Abstract
The occurrence and spread of antibiotic resistance have become a pressing global health concern. Understanding the genetic elements that facilitate the dissemination of antibiotic resistance genes (ARGs) in marine environments is crucial for effective microbial surveillance and management strategies. This study aimed to reveal the presence of mobilizable multiresistance clusters, consisting of ARGs associated with mobile genetic elements (MGEs), in marine bacterial communities. Water samples were collected from two beaches in Jeju, South Korea, and screened to identify multi-drug resistant bacteria. A total of 20 bacterial isolates were selected for whole genome sequencing, and through comprehensive genomic analysis, we identified and characterized nine such clusters primarily composed of betalactams, aminoglycosides, and tetracycline ARGs associated with MGEs like IS6, IS9, and Tn3. Additionally, an extensive analysis of 900 marine bacterial genomes from the National Center for Biotechnology Information (NCBI) database was conducted to gain a broader perspective. Our results provide valuable insights into the prevalence and diversity of mobilizable multiresistance clusters in marine bacterial communities.
1. Introduction
Antibiotic resistance is a global concern in the realm of human health. The emergence of community-acquired infections caused by resistant bacteria has amplified interest in natural environments [1]. These natural environments encompass a diverse range of ecosystems, including animals, soils, glaciers, and marine habitats, all of which serve as pivotal reservoirs for antibiotic resistance genes (ARGs). Despite the typically dilute nature of marine waters, they have evolved into significant reservoirs for antibiotic resistance. This phenomenon is primarily driven by escalating anthropogenic activities, which facilitate the introduction of antibiotic-resistant bacteria and residual antibiotics into marine ecosystems. Consequently, it is imperative to acquire a comprehensive understanding of the dissemination patterns of antibiotic resistance among marine bacteria. This knowledge is fundamental for the implementation of effective antibiotic control measures and the development of strategies to address this pressing issue [1].
Mobile genetic elements (MGEs), such as insertion sequences and transposons, possess the remarkable capacity to capture ARGs from bacterial chromosomes and subsequently transfer them horizontally, either through plasmids or phages, to other bacterial species. In this study, our objective is to illuminate the prevalence of ARGs within marine bacteria and their genomic colocation with MGEs. This research promises to yield valuable insights into the potential for ARG dissemination among marine bacteria, which, in turn, may exacerbate the spread of antibiotic resistance within marine ecosystems.
2. Materials and Methods
Water samples were collected in triplicate from Jungmun beach, located in Jeju, South Korea. Culturable bacteria were isolated from these samples using Marine agar (BD Difco, Difco Laboratory, Detroit, MI, USA) by incubating at 30 °C. Subsequently, isolates were sub-cultured on trypticase soy agar (BD Difco) with 3% additional salt content at 30 °C. A total of 100 distinct colonies were selected for plasmid carriage determination based on their resistance to 14 different antibiotics, as determined through a disk diffusion test. Isolates exhibiting resistance to three or more distinct antibiotics were then screened for plasmids using the alkaline lysis method. This process identified 60 plasmid-carrying isolates whose plasmid sizes were estimated using gel electrophoresis. Twenty isolates were selected from this group based on the distinct sizes of their plasmids for further analysis.
DNA from these 20 isolates was extracted using the QIAamp DNA Mini kit (Qiagen, Hilden, Germany), and the DNA quantity was assessed using QubitTM (Thermo Fisher Scientific, Waltham, MA, USA). Short-read sequencing was conducted using Illumina Hiseq 2500 (Macrogen, Seoul, Republic of Korea), while long-read sequencing employed MinION (Oxford nanopore technology). Raw reads from MinION were subjected to quality trimming using fastp [2] and Filtlong (https://github.com/rrwick/Filtlong/, accessed on 23 January 2022), respectively. Assembly was performed using Unicycler [3], and taxonomic identification was carried out via the RDP classifier [4]. Further, functional analysis for antibiotic resistance genes was executed using AMRFinder [5], and mobile genetic elements (MGEs), including plasmids, prophages, transposons, and integrases were identified through plasmidFinder [6], phaster [7], and Isescan [8].
To complement our findings, we retrieved 900 marine bacterial genomes from the National Center for Biotechnology Information (NCBI) for additional analysis (Figure 1). The assembly and analysis of these genomes followed the same procedures mentioned above.
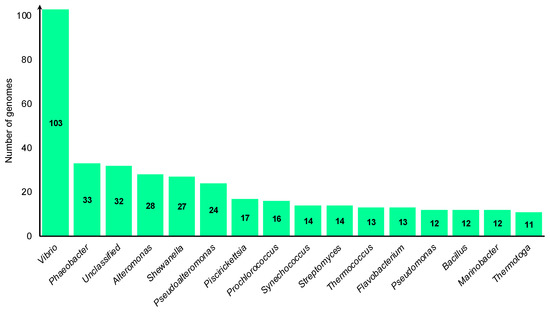
Figure 1.
Graph representing prominent bacterial genomes at the genus level, featuring more than 10 species, retrieved from NCBI.
3. Results and Discussion
Taxonomic analysis revealed that the isolated bacteria belonged to various species, including Vibrio cholera (6), Vibrio alginolyticus (4), Vibrio parahaemolyticus (3), Phaeobacter inhibens (3), Aeromonas salmonicida (2), and Edwardsiella anguillarum (2). Antibiotic susceptibility testing (AST) showed increased resistance to beta-lactam, aminoglycoside, and tetracycline antibiotics, particularly within the Vibrio species, aligning with genotypically identified ARGs as shown in Table 1.

Table 1.
The alignment of phenotypic resistance with the corresponding genotypic resistance in MDR marine bacteria.
These antibiotics are extensively used in livestock farms for treatment and prophylactic purposes [9]. This suggests that the antibiotic residues from neighboring livestock farms may intensify the selective pressure on marine bacteria, leading to heightened resistance to these antibiotics. Moreover, multi-drug resistant (MDR) Vibrio species are significant pathogens for aquatic life and humans. Therefore, the resistance observed in these isolates may pose an increased risk to both human and animal health [10].
The concordance observed between phenotypic and genotypic resistance implies the potential utility of whole genome sequencing for resistance surveillance across diverse environments [11]. Genomic analysis unveiled the presence of 25 intact prophages in the MDR isolates, with PHAGE_Vibrio_K139 (n = 3) and PHAGE_Entero_mEp237 (3) being the most prevalent. The majority of these prophages were classified within the Siphoviridae family. (Figure 2). These findings inspired us to further investigate the functional roles of these prophages and their potential influence on the prevalence of antibiotic resistance within marine bacterial species.
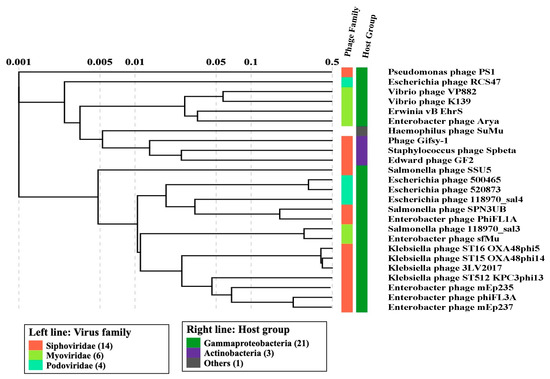
Figure 2.
The taxonomic classification and host range of prophages encoded by marine bacterial genomes (900).
Furthermore, our analysis unveiled the presence of 24 plasmid sequences encoded within these bacterial genomes. These plasmids exhibited diversity, with the majority categorized as Unnamed plasmids (17), while others belonged to the IncFII_1 (4), IncH12A-1 (2), and p3442-IMI-2c (1) categories (see Table 1). The co-occurrence of plasmids and prophages within these genomes is a noteworthy observation, as it implies a potential interplay between mobile genetic elements and their role in shaping the genetic landscape of these marine bacterial species [12].
We identified a total of nine distinct associations between ARGs and MGEs within both prophage-like sequences (Figure 3) and plasmid contigs (Figure 4). Notably, MGEs such as IS6, IS5, and IS110 exhibited predominant associations with ARGs. These specific MGEs are recognized for their capacity to facilitate the transposition of associated genes within genomes, subsequently transferring them to other horizontally gene transfer (HGT) vehicles, including plasmids and prophages [13].
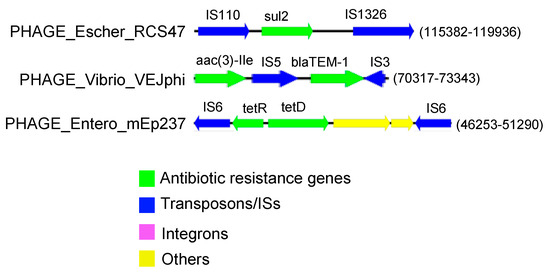
Figure 3.
Three distinct associations between ARGs and MGEs were identified, each encoded within a unique prophage-like sequence. Notably, all three of these sequences were found to be prevalent among Vibrio alginolyticus isolates.
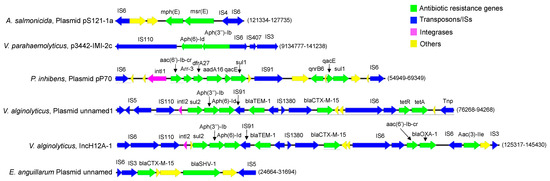
Figure 4.
Mobile resistance clusters encoded by plasmid and prophage-like sequences: six distinctive ARG and MGE associations found within five distinct bacterial species.
Significantly, we have identified two prophage-like sequences, Vibrio phage VP882 and Vibrio phage VFO3k6, each encoding betalactam (blaTEM-1) and aminoglycoside (aph(3″)-Ib) genes, respectively, within various marine bacterial species (Figure 5). Notably, these genes exhibited 100% identical sequences across these diverse species. This highlights transduction as a key driver in the widespread distribution of ARGs by these prophages, showcasing their remarkable conservation [14].
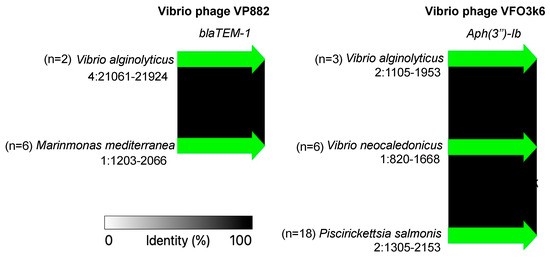
Figure 5.
The sequence conservation of two aminoglycoside resistance genes encoded within prophages.
4. Conclusions
Our study sheds light on the intricate dynamics of antibiotic resistance in marine bacterial communities, revealing the significant role of plasmids and prophages in ARGs’ spread. Future research should delve deeper into the functional roles of these MGEs.
Author Contributions
All authors contributed equally to this work. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Bacterial genomes have been submitted to NCBI under bioproject accession number PRJNA716757 (https://www.ncbi.nlm.nih.gov/bioproject/), accessed on 12 March 2022.
Acknowledgments
The authors would like to thank every person who supported us in conducting this research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Marti, E.; Variatza, E.; Balcazar, J.L. The role of aquatic ecosystems as reservoirs of antibiotic resistance. Trends Microbiol. 2014, 22, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one fastq preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLOS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Wang, Q.; Cole, J.R.; Rosen, G.L. Using the rdp classifier to predict taxonomic novelty and reduce the search space for finding novel organisms. PLoS ONE 2012, 7, e32491. [Google Scholar] [CrossRef] [PubMed]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.H.; McDermott, P.F.; et al. Validating the amrfinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. Phaster: A better, faster version of the phast phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Xie, Z.; Tang, H. Isescan: Automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 2017, 33, 3340–3347. [Google Scholar] [CrossRef] [PubMed]
- Ghimpețeanu, O.M.; Pogurschi, E.N.; Popa, D.C.; Dragomir, N.; Drăgotoiu, T.; Mihai, O.D.; Petcu, C.D. Antibiotic use in livestock and residues in food-a public health threat: A review. Foods 2022, 11, 1430. [Google Scholar] [CrossRef]
- Huang, Z.; Anokyewaa, M.A.; Wang, J.; Jian, J.; Lu, Y. Pathogenicity and antibiotic resistance analysis of vibrio species found in coastal water at mainly beach of shenzhen, china. Front. Mar. Sci. 2022, 9, 980593. [Google Scholar] [CrossRef]
- Calero-Cáceres, W.; Ortuño-Gutiérrez, N.; Sunyoto, T.; Gomes-Dias, C.A.; Bastidas-Caldes, C.; Ramírez, M.S.; Harries, A.D. Whole-genome sequencing for surveillance of antimicrobial resistance in ecuador: Present and future implications. Rev. Panam. De Salud Pública 2023, 47, e8. [Google Scholar] [CrossRef] [PubMed]
- Khedkar, S.; Smyshlyaev, G.; Letunic, I.; Maistrenko, O.M.; Coelho, L.P.; Orakov, A.; Forslund, S.K.; Hildebrand, F.; Luetge, M.; Schmidt, T.S.; et al. Landscape of mobile genetic elements and their antibiotic resistance cargo in prokaryotic genomes. Nucleic Acids Res. 2022, 50, 3155–3168. [Google Scholar] [CrossRef] [PubMed]
- Haudiquet, M.; de Sousa, J.M.; Touchon, M.; Rocha, E.P.C. Selfish, promiscuous and sometimes useful: How mobile genetic elements drive horizontal gene transfer in microbial populations. Philos. Trans. R. Soc. B 2022, 377, 20210234. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.L. Bacteriophage-mediated horizontal gene transfer: Transduction. In Bacteriophages: Biology, Technology, Therapy; Harper, D.R., Abedon, S.T., Burrowes, B.H., McConville, M.L., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 151–192. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).