.; Janeway, K.A.; Livingston, J.A.; Roberts, R.D.; Strauss, S.J.; Gorlick, R. Author Correction: Osteosarcoma. Nat. Rev. Dis. Prim. 2022, 8, 82. [Google Scholar] [CrossRef]
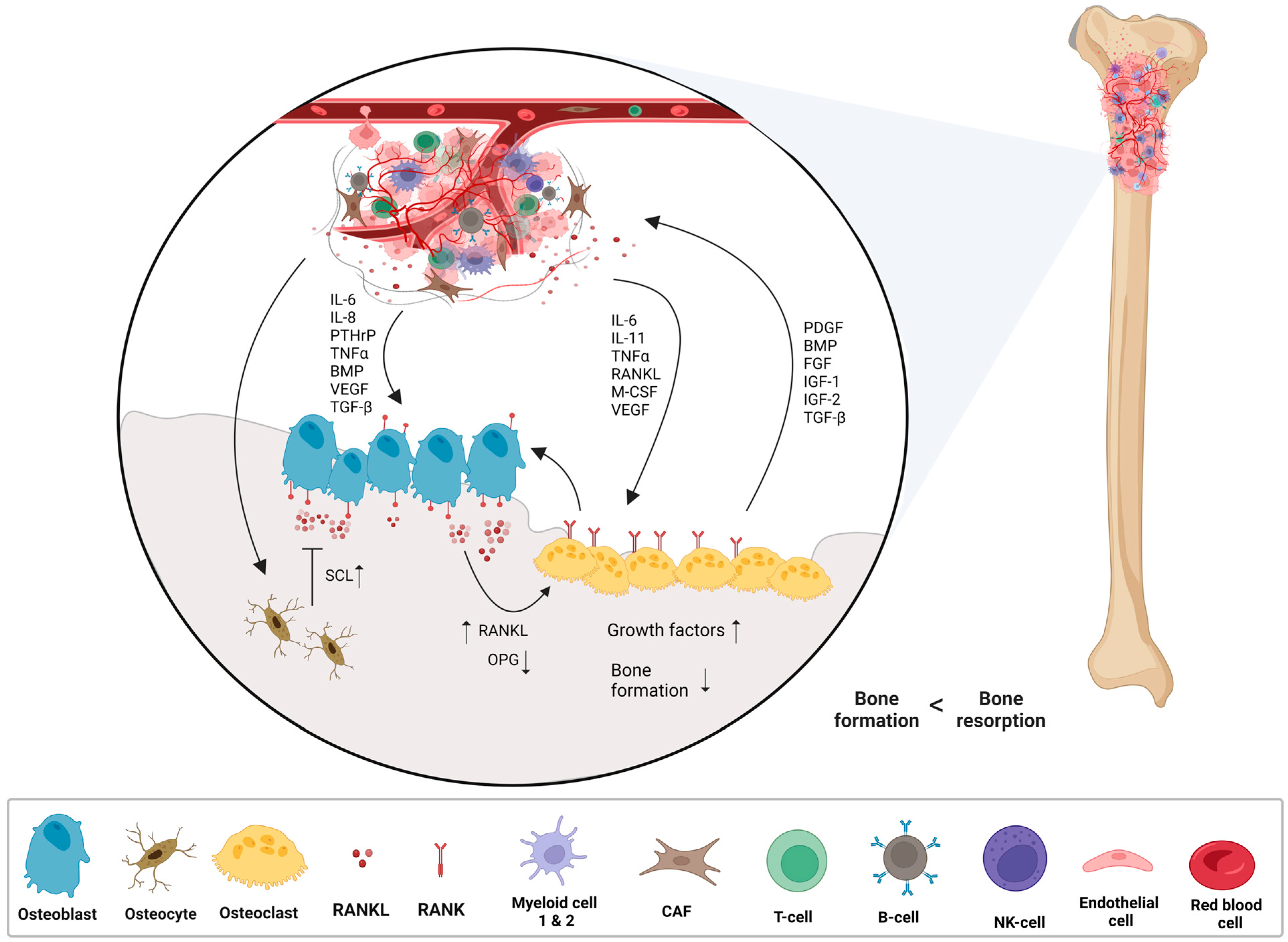
- Corre, I.; Verrecchia, F.; Crenn, V.; Redini, F.; Trichet, V. The Osteosarcoma Microenvironment: A Complex But Targetable Ecosystem. Cells 2020, 9, 976. [Google Scholar] [CrossRef]
- Ara, T.; DeClerck, Y.A. Interleukin-6 in bone metastasis and cancer progression. Eur. J. Cancer 2010, 46, 1223–1231. [Google Scholar] [CrossRef]
- Zhu, T.; Han, J.; Yang, L.; Cai, Z.; Sun, W.; Hua, Y.; Xu, J. Immune Microenvironment in Osteosarcoma: Components, Therapeutic Strategies and Clinical Applications. Front. Immunol. 2022, 13, 907550. [Google Scholar] [CrossRef] [PubMed]
- Lokau, J.; Schoeder, V.; Garbers, C. The role of interleukin-11 in osteosarcoma. Pathologe 2020, 41, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Rothzerg, E.; Feng, W.; Song, D.; Li, H.; Wei, Q.; Fox, A.; Wood, D.; Xu, J.; Liu, Y. Single-Cell Transcriptome Analysis Reveals Paraspeckles Expression in Osteosarcoma Tissues. Cancer Inform. 2022, 21, 11769351221140101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, J.; Zhao, N.; Wang, C.; Kamar, S.; Zhou, Y.; He, Z.; Yang, J.; Sun, B.; Shi, X.; et al. Progress in the chemotherapeutic treatment of osteosarcoma. Oncol. Lett. 2018, 16, 6228–6237. [Google Scholar] [CrossRef]
- Ritter, J.; Bielack, S.S. Osteosarcoma. Ann. Oncol. 2010, 21 (Suppl. S7), vii320–vii325. [Google Scholar] [CrossRef]
- Simpson, E.; Brown, H.L. Understanding osteosarcomas. J. Am. Acad. Physician Assist. 2018, 31, 15–19. [Google Scholar] [CrossRef]
- Durfee, R.A.; Mohammed, M.; Luu, H.H. Review of Osteosarcoma and Current Management. Rheumatol. Ther. 2016, 3, 221–243. [Google Scholar] [CrossRef]
- Ando, K.; Heymann, M.F.; Stresing, V.; Mori, K.; Redini, F.; Heymann, D. Current therapeutic strategies and novel approaches in osteosarcoma. Cancers 2013, 5, 591–616. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, P.; Xu, Y.; Yan, J.; Liu, Z.; Lau, W.B.; Lau, B.; Li, Y.; Zhao, X.; Wei, Y.; et al. Surgical stress and cancer progression: The twisted tango. Mol. Cancer 2019, 18, 132. [Google Scholar] [CrossRef]
- Gottschalk, A.; Sharma, S.; Ford, J.; Durieux, M.E.; Tiouririne, M. Review article: The role of the perioperative period in recurrence after cancer surgery. Anesth. Analg. 2010, 110, 1636–1643. [Google Scholar] [CrossRef]
- Tohme, S.; Simmons, R.L.; Tsung, A. Surgery for cancer: A trigger for metastases. Cancer Res. 2017, 77, 1548–1552. [Google Scholar] [CrossRef] [PubMed]
- Carrle, D.; Bielack, S.S. Current strategies of chemotherapy in osteosarcoma. Int. Orthop. 2006, 30, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, S.; Tripathi, D.N.; Vikram, A.; Ramarao, P.; Jena, G.B. Methotrexate-induced cytotoxicity and genotoxicity in germ cells of mice: Intervention of folic and folinic acid. Mutat. Res. 2009, 673, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Wittig, J.C.; Bickels, J.; Priebat, D.; Jelinek, J.; Kellar-Graney, K.; Shmookler, B.; Malawer, M.M. Osteosarcoma: A multidisciplinary approach to diagnosis and treatment. Am. Fam. Physician 2002, 65, 1123–1132. [Google Scholar]
- Janeway, K.A.; Grier, H.E. Sequelae of osteosarcoma medical therapy: A review of rare acute toxicities and late effects. Lancet Oncol. 2010, 11, 670–678. [Google Scholar] [CrossRef]
- Crews, K.R.; Liu, T.; Rodriguez-Galindo, C.; Tan, M.; Meyer, W.H.; Panetta, J.C.; Link, M.P.; Daw, N.C. High-dose methotrexate pharmacokinetics and outcome of children and young adults with osteosarcoma. Cancer 2004, 100, 1724–1733. [Google Scholar] [CrossRef]
- Xu, M.; Xu, S.; Yu, X. Clinical Analysis of Osteosarcoma Patients Treated with High-Dose Methotrexate-Free Neoadjuvant Chemotherapy. Curr. Oncol. 2014, 21, 678–684. [Google Scholar] [CrossRef]
- Graf, N.; Winkler, K.; Betlemovic, M.; Fuchs, N.; Bode, U. Methotrexate pharmacokinetics and prognosis in osteosarcoma. J. Clin. Oncol. 1994, 12, 1443–1451. [Google Scholar] [CrossRef]
- Jaffe, N.; Gorlick, R. High-dose methotrexate in osteosarcoma: Let the questions surcease—Time for final acceptance. J. Clin. Oncol. 2008, 26, 4365–4366. [Google Scholar] [CrossRef]
- Arcamone, F.; Cassinelli, G.; Fantini, G.; Grein, A.; Orezzi, P.; Pol, C.; Spalla, C. Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius. Biotechnol. Bioeng. 2000, 67, 704–713. [Google Scholar] [CrossRef]
- Cortés-Funes, H.; Coronado, C. Role of anthracyclines in the era of targeted therapy. Cardiovasc. Toxicol. 2007, 7, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Luetke, A.; Meyers, P.A.; Lewis, I.; Juergens, H. Osteosarcoma treatment—Where do we stand? A state of the art review. Cancer Treat. Rev. 2014, 40, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Rothzerg, E.; Pfaff, A.L.; Koks, S. Innovative approaches for treatment of osteosarcoma. Exp. Biol. Med. 2022, 247, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Longhi, A.; Ferrari, S.; Bacci, G.; Specchia, S. Long-term follow-up of patients with doxorubicin-induced cardiac toxicity after chemotherapy for osteosarcoma. Anti-Cancer Drugs 2007, 18, 737–744. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Sprangers, B.; Lapman, S. The growing pains of ifosfamide. Clin. Kidney J. 2020, 13, 500–503. [Google Scholar] [CrossRef]
- Sarbay, H.; Demir, U.F.; Yilmaz, G.; Atay, A.A.; Malbora, B. Ifosfamide induced encephalopathy in a child with osteosarcoma. J. Oncol. Pharm. Pract. 2021, 27, 1302–1306. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Harstrick, A.; Beyer, J.; Metzner, B.; Ruther, U.; Hartmann, J.T.; Holstein, K.; Derigs, H.G.; de Wit, R.; Casper, J.; et al. The use of dose-intensified chemotherapy in the treatment of metastatic nonseminomatous testicular germ cell tumors. German Testicular Cancer Study Group. Semin. Oncol. 1998, 25 (Suppl. S4), 24–32, discussion 45–48. [Google Scholar] [PubMed]
- Hu, Z.; Wen, S.; Huo, Z.; Wang, Q.; Zhao, J.; Wang, Z.; Chen, Y.; Zhang, L.; Zhou, F.; Guo, Z.; et al. Current Status and Prospects of Targeted Therapy for Osteosarcoma. Cells 2022, 11, 3507. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, J.; Chen, Y.; Kang, Y.; Liao, Z.; He, Y.; Zhang, C. Novel Immunotherapies for Osteosarcoma. Front. Oncol. 2022, 12, 830546. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rothzerg, E.; Xu, J.; Wood, D. Different Subtypes of Osteosarcoma: Histopathological Patterns and Clinical Behaviour. J. Mol. Pathol. 2023, 4, 99-108. https://doi.org/10.3390/jmp4020011
Rothzerg E, Xu J, Wood D. Different Subtypes of Osteosarcoma: Histopathological Patterns and Clinical Behaviour. Journal of Molecular Pathology. 2023; 4(2):99-108. https://doi.org/10.3390/jmp4020011
Chicago/Turabian StyleRothzerg, Emel, Jiake Xu, and David Wood. 2023. "Different Subtypes of Osteosarcoma: Histopathological Patterns and Clinical Behaviour" Journal of Molecular Pathology 4, no. 2: 99-108. https://doi.org/10.3390/jmp4020011
APA StyleRothzerg, E., Xu, J., & Wood, D. (2023). Different Subtypes of Osteosarcoma: Histopathological Patterns and Clinical Behaviour. Journal of Molecular Pathology, 4(2), 99-108. https://doi.org/10.3390/jmp4020011