
Structural Study of a Pd–Fe Hetero-Trinuclear Compound †


Abstract
:1. Introduction
2. Materials and Methods
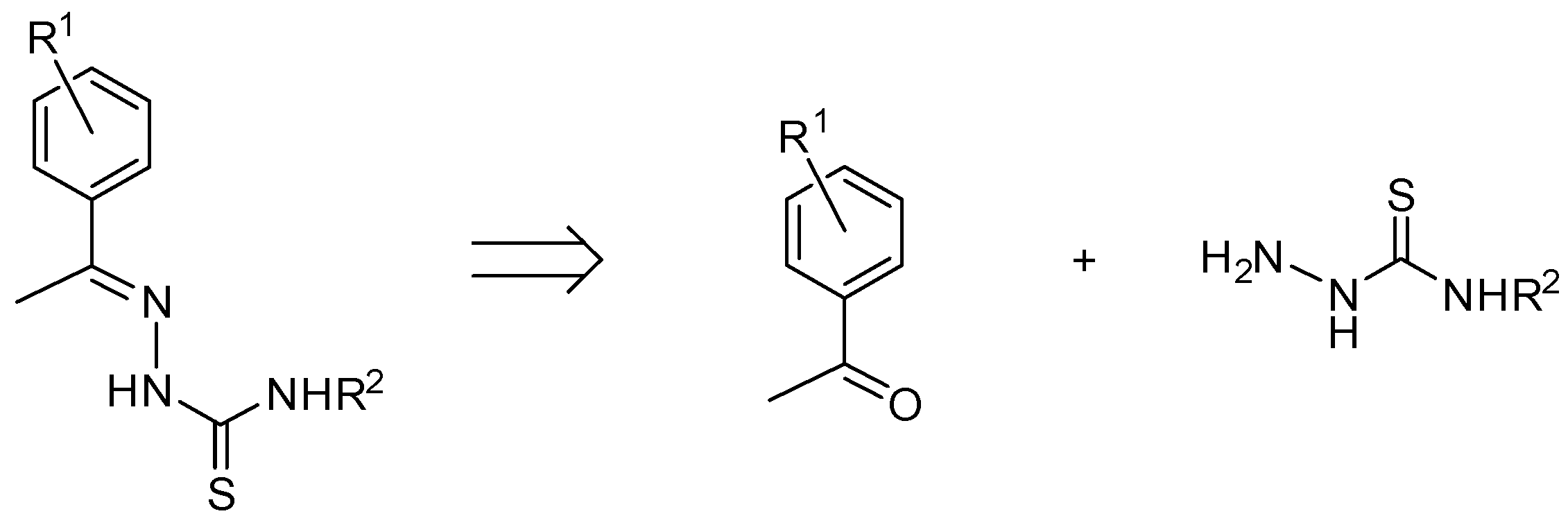
2.1. Ligand Synthesis Procedure
2.2. Syntheis of Palladium Organometallic Compounds
2.3. Synthesis of Pd–Fe Hetero-Trimetallic Compounds
3. Results and Discussion
3.1. H and 31P-{1H}-NMR Spectroscopy Study
3.1.1. H-NMR for Ligands (1–4)
3.1.2. H-NMR for Palladium Organometallic Compounds (5–8)
3.1.3. H-NMR and 31P-{1H}-NMR for Pd–Fe Organometallic Compounds (9–12)
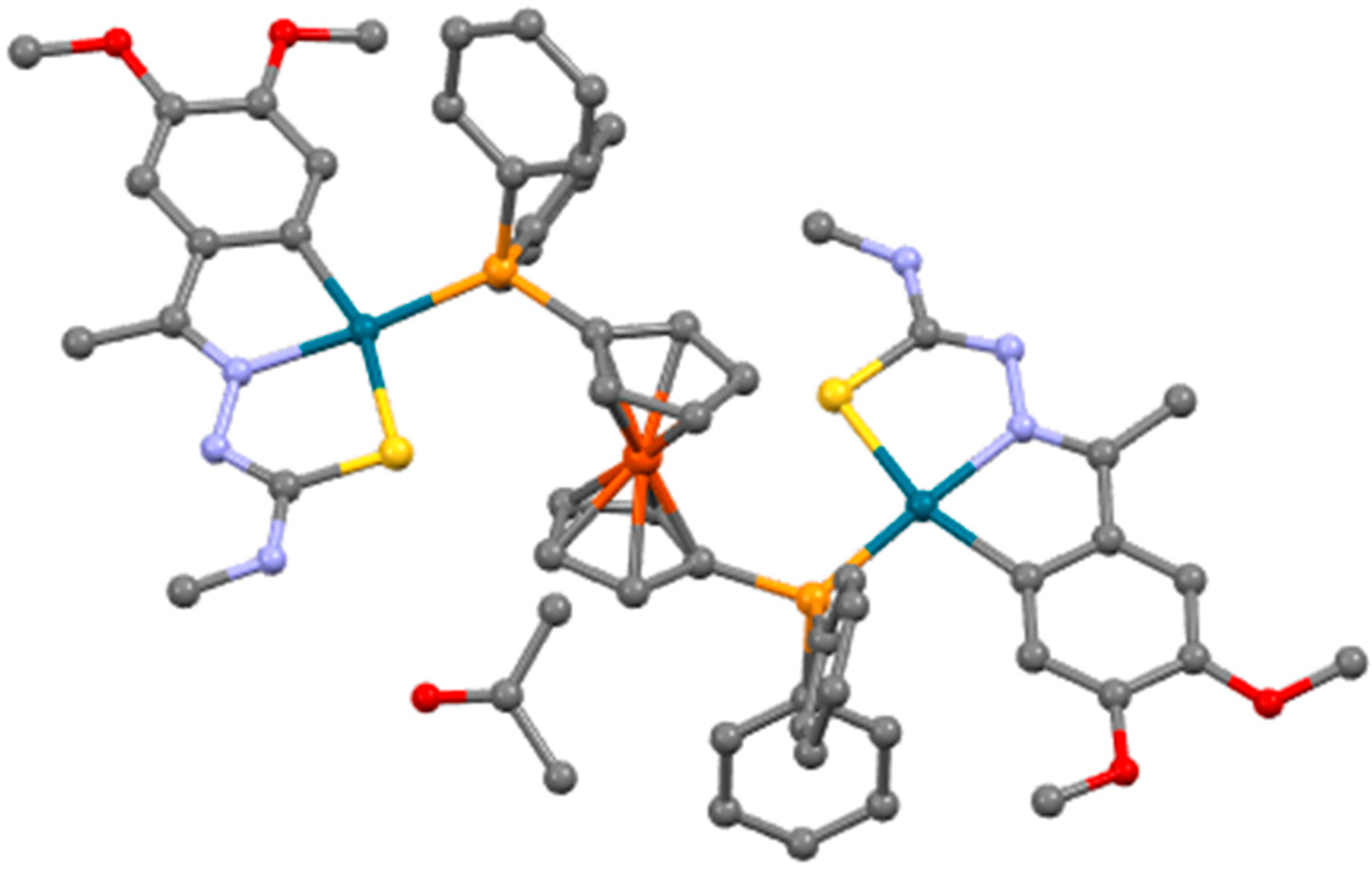
3.2. X-ray Spectroscopy Discussion for Compound 10
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shakya, B.; Yadav, P.N. Thiosemicarbazones as Potent Anticancer Agents and their Modes of Action, Mini-Rev. Med. Chem. 2020, 20, 638–661. [Google Scholar] [CrossRef]
- Hendricks, M.E.; Xu, X.; Boller, T.R.; Samples, M.E.; Johnson, R.A.; Nataro, C. Synthesis, characterization and electrochemistry of [Pd(PP)MeCl] compounds with 1,1′-bis(phosphino)ferrocene ligands. Polyhedron 2021, 199, 115104–115111. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ketone (mmol) | Thiosemicarbazide (mmol) | R2 |
|---|---|---|---|
| 1 | 5.5 | 5.5 | H |
| 2 | 5.5 | 5.5 | Me |
| 3 | 5.5 | 5.5 | Et |
| 4 | 5.5 | 5.5 | Ph |
| Compound | Ligand Amount | R2 |
|---|---|---|
| 5 | 1—0.67 mmol | H |
| 6 | 2—0.67 mmol | Me |
| 7 | 3—0.67 mmol | Et |
| 8 | 4—0.67 mmol | Ph |
| Compound | dppf | R2 |
|---|---|---|
| 9 | 0.20 mmol | H |
| 10 | 0.20 mmol | Me |
| 11 | 0.20 mmol | Et |
| 12 | 0.19 mmol | Ph |
| 1H NMR Data | |
|---|---|
| 1 | 1H NMR (250 MHz, CDCl3) δ (ppm): 8.69 (s, 1H, NNH); 7.31 (m, 2H, NH, H6); 7.23 (d, 4J = 2.1 Hz, 1H, H2); 6.88 (d, 3J = 8.3 Hz, 1H, H5); 6.32 (s, 1H, NH); 3.93 (s, 3H, OMe); 3.92 (s, 3H, OMe); 2.28 (s, 3H, MeC=N). |
| 2 | 1H NMR (250 MHz, CDCl3) δ (ppm): 8.61 (s, 1H, NNH); 7.57 (s, 1H, NHMe); 7.27 (d, 1H, 3J = 8.7 Hz, H6); 7.24 (d, 4J=2,1 Hz, 1H, H2); 6.89 (d, 1H, 3J=8,7 Hz, H5); 3.95 (s, 3H, OMe); 3.93 (s, 3H, OMe); 3.29 (d, 3J = 5.6 Hz, 3H, NHMe); 2.26 (s, 3H, MeC=N). |
| 3 | 1H NMR (250 MHz, CDCl3) δ (ppm): 8.59 (s, 1H, NNH); 7.54 (m, 1H, NHEt); 7.23 (m, 1H, H6); 7.21 (d, 4J = 2.1 Hz, 1H, H2); 6,87 (d, 1H, 3J = 8.8 Hz, H5); 3.92 (s, 3H, OMe); 3.91 (s, 3H, OMe); 3.76 (m, 2H, NHCH2CH3); 2.25 (s, 3H, MeC=N); 1.29 (t, 3J = 7.3 Hz, 3H, NHCH2CH3). |
| 4 | 1H NMR (250 MHz, CDCl3) δ (ppm): 9.34 (s, 1H, NHPh); 8.69 (s, 1H, NNH); 7.66 (d, 3J = 7.8 Hz, 2H, HaHa’); 7.38 (t, 3J = 7.8 Hz, 2H, HbHb’); 7.26 (m, 3H, H2H6Hc); 6.88 (d, 3J = 8.3 Hz, 1H, H5); 3.92 (s, 3H, OMe); 3.91 (s, 3H, OMe); 2.30 (s, 3H, MeC=N). |
| 1H NMR Data | |
|---|---|
| 5 | 1H NMR (250 MHz, CDCl3) δ (ppm): 7.04 (s, 1H, H5); 6.23 (s, 1H, H2); 5.22 (s, 2H, NH2); 3.95 (s, 3H, OMe); 3.78 (s, 3H, OMe); 1.92 (s, 3H, MeC=N). |
| 6 | 1H NMR (250 MHz, CDCl3) δ (ppm): 7.09 (s, 1H, H5); 6.27 (s, 1H, H2); 5,00 (m, 1H, NHMe); 3.97 (s, 3H, OMe); 3.81 (s, 3H, OMe); 2.96 (d, 3J = 4,9 Hz, 3H, NHMe); 1.93 (s, 3H, MeC=N). |
| 7 | 1H NMR (250 MHz, CDCl3) δ (ppm): 7.08 (s, 1H, H5); 6.26 (s, 1H, H2); 5.03 (m, 1H, NHCH2CH3); 3.97 (s, 3H, OMe); 3.81 (s, 3H, OMe); 3.43 (m, 2H, NHCH2CH3); 1.95 (s, 3H, MeC=N); 1.22 (t, 3H, 3J = 7.2 Hz, NHCH2CH3). |
| 8 | 1H NMR (250 MHz, CDCl3) δ (ppm): 7.52 (d, 3J = 7.6 Hz, 2H, HaHa’); 7.27 (t, 3J = 7.6 Hz, 2H, HbHb’); 7.01 (t, 3J = 7.6 Hz, 1H, Hc); 7.00 (s, 1H, NHPh); 6.94 (s, 1H, H5), 6.41 (s, 1H, H2); 3.86 (s, 3H, OMe); 3.51 (s, 3H, OMe); 1,91 (s, 3H, MeC=N). |
| 1H NMR Data | |
|---|---|
| 9 | 1H NMR (400 MHz, dmso-d6) δ (ppm): 7.48 (m, 12H, m‒PPh2, p‒PPh2); 7.37 (m, 8H, o‒PPh2); 6.74 (s, 2H, H2); 6.55 (s, 4H, 2×NH2); 5.77 (d, 4JHP = 4.1 Hz, 2H, H5); 5.11 (s, 4H, Cp); 4.19 (s, 4H, Cp); 3.67 (s, 6H, 2×OMe); 2.80 (s, 6H, 2×OMe); 2.27 (s, 6H, 2×MeC=N). 31P‒{1H} NMR (400 MHz, dmso-d6) δ (ppm): 32.45 (s). |
| 10 | 1H NMR (400 MHz, CDCl3) δ (ppm): 7.57 (m, 8H, m‒PPh2); 7.39 (t, 3J = 7.0 Hz, 4H, p‒PPh2); 7.30 (m, 8H, o‒PPh2); 6.69 (s, 2H, H2); 5.91 (d, 4JHP = 4.3 Hz, 2H, H5); 5.17 (s, 4H, Cp); 4.71 (s, 2H, 2×NHMe); 4.28 (s, 4H, Cp); 3.81 (s, 6H, 2×OMe); 2.97 (d, 3J = 4.9 Hz, 6H, 2×NHMe); 2.92 (s, 6H, 2×OMe); 2.40 (s, 6H, 2×MeC=N). 31P‒{1H} NMR (400 MHz, CDCl3) δ (ppm): 28.25 (s). |
| 11 | 1H NMR (400 MHz, CDCl3) δ (ppm): 7.43 (m, 20H, PPh2); 6.66 (s, 2H, H2); 5.88 (s, 2H, H5); 5.15 (s, 4H, Cp); 4.68 (s, 2H, 2×NHCH2CH3); 4.25 (s, 4H, Cp); 3.78 (s, 6H, OMe); 3.37 (m, 4H, 2×NHCH2CH3); 2.90 (s, 6H, 2×OMe); 2.35 (s, 6H, 2×MeC=N); 1.14 (t, 6H, 3J = 6.8 Hz, 2×NHCH2CH3). 31P‒{1H} NMR (400 MHz, CDCl3) δ (ppm): 28.25 (s). |
| 12 | 1H NMR (250 MHz, CDCl3) δ (ppm): 7.47 (m, 28H, PPh2, HaHa’HbHb’); 6.97 (t, 3J = 7.1 Hz, 2H, Hc); 6.75 (s, 2H, H2); 6.65 (s, 2H, 2×NHPh); 5.95 (d, 4JHP = 3.6 Hz, 2H, H5); 5.23 (s, 4H, Cp); 4.32 (s, 4H, Cp); 3.83 (s, 6H, 2×OMe); 2.93 (s, 6H, 2×OMe); 2.47 (s, 6H, 2×MeC=N). 31P‒{1H} NMR (400 MHz, CDCl3) δ (ppm): 28.22 (s). |
| Compound | 10 |
|---|---|
| Empirical formula | C58H58FeN6O4P2Pd2S2 · C3H6O |
| Formula weight | 1355.89 |
| Temperature | 100 (2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Triclinic |
| Space group | P‒1 |
| Unit cell dimensions | a = 11,148 (5)Å; α = 86,251 (5)° b = 12,652 (5)Å; β = 84,791 (5)° c = 20,736 (5)Å; γ = 78,360 (5)° |
| Volume | 2849 (18)Å3 |
| Z | 2 |
| Calculated density | 1.580 Mg/m3 |
| Absorption coefficient | 1.061 mm−1 |
| F(000) | 1384 |
| Crystal size | 0.22 × 0.15 × 0.04 mm3 |
| Theta range for data collection | 0.987–26,373° |
| Limiting indexes | −13 ≤ h ≤ 13 −15 ≤ k ≤ 15 −25 ≤ l ≤ 25 |
| Reflections collected | 87,669 |
| Reflections unique | 11,617 [R(int) = 0.0444] |
| Data/restraints/parameters | 11,617/0/888 |
| Goodness-of-fit on F2 | 1.028 |
| Final R indexes [I > 2sigma(I)] | R1 = 0.0273; wR2 = 0.0588 |
| R indexes (all data) | R1 = 0.0405; wR2 = 0.0632 |
| Largest diff. peak and hole | 0.675 y − 0.662 e/Å3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munín-Cruz, P.; Rúa-Sueiro, M.; Ortigueira, J.M.; Pereira, M.T.; Vila, J.M. Structural Study of a Pd–Fe Hetero-Trinuclear Compound. Chem. Proc. 2022, 9, 5. https://doi.org/10.3390/IOCC_2022-12145
Munín-Cruz P, Rúa-Sueiro M, Ortigueira JM, Pereira MT, Vila JM. Structural Study of a Pd–Fe Hetero-Trinuclear Compound. Chemistry Proceedings. 2022; 9(1):5. https://doi.org/10.3390/IOCC_2022-12145
Chicago/Turabian StyleMunín-Cruz, Paula, Marcos Rúa-Sueiro, Juan M. Ortigueira, María Teresa Pereira, and José M. Vila. 2022. "Structural Study of a Pd–Fe Hetero-Trinuclear Compound" Chemistry Proceedings 9, no. 1: 5. https://doi.org/10.3390/IOCC_2022-12145
APA StyleMunín-Cruz, P., Rúa-Sueiro, M., Ortigueira, J. M., Pereira, M. T., & Vila, J. M. (2022). Structural Study of a Pd–Fe Hetero-Trinuclear Compound. Chemistry Proceedings, 9(1), 5. https://doi.org/10.3390/IOCC_2022-12145