Abstract
The influence of alkali metals (Na, K) or transition metals (Co, Cr, Cu, and Y) incorporated into perovskite crystal on the electronic structures, spectroscopic, and magnetic properties, and thermodynamic properties was investigated by first-principles calculation. Incorporation of Na or K into the perovskite crystal generated 3s, 3p, 4s, and 4p orbitals of Na or K above the conduction band, which promoted the charge transfer from alkali metal to the conduction band, accelerating the electron diffusion related to the photovoltaic properties. For the Cr, Cu, and Y-incorporated perovskite crystals, the electron density distribution of d-p hybrid orbital on the transition metal and iodine halogen ligand were delocalized at frontier orbital. The electronic correlation worked in between the localized spin on 3d orbital of the metal, and the itinerant carriers on the 5p orbital of the iodine halogen ligand and the 6p orbital of the lead atom in the perovskite crystal. The vibration behavior of the Raman and infrared spectra were associated with change of polarization and slight distortion near the coordination structure. The considerable splitting of chemical shift of 127I-NMR and 207Pb-NMR in the magnetic field was caused by crystal field splitting with the Jahn-Teller effect with nearest-neighbor nuclear quadrupole interaction based on the charge distribution. Decrease of the Gibbs free energy and entropy indicated the thermodynamic stabilization without scattering carrier diffusion as phonon effectiveness. The decrease of the entropy was based on a slight change of stretching vibration mode of Pb–I bond with vending mode of N–H and C–H bonds in the infrared and Raman spectra. The minor addition of alkali metal or transition metal into the perovskite crystal would improve the photovoltaic properties, open voltage related to band gap, and short-circuit current density based on the carrier diffusion with phonon effectiveness.
Keywords:
perovskite; solar cell; photovoltaic device; First-principles calculation; K; Na; Cu; Co; Cr 1. Introduction
Organic–inorganic lead halide perovskite crystals have great advantages to apply in electronic application in photovoltaic devices with excellent photovoltaic performance of open voltage, conversion efficiency, and optical absorption [1,2,3,4]. The photovoltaic properties depend on perovskite crystal structure, molar ratio of chemical element composition, electronic structure, surface morphology, and crystallinity in active layer, hole-transporting layer using organic semi-conductive materials, and electron-transporting layer using mesoporous structure of titanium oxide. The general chemical formula of perovskite compounds is ABX3, where ‘A’ and ‘B’ are two cations, and ‘X’ is an anion. The perovskite crystal with cubic system has the B cations in six fold coordination, surrounded by an octahedron of X anions and the A cation in octahedral coordination. The perovskite crystals are constructed with lead (Pb) atom at B-site, halogen anions at X-site, and organic cation such as methyl ammonium (MA: CH3NH3), formamidinium (FA: CH3(NH2)2), ethylammonium (EA: CH3CH2(NH2)2), and guanidinium (GA: C(NH2)3) at A-site. Partial substitution of organic cation with alkali metal ion (lithium, sodium, potassium, rubidium, and cesium) at A-site [5,6,7,8], lead metal, transition metal (tin, copper, cobalt, nickel, and chromium) [9,10,11] at B-site, and halogen anion (iodine, chloride, and bromide) at X-site in the cubic crystal was performed for improving the photovoltaic performance and long-term stability. In particular, the correlative relationship between the photovoltaic and optical properties, and electronic structure was discussed on the basis of experimental results and quantum calculation. The photovoltaic performance depends on surface morphology, crystal growth, domain size, defects, pinholes, and trap state in the perovskite layer [12,13,14,15,16]. For example, partial substitution of organic cation with alkali ion controlled the crystal structure and smoothness of surface morphology with the crystal growth and enlarged domain, which promoted the photo-induced carrier generation and diffusion in the perovskite layer [17,18,19,20]. The photovoltaic and optical properties of the perovskite compounds incorporated with alkali ions such as lithium, potassium (K), sodium (Na), rubidium (Rb), and cesium (Cs) were considered by the experimental results. The photovoltaic performance was improved by enhancement of carrier diffusion in the perovskite crystal with addition of K. The photovoltaic and optical properties were based on the electronic structure and band gap related by the crystal structure and lattice constant with addition of alkali metal. The electronic structures of perovskite compounds incorporated with alkali metals were discussed in detail [21,22,23].
Experimental investigation on the incorporation of transition metals on lead (Pb) site of perovskite crystal was performed for optimization with tuning the electronic structure, energies levels, total density of state within the band gap, and optical absorption [24,25,26,27,28,29]. For example, the photovoltaic and optical properties of the Cu-incorporated perovskite solar cell were discussed on the basis of the electron correlation near the metal-ligand structure in the perovskite crystal [26,27]. The Cu-based perovskite solar cell had the slight perturbation of coordination structure, such as the Jahn-Teller effect with tuning the crystal field splitting, which influenced the magnetic and optical properties in the ultraviolet and near-infrared (NIR) region. The transition metal-based perovskite solar cells had the high potential to promote the photocurrent generation, transporting, and optical properties with a wide range of optical absorption near NIR [28,29]. Incorporation of the transition metal into the perovskite crystal modified the crystal structure, phase, work function, and band gap. Raman and optical spectra of the Co-based perovskite solar cell using mixed halides compounds were characterized. Experimental investigations on the photovoltaic properties of methylammonium lead perovskite solar cell with partially replacing lead atom with transition metals were performed. In particular, adjusting the molar ratio of Pb and Co in the mixed-metal perovskite crystal modified the perovskite work function, the photovoltaic performance with the power conversion efficiencies, and open-circuit voltages. The transition metal incorporated into the perovskite crystal had the electron correlation between itinerant electron at conduction state and localized spin on 3d orbital in the transition metal at multiple states [30,31]. The electron correlations were based on extent of hybrid orbitals between the central metal and halogen ligand with crystal splitting of the 3d and 4d orbitals in coordination structure, which influences the carrier generation, mobility, photovoltaic, magnetic, and optical properties. Incorporation of the transition metals into the perovskite crystal is an important role in optimizing the tuning of the electronic structure and electronic correlation related to the photovoltaic, optical, and magnetic properties [32,33,34].
The purpose of this work is to investigate the electronic structures and the optical and spectroscopic properties of Na, K, Co, Cr, Cu, and Y incorporated perovskite crystals for solar cell applications. Effects of minor addition of Na or K into the MAPbI3 perovskite crystal on total DOS near frontier orbital, chemical shifts of 207Pb and 127I-NMR spectra, optical and IR/Raman spectra, and thermal dynamics will be investigated by first-principles calculation using DFT. The additive effects of transition metal such as Co, Cr, Cu, and Y in the FAPbI3 perovskite crystal on the electronic structures, chemical shifts of 207Pb-NMR and 127I-NMR spectra, optical absorption, vibration mode of Raman and IR spectra will be investigated by first-principles calculation using DFT. Electron correlation near the coordination structure with charge bias will be discussed by chemical shift of 127I-NMR and IR/Raman spectra. The thermodynamic stability will be considered by thermodynamic parameters of enthalpy, Gibbs free energy, and entropy related to the Pb–I stretching vibration of IR and Raman spectra. The electron–lattice interaction as phonon effect with scattering the conduction electrons in the perovskite crystal will be considered for improving the photovoltaic properties related to the carrier diffusion behavior. The photovoltaic and optical properties, and magnetic interaction will be discussed on the basis of the electronic structures including the total and partial density of state (DOS and pDOS), the electron density distribution, occupancy and energy levels of the 5p orbital of the iodine halogen atom, the 3d and 4d orbitals of Co2+, Cr2+, Cu2+, Y3+ and the 6p orbital of the Pb atom near frontier orbital.
2. Quantum Calculation
The electronic structures of the perovskite crystal were single-point calculated by ab initio quantum calculation based on the restricted Hartree–Fock (HF) method, the hybrid DFT using restricted B3LYP (B3LYP) with LANL2MB as the basis set (Gaussian 09). The MAPbI3 perovskite crystals with supercells of 2 × 2 × 2 as cluster model were used on the basis of the experimental results using X-ray diffraction data [35]. The perovskite compounds formed in a cubic crystal phase with a lattice constant of 6.391 Å. The perovskite crystal as cluster model was fixed at +8 as positive charge. In the case of the element-substitution system, the cluster model was used for the quantum calculation. The quantum calculation was employed by hybrid DFT using B3LYP with LANL2MB as the basis set. When a slight incorporation of K or Na in the MAPbI3 perovskite crystal was performed, the lattice constant was fixed. The Na- or K-incorporated MAPbI3 cubic structures as cluster model with supercells of 2 × 2 × 2 were fixed to be +8 as the positive charge. As the isolated dilution system, the mole ratio of the Na or K ions to amino cation ion was adjusted to be 1:7.
For the metal incorporated perovskite crystal with supercells of 2 × 2 × 2 as cluster model and the FAPbI3 perovskite crystal as cluster model and periodic structure with cell range of 100, we employed the quantum calculation using hybrid DFT and PBEPBE using UB3LYP with LANL2MB as the basis set. As reference, the FAPbI3 perovskite crystal with supercells of 2 × 2 × 2 as cluster model were fixed at +8 as positive charge. The effect of ionic radius will be weak in the isolated dilution system. The single-point energy calculation of the cluster model with supercells of 2 × 2 × 2 and positive charge of + 8 was performed as not to disturb the cubic structure, similarly to the standard case using the FAPbI3 perovskite crystal based on the experimental results.
The total DOS (TDOS) around the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), and the HOMO–LUMO gap (Eg) of the Na- or K-incorporated MAPbI3 perovskite cubic structure with supercells of 2 × 2 × 2 at charge of + 8 were calculated. The electrostatic potential (ESP) of the perovskite crystals was estimated by Mulliken population analysis. The isotropic chemical shifts of 127I-NMR and 207Pb-NMR were calculated by DFT using NMR and gauge-including atomic orbitals (GIAO) with B3LYP and LANL2MB as the basis set. The vibration modes of IR and Raman spectra were calculated by DFT using frequency mode. The thermodynamic parameters of enthalpy, Gibbs free energy, and entropy were calculated by DFT using frequency mode. Effect of incorporation of alkali metal, or transition metal into the perovskite crystal on the photovoltaic properties was discussed on the basis of the electronic structure, optical and magnetic properties, and thermodynamic properties.
3. Results and Discussion
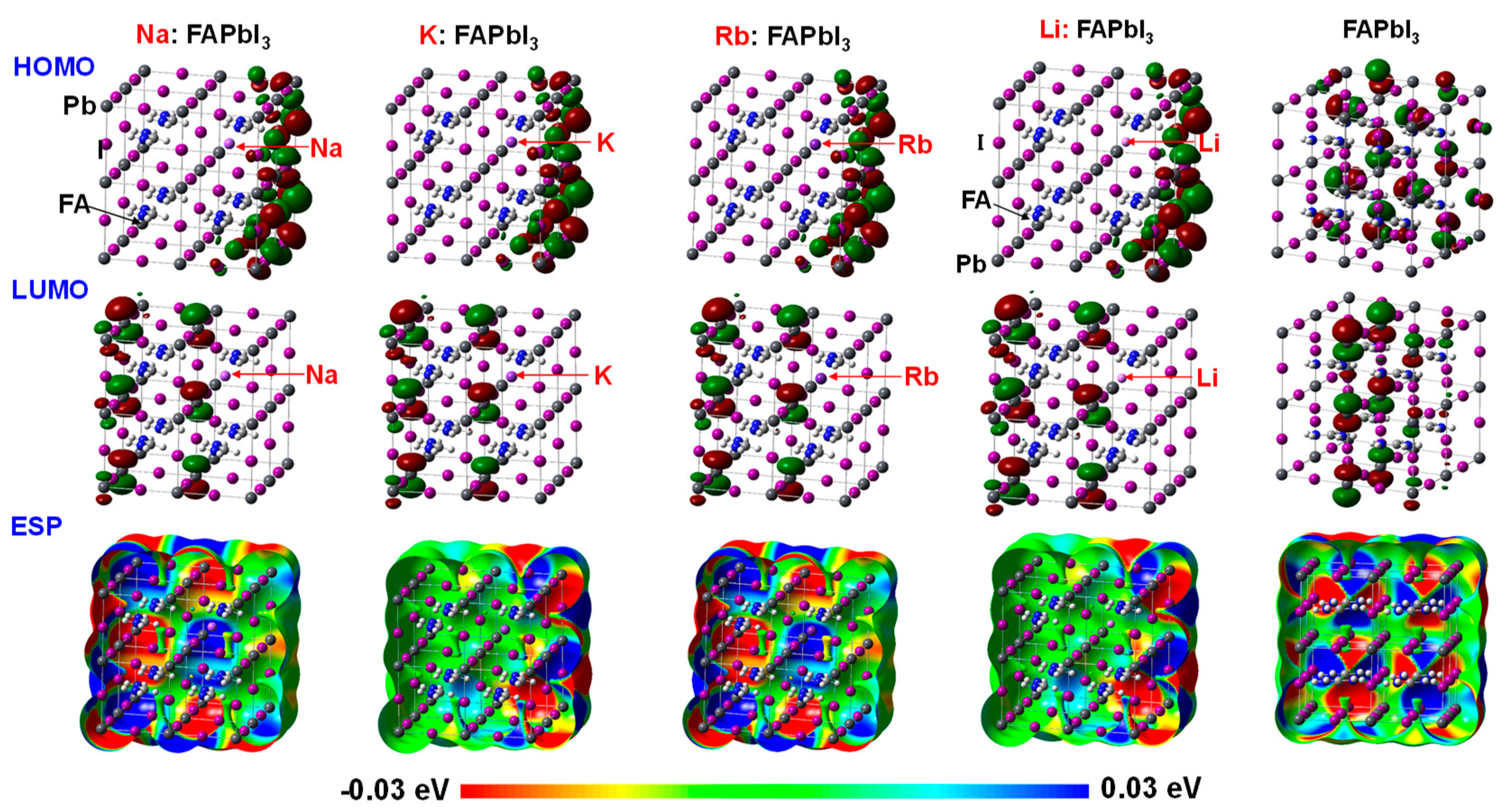
The electronic density distribution at HOMO and LUMO and electrostatic potential of the 12.5% Na- or K-incorporated MAPbI3 perovskite crystal with supercells of 2 × 2 × 2 were investigated, as shown in Figure 1. A part of MA was replaced by the alkali metal of Na and K in the perovskite crystal with the cubic structure as the 2 × 2 × 2 supercell. In the standard case using the MAPbI3 perovskite crystal, the electron density distribution on the 5p orbital of I atom was slightly localized at HOMO. The electron density distribution on the 6p orbital was delocalized at LUMO. Incorporation of Na or K slightly influenced the electron density distribution of 5p orbital on I atom, although the change of the electron density distribution of 6p orbital on Pb atom did not occur. The orbitals have an important role of charge transfer at valence and conduction band state.
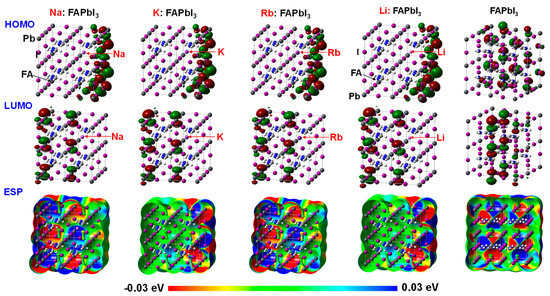
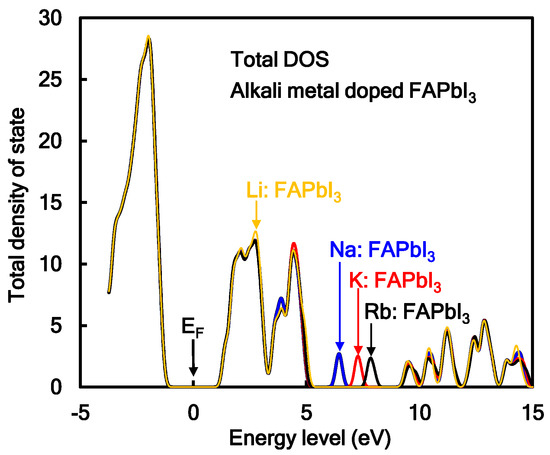
Figure 1.
Electronic density distribution at highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), electrostatic potential (ESP) and total density of states (DOS) near fermi energy level (EF) of alkali metal such as sodium (Na), potassium (K), rubidium (Rb) and lithium (Li) incorporated FAPbI3 perovskite crystals.
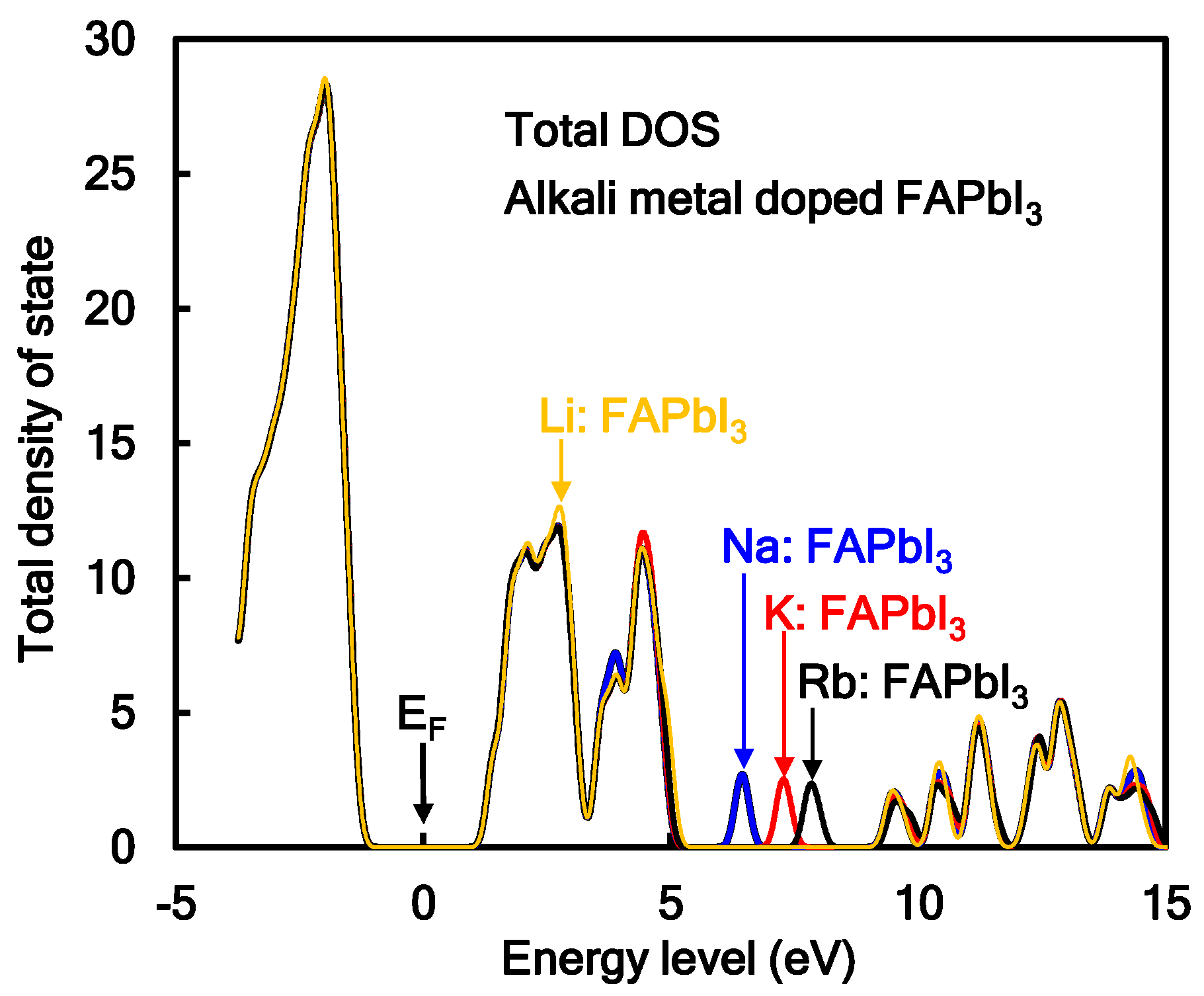
Additive effect of Na or K into the perovskite crystal on total density of state (TDOS) was investigated, as shown in Figure 1. In the case of the Na- or K-incorporated perovskite crystal, the TDOS and energy levels at HOMO and LUMO were slightly changed. The electrons charge-transferred from 3s and 4s orbitals of Na and K to 6p orbital of Pb atom near the conduction levels, promoting the electron concentration and diffusion related to mobility in the perovskite crystal. The energy levels at LUMO, HOMO, band gap, and Fermi level were estimated. The band gaps for MAPbI3, MA(Na)PbI3, and MA(K)PbI3 were obtained to be 2.70, 2.74, and 2.73 eV, which corresponded to wavelengths of 459, 452 and 454 nm, respectively. The calculated band gaps were larger than the experimental results of 1.63 eV, converted from the wavelength of 765 nm in photoluminescence spectra [7]. The reason of increasing the band gap was surface effect of the perovskite cluster rather than periodic effect of the perovskite crystal. Incorporation of alkali metal slightly influenced the DOS, the energy levels at HOMO and LUMO, and band gap by slight perturbation of the coordination structure under electrostatic bias. The optical spectra and excitation process from the ground state will be influenced by incorporation of the alkali metal.
The electronic structures at HOMO and LUMO of the transition metal (M)-incorporated FAPbI3 perovskite crystals were also investigated. The single metal atom (M = Co2+, Cr2+, Cu2+, and Y3+) was incorporated into a FAPbI3 perovskite crystal with the cubic structure as the 2 × 2 × 2 supercell. In the case of the Co-incorporated FAPbI3 perovskite crystal, the electron density distribution of the 6p orbital of the Pb atom was localized at LUMO. In the case of the Cr-incorporated FAPbI3 perovskite crystal, the 3d orbital of Cr atom conjugated with the 5p orbital of I atom as ligand was localized at LUMO. The 6p orbital of the Pb atom was slightly existed. In the case of the Cu and Y-incorporated FAPbI3 perovskite crystals, the electron density distribution at HOMO was delocalized on the 3d-p hybrid orbital of the Cu, Y, and I atoms. The electron density distribution on the 6p orbital of the Pb atom was localized at LUMO. The orbitals work function in the charge transfer as the electron-accepting orbital at the conducting band state.
The TDOSs of the transition metal-incorporated FAPbI3 perovskite crystals with the 2 × 2 × 2 supercell structure were investigated. Comparison of TDOSs and the energy levels in the transition metal-incorporated FAPbI3 perovskite crystals was investigated. In the case of the Co and Cu-incorporated FAPbI3 perovskite crystals, TDOSs with the energy levels at HOMO and LUMO did not change. In the case of the Cr and Y-incorporated FAPbI3 perovskite crystals, the TDOSs and energy levels at HOMO and LUMO appeared the DOS of the 3d and 4d orbitals of the transition metal near frontier orbital. The energy levels at HOMO and LUMO in the Y-incorporated FAPbI3 perovskite crystal were lowered by the slight large amount of positive charges on the Y metal, compared with the other case. In the case of the Cr and Y-incorporated FAPbI3 perovskite crystal, the degenerated 3d upper (α) spin orbitals of the Cr atom and 4d upper (α) and lower (β) spin orbitals of Y atom were above and below the Fermi level. The 4s orbitals of the Cr and Y atom were mixed with the 6p orbital of the Pb atom above LUMO. The band gap between the 5p orbital on the I atom conjugated with the upper (α) spin of the 3d orbital on the Cr atom, upper spin of the 4d orbital on the Y atom and the 6p orbital of the Pb atom was narrowed. The narrowed band gap corresponded to a broad absorption in the near-infrared region. The occupancies and energy levels of the 6s and 6p orbitals of the Pb atom, the 3d, 4s, and 4p orbitals of the transition atom, and the 5p orbital of the I atom, and crystal splitting 3d orbitals near frontier orbital in the transition metal-incorporated FAPbI3 perovskite crystal were reported. In the case of the transition metal-incorporated FAPbI3 perovskite crystals, there was a sixfold full occupancy on the degenerated upper spin of the 3d orbital on the transition metal. The occupancies, the partial density of state (PDOS), energy levels, and crystal splitting provided significant information with the electron correlation based on the extent of intermixing of hybrid orbitals near the frontier orbital.
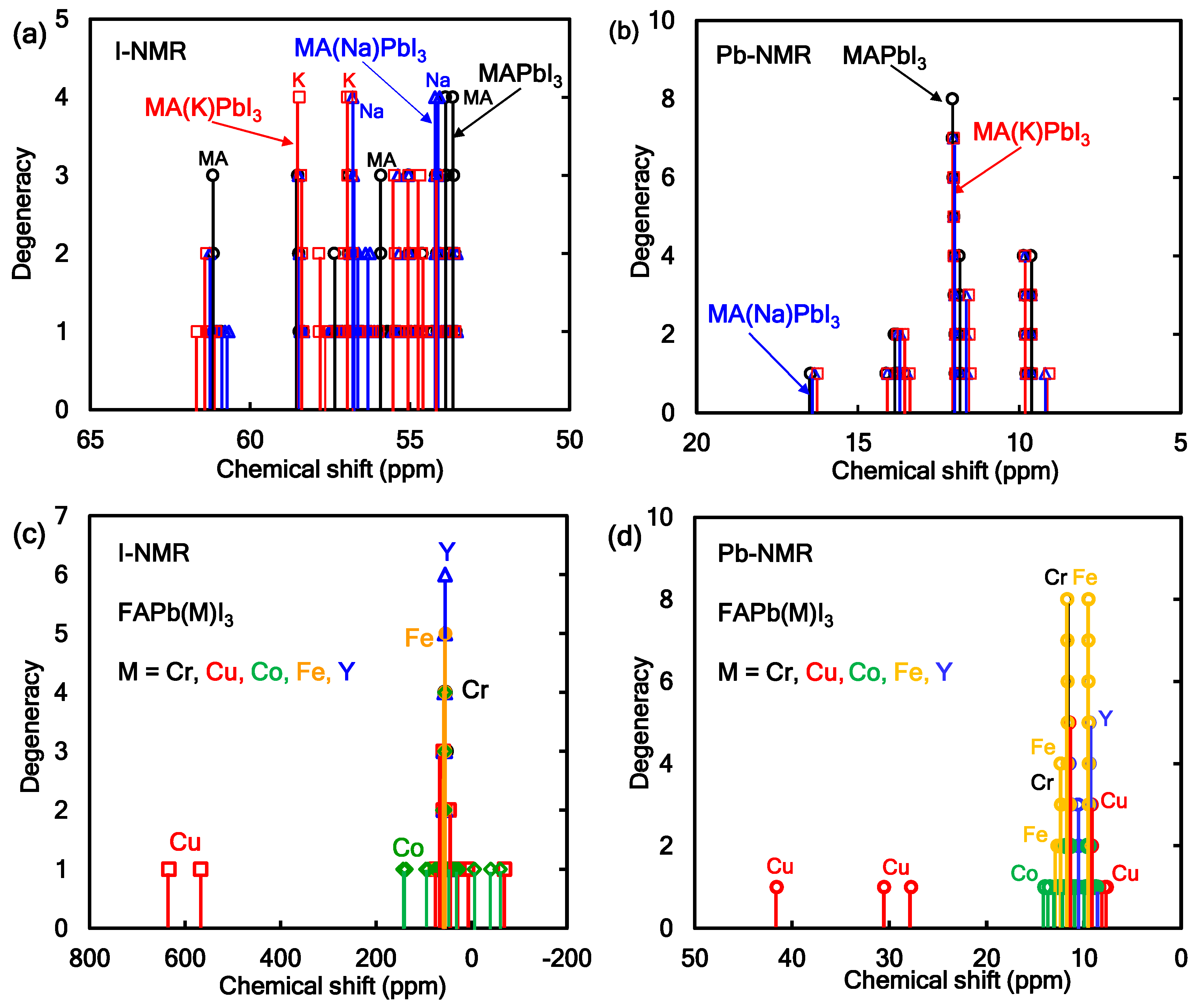
The NMR active atoms such as 207Pb, 127I, 23Na, 39K, and 41K in the perovskite crystal have nuclear spin with nuclear number of 1/2, 5/2, 3/2, 3/2, and 3/2, respectively. The nuclear magnetic interaction between the nuclear spin in halogen atom of 127I and 23Na, 39K, or 41K works as nuclear magnetic interaction and nuclear quadrupole interaction, which will cause energy level splitting with splitting of chemical shift in a wide range of magnetic field. Additive effect of 23Na or 41K in the MAPbI3 perovskite crystals on the chemical shift of 127I-NMR and 207Pb-NMR was investigated. The chemical shift of 127I-NMR and 207Pb-NMR and spectral assignment of 127I, 39K, and 23Na in 127I-NMR for the perovskite crystals were shown in Figure 2a,b. The chemical shift of 127I-NMR in the 39Na- or 39K-incorporated MAPbI3 crystal was markedly split and widened in the range of the magnetic field. In particular, the chemical shift of 127I-NMR at IK1 and IK2 was originated in the nearest-neighbor nuclear magnetic interaction around 39K or 23Na. The electron correlation between potassium and nearest neighbor iodine IK3 and IK4 depended on the nuclear quadrupole interaction based on the electron field graduate on the ligand near nitrogen atom in the methyl ammonium cation. As shown in Figure 2b, the chemical shift of 207Pb-NMR of the 39K-incorporated MAPbI3 perovskite crystal was slightly shifted in the strong magnetic field side. The slight splitting of the chemical shift was based on the nuclear magnetic interaction between Pb and K in the coordination structure. Excessive introduction will cause distortion of the coordination structure, yielding a wide spitting of the chemical shift.
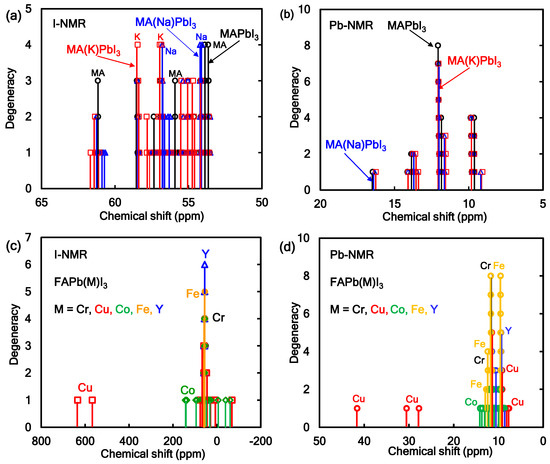
Figure 2.
Chemical shifts of 127I-NMR and 207Pb-NMR of the FAPbI3 perovskite crystal with (a,b) alkali metal or (c,d) transition metal.
The chemical shifts of 127I-NMR and 207Pb-NMR in the metal-incorporated FAPbI3 crystals were calculated as shown in Figure 2c,d. The effect of the incorporation of transition-metal atoms such as Cr, Cu, Co, Fe, and Y atoms in the FAPbI3 perovskite crystals on the chemical shift of 127I-NMR and 207Pb-NMR was investigated. As shown in Figure 2c, the chemical shifts of 127I-NMR in the Cr, Fe, and Y-incorporated FAPbI3 perovskite crystals were slightly split, and the degeneracy was increased by the symmetry crystal field effect. In the case of the Co and Cu-incorporated FAPbI3 crystals, the chemical shifts of 127I-NMR were markedly split in a wide range from 32 to 68 ppm. The chemical shifts of 127I-NMR were based on the magnetic interaction near the nearest iodine ligand conjugated with the atom in the perovskite crystal. As shown in Figure 2d, the chemical shifts of 207Pb-NMR in the Cr, Fe, and Y-incorporated FAPbI3 crystals were slightly split by the symmetry effect. The chemical shifts of 207Pb-NMR in the Co and Cu incorporated FAPbI3 crystals were widely split in the range of 8–42 ppm. The chemical shifts of 207Pb-NMR were based on the magnetic interaction near the ligand field in the perovskite crystal.
The magnetic behavior originated in the nearest-neighbor nuclear magnetic interaction of the nuclear quadrupole interaction based on electronic field graduate (EFG) with asymmetry parameter (η) of the Co and Cu metals and I atom with multi-nuclear spins of 7/2, 3/2, and 5/2. The magnetic behavior of the chemical shift of 207Pb-NMR and 127I-NMR was directly related to the symmetry effect of the coordination structure near the ligand field in the perovskite crystal. The chemical shifts of 127I-NMR and 207Pb-NMR in the perovskite crystals will depend on the electronic structure near frontier orbital in the perovskite crystal with the crystal phase including cubic, tetragonal and orthorhombic phases. As the calculation model was based on the experimental results, the crystal structure was assumed to be a cubic crystal system. The magnetic properties depended on the perturbation of coordination structure with the nearest-neighbor magnetic interaction in the perovskite crystals. The magnetic properties were reflected by extent of the electron correlation based on the magnetic interaction.
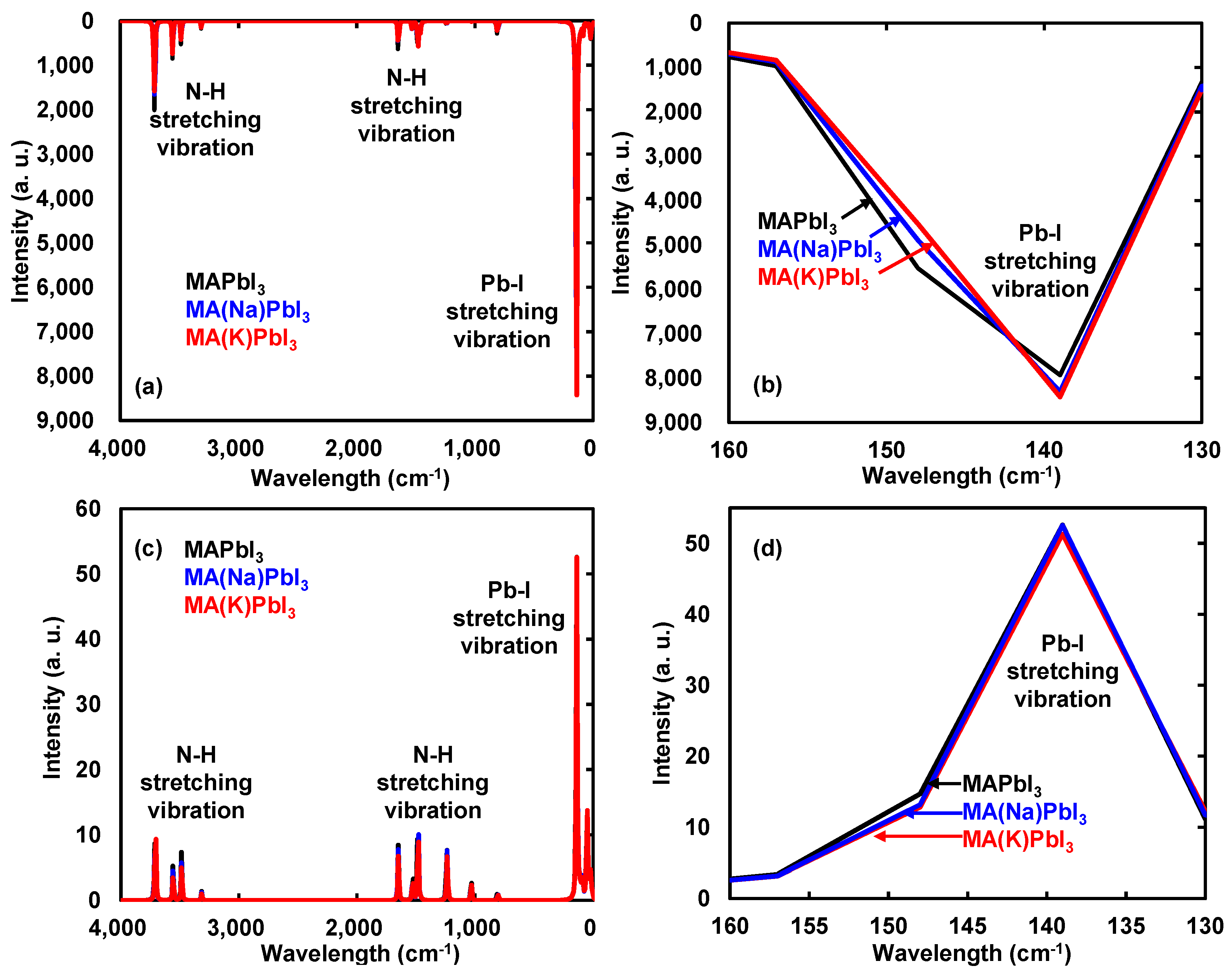
The calculated IR, Raman spectra, and vibration modes of the Na- or K-incorporated perovskite crystal were investigated, as shown in Figure 3a–d. The vibration modes indicated in the direction of the arrow as shown in Figure 3. As shown in Figure 3a,c, the vibration modes in IR and Raman spectra of 3712, 3703, 3559, 3487, 3307, 1651, 1480, 1237, 1030, and 814 cm−1 were assigned to the bending modes of N–H bonds of MA in the perovskite crystal. As shown in Figure 3b,d, the strong stretching vibration modes of Pb–I bonds in IR and Raman spectra of 139 and 147 cm−1 were slightly shifted and changed, as compared with those of the MAPbI3 perovskite crystal. The vibration modes in IR and Raman spectra of 139 and 147 cm−1 were identified with stretching modes of Pb–I bands combined with bending modes of N–H and C–H bonds of MA in the perovskite crystal.
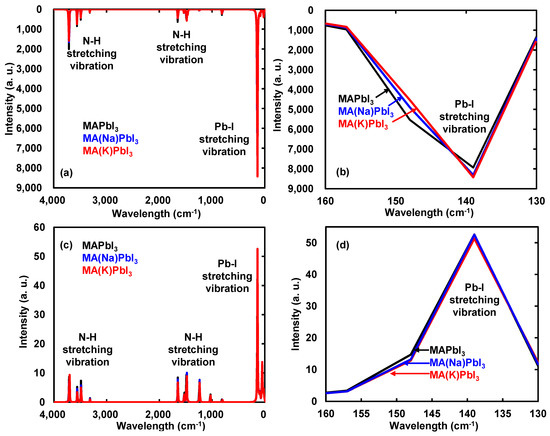
Figure 3.
(a,b) Infrared (IR) and (c,d) Raman spectra of the perovskite crystals incorporated with Na or K. (b,d) Enlarged view of (a,c), respectively.
The vibration modes of IR and Raman spectra were similar to the experimental results. Incorporation of Na or K into the perovskite crystal slightly changed charge bias, yielding a slight shift to low region of wavenumber in IR spectra. The vibration modes of Raman spectra based on difference in polarizability were slightly changed by incorporation of Na and K. The vibration behavior of IR and Raman spectra was based on distortion of the coordination structure with slight change of charge distribution near the ligand field. The slight perturbation with incorporation of Na or K in the perovskite crystal will not affect scattering of carrier diffusion through electron–lattice interaction as phonon effectiveness. The vibration modes in IR and Raman spectra as electron–lattice interaction will be correlated to thermodynamic parameters of entropy and Gibbs free energy.
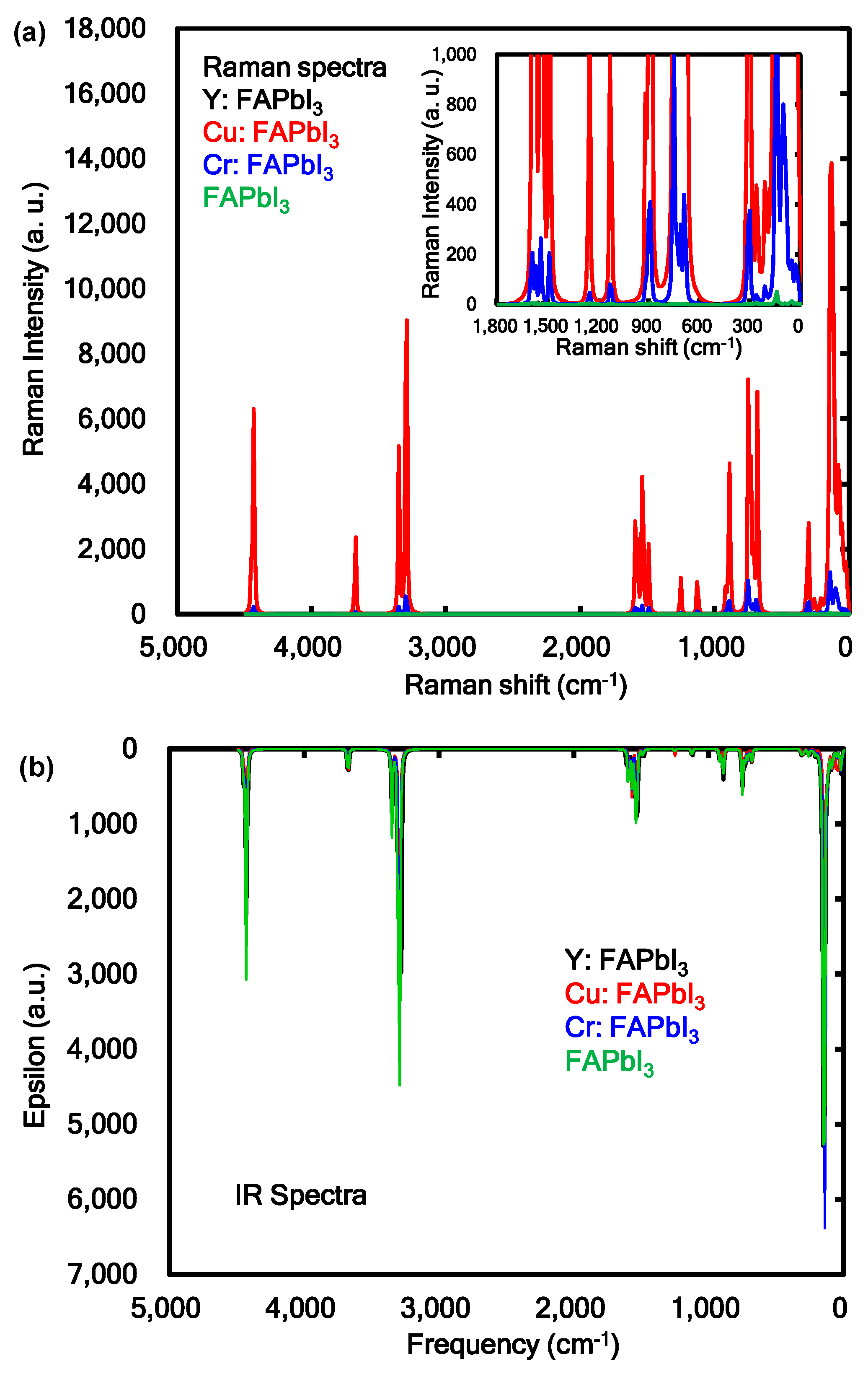
The Raman and IR spectra of the transition metal incorporated perovskite crystal were calculated as shown in Figure 4a,b. As shown in Figure 4a, the strong intensity of vibration modes in Raman spectra of 100, 140, 300, 680, 750, 890, 1530, 3290, 3350, 3670, and 4430 cm−1 were assigned to the bending and stretching mode of FA in the Cu and Cr incorporated FAPbI3 perovskite crystal. The medium intensity of vibration modes in Raman of 30, 100, and 140 cm−1 were identified with asymmetric stretching mode between Cr, Cu, and I atoms as ligand in the coordination cubic structure. The intensities on the vibration modes in the Cu and Cr incorporated FAPbI3 perovskite crystal were strongly increased, as compared with those of Y incorporated FAPbI3 crystal and standard reference using the FAPbI3 perovskite crystal. As shown in Figure 4b, the vibration modes in IR spectra of 23, 100, 133, 140, 300, 678, 732, 746, 752, 882, 1252, 1533, 1570, 1573, 1590, 3290, 3298, 3300, 3345, 3393, and 4430 cm−1 were assigned to the bending mode of N-C and N-H bonds of FA in the perovskite crystal. The strong intensity of vibration modes in IR spectra of 142 and 150 cm−1 were identified with stretching mode between Pb and I atom as ligand in the coordination structure. The vibration modes in IR spectra of 5, 42, and 141 cm−1 were based on stretching mode between Cu and I atom as ligand in the coordination structure. The calculated vibration modes in Raman and IR spectra were similar to the experimental results [36]. The vibration behavior of the Raman and IR spectra were associated with change of polarization and distortion of the coordination structure near the ligand field. The electron-lattice interaction as phonon effect in the transition-metal incorporated perovskite crystal will influence the photovoltaic properties based on the carrier diffusion.
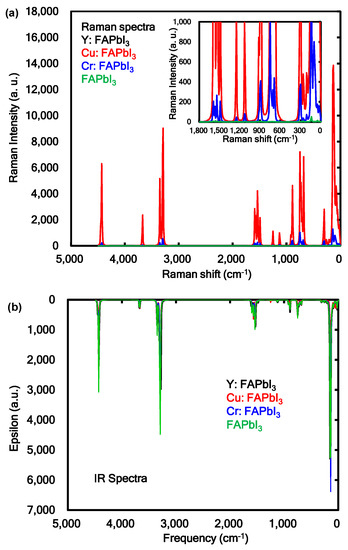
Figure 4.
(a) Raman and (b) infrared (IR) spectra of the transition metal, yttrium (Y), copper (Cu), chromium (Cr) incorporated FAPbI3 perovskite crystal.
The thermodynamic parameters of Gibbs free energy and entropy of the Na- and K-incorporated perovskite crystal were investigated. The thermodynamic parameters of enthalpy, entropy, and Gibbs free energy in quantitative investigation are estimated to be 2081 kJ mol−1, 4417 J K−1 mol−1, 760 kJ mol−1 for Na-incorporated perovskite crystal, and 2085 kJ mol−1, 4438 J K−1 mol−1, 753 kJ mol−1 for K-incorporated perovskite crystal. As reference, the thermal parameters of enthalpy, entropy, Gibbs free energy for MAPbI3 perovskite crystal were estimated to be 2326 kJ mol−1, 4610 J K−1 mol−1, 946 kJ mol−1. Incorporation of Na and K into the perovskite crystal decreased the enthalpy, entropy, and Gibbs free energy, which indicated thermal stabilization. The contribution of Gibbs free energy was dominated by decrease of enthalpy and entropy related to the Pb–I stretching vibration mode combined with bending modes of N–H and C–H bonds of MA in the perovskite crystal. The incorporation of Na or K induced to be the thermal stabilization, promotion of carrier generation, and transfer without scattering of carrier diffusion through the electron–lattice interaction as the phonon effectiveness based on the stretching vibration mode of Pb–I bond combined with bending modes of N–H and C–H bonds of MA in the perovskite crystal. Minor partial replacement of Na or K promoted the electron diffusion while suppressing the phonon effectiveness. The minor addition of Na or K will improve the photovoltaic performance of conversion efficiency, short-circuit current density based on the carrier diffusion, and open voltage related to band gap.
4. Conclusions
Electronic structures, spectroscopic properties, and thermodynamics on the Na- or K-incorporated MAPbI3 perovskite crystal were characterized for improving the photovoltaic and optical properties. Influence of Na- or K-incorporated into the MAPbI3 perovskite crystal on the electronic structure, optical, magnetic, and thermodynamic properties was investigated by first-principles calculation. Incorporation of Na or K into the perovskite crystal generated 3s, 3p, 4s, and 4p orbitals of Na or K above the conduction band, which promoted the charge transfer from alkali metal to the conduction band, accelerating the electron diffusion related to the photovoltaic properties. In the Na- or K-incorporated perovskite crystal, the electron conductivity was associated with the delocalization of the electronic density distribution of 6p orbital on Pb at LUMO, and the hole-transporting was related to the electronic density distribution of 5p orbital on iodine at HOMO. In the K-incorporated MAPbI3 perovskite crystal, the electrostatic potential near iodine was slightly changed. In the both cases using incorporation of Na or K, the energy levels on HOMO, LUMO, and Eg were slightly changed, yielding blue shift of the optical absorption. The splitting of chemical shift of 127I-NMR was originated in the nearest-neighbor nuclear magnetic interaction and nuclear quadrupole interaction based on deviation of charge near iodine as ligand in the coordination structure. The stretching vibration mode of Pb–I bond with vending mode of N–H and C–H bonds in IR and Raman spectra was slightly influenced by incorporation of alkali metal. The slight perturbation of the atomic coordination will not affect the carrier diffusion with scattering the conductive electron as phonon effectiveness based on stretching vibration mode of Pb–I bond with vending mode of N–H and C–H bonds in MA cation. The thermodynamic parameters with decrease of enthalpy, entropy, and Gibbs free energy indicate the thermal stabilization. Minor partial replacement of Na or K gave a slight perturbation to the electronic structure, promoting the electron diffusion while suppressing the phonon effectiveness. The minor addition of Na or K will improve the photovoltaic performance, open voltage related to band gap, and short-circuit current density based on the carrier diffusion with scattering the conductive electron as phonon effectiveness. The incorporation of the transition metals in the perovskite crystal influenced the electronic structure, optical and magnetic properties. The slight perturbation of coordination structure in the metal-incorporated FAPbI3 perovskite crystals as the isolated dilution system is an important factor in designing photovoltaic solar cells and photo-induced magnetic devices. The effects of slight incorporation of the metals such as Cr2+, Co2+, Cu2+ and Y3+ atoms into the perovskite crystal on the electronic structures, chemical shifts, and optical absorption spectra were investigated by the first-principle calculation. In the case of the Cr2+, Cu2+ and Y3+-incorporated FAPbI3 perovskite crystal, the electron density distribution of d-p hybrid orbital on the transition metal and iodine halogen ligand were delocalized at the frontier orbital. The total and partial density of state showed the 3dp hybrid orbital near frontier orbital, yielding the narrowing band gap. The magnetic properties were originated in the coordination structure and the electronic correlation between the transition metal, the Pb and I atoms in the perovskite crystal. The vibration modes of the Raman and IR spectra were associated with change of polarization and slight distortion of the coordination structure in the perovskite crystal. The photovoltaic properties were influenced by electronic correlation with electron lattice interaction as phonon effect. Control of the coordination structure, the electronic correlation and the magnetic interactions in the perovskite crystal are important factors in optimizing the tuning of photovoltaic and optical properties.
Author Contributions
Conceptualization, A.S.; methodology, A.S.; software, A.S.; validation, A.S.; formal analysis, A.S.; investigation, A.S.; resources, A.S.; data curation, A.S.; writing—original draft preparation, A.S.; writing—review and editing, A.S.; visualization, A.S.; supervision, T.O.; project administration, T.O.; funding acquisition, T.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhumekenov, A.A.; Saidaminov, M.I.; Haque, M.A.; Alarousu, E.; Sarmah, S.P.; Murali, B.; Dursun, I.; Miao, X.H.; Abdelhady, A.L.; Wu, T.; et al. Formamidinium lead halide perovskite crystals with unprecedented long carrier dynamics and diffusion length. ACS Energy Lett. 2016, 1, 32–37. [Google Scholar] [CrossRef]
- Umemoto, Y.; Suzuki, A.; Oku, T. Effects of halogen doping on the photovoltaic properties of HC(NH2)2PbI3 perovskite solar cells. AIP Conf. Proc. 2017, 1807, 020011. [Google Scholar]
- Suzuki, K.; Suzuki, A.; Zushi, M.; Oku, T. Microstructures and properties of CH3NH3PbI3-xClxCH3NH3PbI3-xClx hybrid solar cells. AIP Conf. Proc. 2015, 1649, 96–101. [Google Scholar]
- Suzuki, A.; Okada, H.; Oku, T. Fabrication and characterization of CH3NH3PbI3-x-yBrxCly perovskite solar cells. Energies 2016, 9, 376. [Google Scholar] [CrossRef]
- Nishi, K.; Oku, T.; Kishimoto, T.; Ueoka, N.; Suzuki, A. Photovoltaic characteristics of CH3NH3PbI3 perovskite solar cells added with ethylammonium bromide and formamidinium iodide. Coating 2020, 10, 410. [Google Scholar] [CrossRef]
- Zhao, W.; Yao, Z.; Yu, F.; Yang, D.; Liu, S. Alkali metal doping for improved CH3NH3PbI3 perovskite solar cells. Adv. Sci. 2018, 5, 1700131. [Google Scholar] [CrossRef]
- Dimesso, L.; Wussler, M.; Mayer, T.; Mankel, E.; Jaegermann, W. Inorganic alkali lead iodide semiconducting APbI3 (A = Li, Na, K, Cs) and NH4PbI3 films prepared from solution: Structure, morphology, and electronic structure. AIMS Mater. Sci. 2016, 3, 737–755. [Google Scholar] [CrossRef]
- Zhao, Z.; Gu, F.; Rao, H.; Ye, S.; Liu, Z.; Bian, Z.; Huang, C. Metal halide perovskite materials for solar cells with long term stability. Adv. Energy Mater. 2019, 9, 1802671. [Google Scholar] [CrossRef]
- Suzuki, A.; Oku, T. Effects of transition metals incorporated into perovskite crystals on the electronic structures and magnetic properties by first-principles calculation. Heliyon 2018, 4, e00755. [Google Scholar] [CrossRef]
- Ueoka, N.; Oku, T.; Suzuki, A. Additive effects of alkali metals on Cu-modified CH3NH3PbI3-dCld photovoltaic devices. RSC Adv. 2019, 9, 24231–24240. [Google Scholar] [CrossRef]
- Garcı’a, G.; Palacios, P.; Proupin, E.M.; Alejo, A.L.M.; Conesa, J.C.; Wahno´n, P. Influence of chromium hyperdoping on the electronic structure of CH3NH3PbI3 perovskite: A first-principles insight. Sci. Rep. 2018, 8, 2511. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, N.; Wang, L.; Li, L.; Xu, Z.; Jiao, H.; Liu, P.; Zhu, C.; Zai, H.; Sun, M.; et al. Impacts of alkaline on the defects property and crystallization kinetics in perovskite solar cells. Nat. Commun. 2019, 10, 1112. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Wang, Z.; Burlakov, V.M.; Lim, J.; McMeekin, D.; Pathak, S.; Snaith, H.J. Controlling nucleation and growth of metal halide perovskite thin films for high-efficiency perovskite solar cells. Small 2017, 13, 1602808. [Google Scholar] [CrossRef]
- Bi, D.; Yi, C.; Luo, J.; Decoppet, J.D.; Zhang, F.; Zakeeruddin, S.M.; Li, X.; Hagfeldt, A.; Gratzel, M. Polymer-templated nucleation and crystal growth of perovskite films for solar cells with efficiency greater than 21%. Nat. Energy 2016, 1, 16142. [Google Scholar] [CrossRef]
- Suzuki, A.; Kato, M.; Ueoka, N.; Oku, T. Additive effect of formamidinium chloride in methylammonium lead halide compound-based perovskite solar cells. J. Electron. Mater. 2019, 48, 3900–3907. [Google Scholar] [CrossRef]
- Kishimoto, T.; Suzuki, A.; Ueoka, N.; Oku, T. Effects of guanidinium addition to CH3NH3PbI3−xClx perovskite photovoltaic devices. J. Ceram. Soc. Jpn. 2019, 7, 491–497. [Google Scholar] [CrossRef]
- Kandori, S.; Oku, T.; Nishi, K.; Kishimoto, T.; Ueoka, N.; Suzuki, A. Fabrication and characterization of potassium and formamidinium-added perovskite solar cells. J. Ceram. Soc. Jpn. 2020, 128, 805–811. [Google Scholar] [CrossRef]
- Liu, D.; Li, S.; Bian, F.; Meng, X. First-principles investigation on the electronic and mechanical properties of Cs doped CH3NH3PbI3. Materials 2018, 11, 1141. [Google Scholar] [CrossRef]
- Cao, J.; Tao, S.X.; Bobbert, P.A.; Wong, C.P.; Zhao, N. Interstitial occupancy by extrinsic alkali cations in perovskites and its impact on ion migration. Adv. Mater. 2018, 30, 1707350. [Google Scholar] [CrossRef]
- Machiba, H.; Oku, T.; Kishimoto, T.; Ueoka, N.; Suzuki, A. Fabrication and evaluation of K-doped MA0.8FA0.1K0.1- PbI3(Cl) perovskite solar cells. Chem. Phys. Lett. 2019, 730, 117–123. [Google Scholar] [CrossRef]
- Pina, P.; Triati, D.K.; Wungu, H.; Rahmat, H. The characteristics of band structures and crystal binding in allinorganic perovskite APbBr3 studied by the first principle calculations using the density functional theory (DFT) method. Results Phys. 2019, 15, 102592. [Google Scholar]
- Suzuki, A.; Oku, T. First-principles calculation study of electronic structures of alkali metals (Li, K, Na and Rb)-incorporated formamidinium lead halide perovskite compounds. Appl. Surf. Sci. 2019, 483, 912–921. [Google Scholar] [CrossRef]
- Suzuki, A.; Miyamoto, Y.; Oku, T. Electronic structures, spectroscopic properties, and thermodynamic characterization of sodium- or potassium-incorporated CH3NH3PbI3 by first-principles calculation. J. Mater. Sci. 2020, 55, 9728–9738. [Google Scholar] [CrossRef]
- Sampson, M.D.; Park, J.S.; Schaller, R.D.; Chan, M.K.Y.; Martinson, A.B.F. Transition metal-substituted lead halide perovskite absorbers. J. Mater. Chem. 2017, 5, 3578–3588. [Google Scholar] [CrossRef]
- Hoefler, S.F.; Trimmel, G.; Rath, T. Progress on lead-free metal halide perovskites for photovoltaic applications: A review. Mon. Chem. -Chem. Mon. 2017, 148, 795–826. [Google Scholar] [CrossRef]
- Li, X.; Zhong, X.; Hu, Y.; Li, B.; Sheng, Y.; Zhang, Y.; Weng, C.; Feng, M.; Han, H.; Wang, J. Organic-inorganic copper(II)-based material: A low-toxic, highly stable light absorber for photovoltaic application. J. Phys. Chem. Lett. 2017, 8, 1804–1809. [Google Scholar] [CrossRef]
- Cortecchia, D.; Dewi, H.A.; Yin, J.; Bruno, A.; Chen, S.; Baikie, T.; Boix, P.P.; Gr€atzel, M.; Mhaisalkar, S.; Soci, C.; et al. Lead-free MA2CuClxBr4ex hybrid perovskites. Inorg. Chem. 2016, 55, 1044–1052. [Google Scholar] [CrossRef]
- Daub, M.; Stroh, R.; Hillebrecht, H. Synthesis, crystal structure, and optical properties of (CH3NH3)2CoX4 (X ¼ Cl, Br, I, Cl0.5Br0.5, Cl0.5I0.5, Br0.5I0.5). Z. Anorg. Allg. Chem. 2016, 642, 268–274. [Google Scholar] [CrossRef]
- Klug, M.T.; Osherov, A.; Haghighirad, A.A.; Stranks, S.D.; Brown, P.R.; Bai, S.; Wang, J.T.W.; Dang, X.; Bulovic, V.; Snaith, H.J.; et al. Tailoring metal halide perovskites through metal substitution: Influence on photovoltaic and material properties. Energy Environ. Sci. 2017, 10, 236–246. [Google Scholar] [CrossRef]
- Suzuki, A.; Oku, T. First-principles calculation study of electronic structures and magnetic properties of Mn-doped perovskite crystals for solar cell applications. Jpn. J. Appl. Phys. 2018, 57, 02CE04-1-7. [Google Scholar] [CrossRef]
- Xiao, Z.; Yan, Y. Progress in theoretical study of metal halide perovskite solar cell materials. Adv. Energy Mater. 2017, 7, 1701136. [Google Scholar] [CrossRef]
- Suzuki, A.; Oku, T. Electronic structures and magnetic properties of transition metal doped CsPbI3 perovskite compounds by first-principles calculation. Phys. Solid State 2019, 61, 1074–1085. [Google Scholar] [CrossRef]
- Suzuki, A.; Mari Oe, M.; Oku, T. Fabrication and characterization of Ni-, Co-, and Rb-incorporated CH3NH3PbI3 perovskite solar cells. J. Electron. Mater. 2021, 50, 1980–1995. [Google Scholar] [CrossRef]
- Suzuki, A.; Oku, T. Effects of mixed-valence states of Eu-doped FAPbI3 perovskite crystals studied by first-principles calculation. Mater. Adv. 2021, 2, 2609–2616. [Google Scholar] [CrossRef]
- Weller, M.T.; Weber, O.J.; Frost, J.M.; Walsh, A. Cubic perovskite structure of black formamidinium lead iodide, a-[HC(NH2)2]PbI3, at 298 K. J. Phys. Chem. Lett. 2015, 6, 3209–3212. [Google Scholar] [CrossRef]
- Quarti, C.; Grancini, G.; Mosconi, E.; Bruno, P.; Ball, J.M.; Lee, M.M.; Snaith, H.J.; Petrozza, A.; de Angelis, F. The Raman spectrum of the CH3NH3PbI3 hybrid perovskite: Interplay of theory and experiment. J. Phys. Chem. Lett. 2014, 5, 279–284. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).