
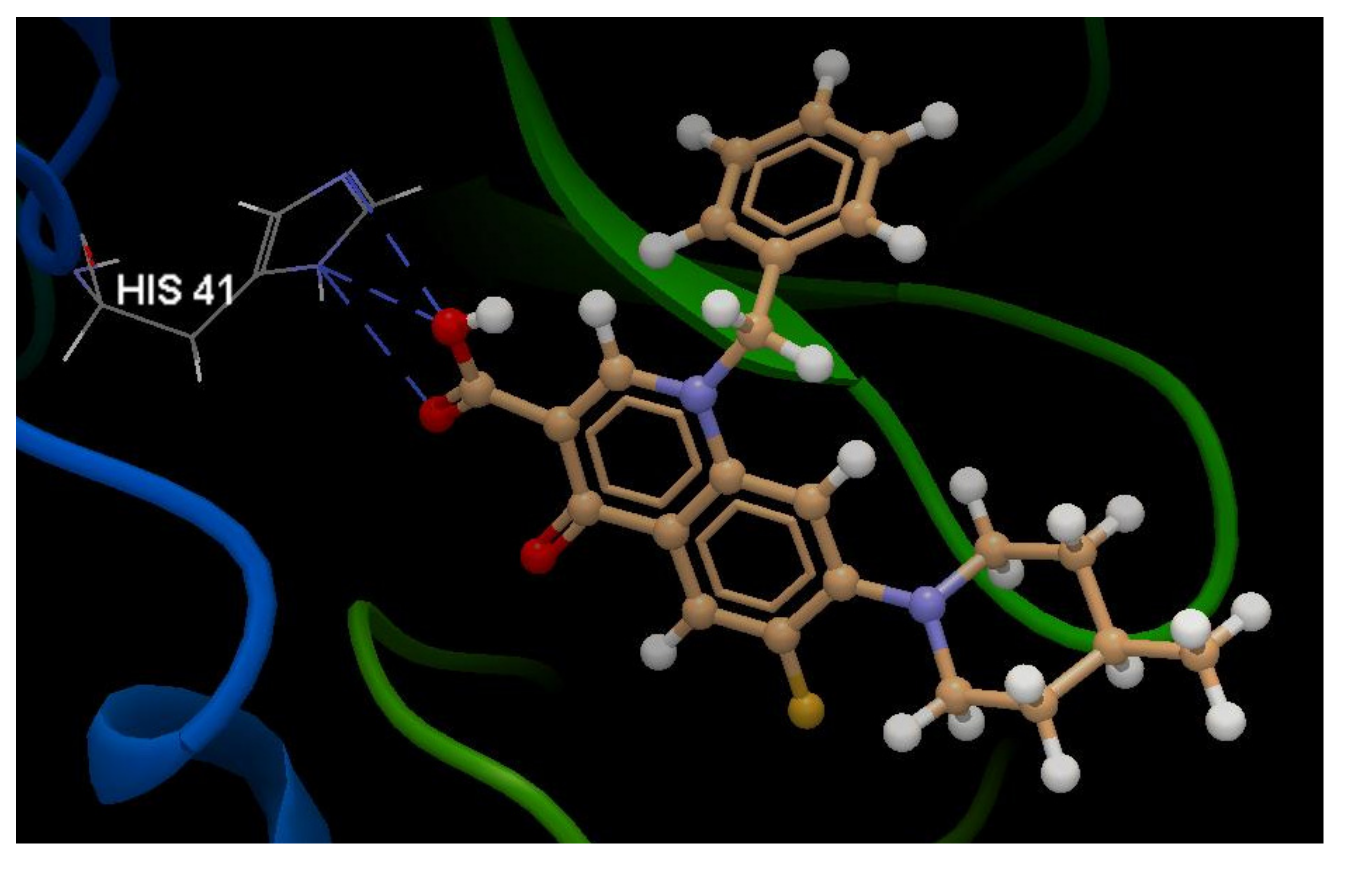
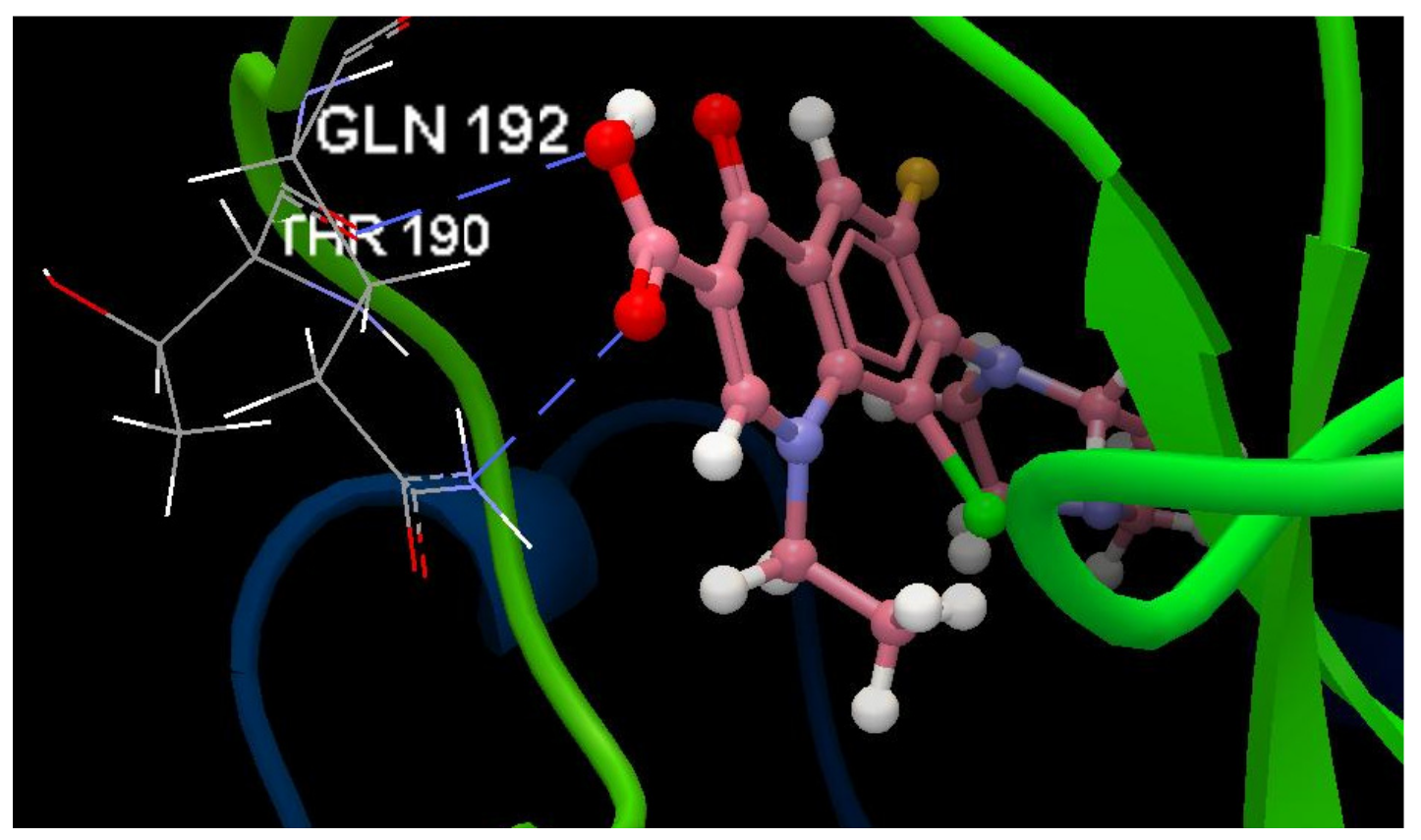
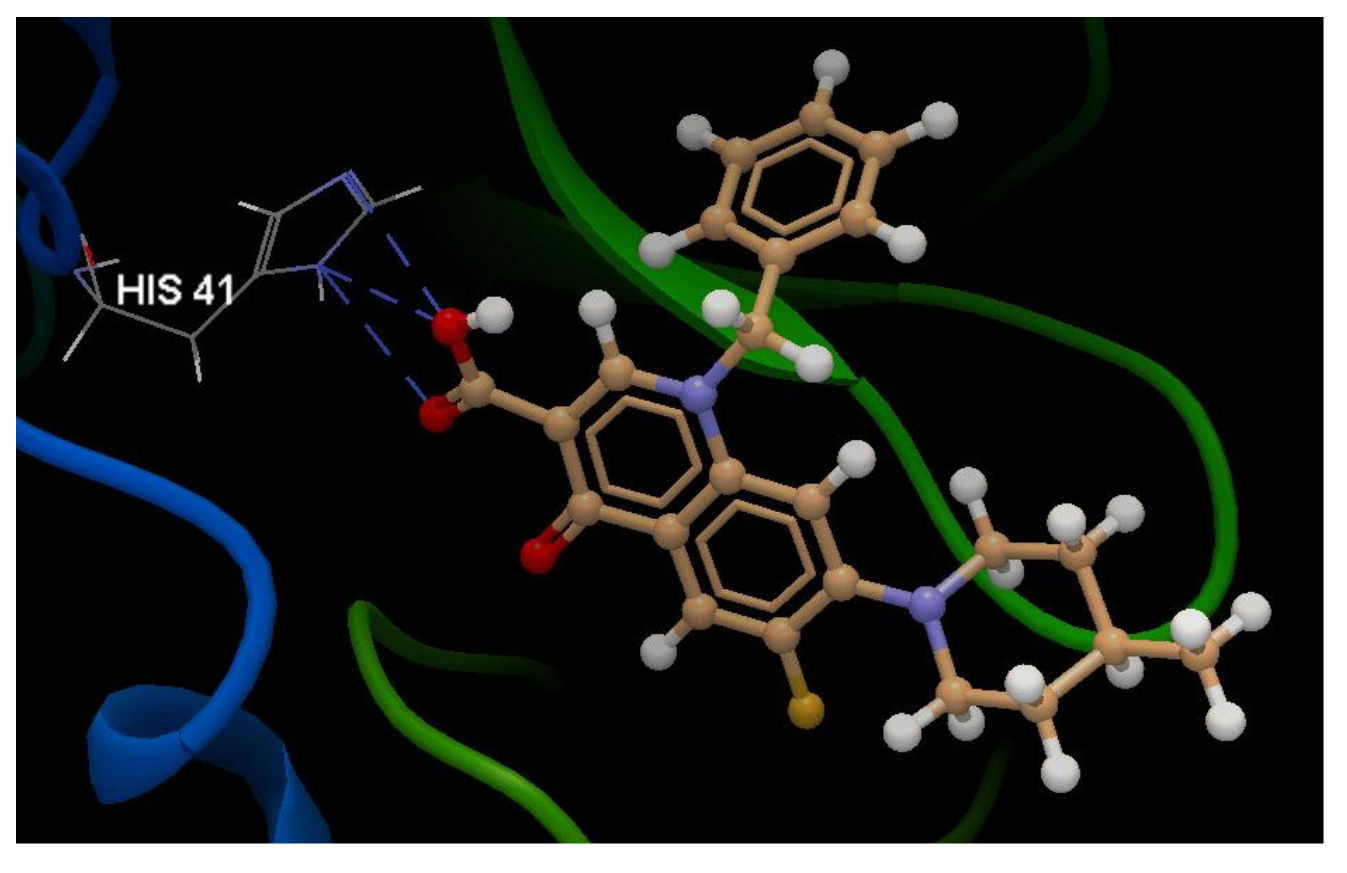
A Computational Study to Identify Some Potential Inhibitors of SARS-CoV-2 Main Protease from Biological Active Quinolones †


{kind=link}
{kind=link}
Funding
Data Availability Statement
Conflicts of Interest
References
- Mohapatra, R.K.; Pintilie, L.; Ashish, K.; Sarangi, A.K.; Das, D.; Sahu, R.; Perekhoda, L. The recent challenges of highly contagious COVID-19; causing respiratory infections: Symptoms, diagnosis, transmission and possible vaccines. Chem. Biol. Drug Des. 2020, 96, 1187–1208. [Google Scholar] [CrossRef] [PubMed]
- Marciniec, K.; Beberok, A.; Pęcak, P.; Boryczka, S.; Wrześniok, D. Ciprofloxacin and moxifloxacin could interact with SARS-CoV-2 protease: Preliminary in silico analysis. Pharmacol. Rep. 2020, 72, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Yacouba, A.; Olowo-okere, A.; Yunusa, I. Repurposing of antibiotics for clinical management of COVID-19: A narrative review. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 37. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pintilie, L. A Computational Study to Identify Some Potential Inhibitors of SARS-CoV-2 Main Protease from Biological Active Quinolones. Chem. Proc. 2022, 7, 10. https://doi.org/10.3390/chemproc2022007010
Pintilie L. A Computational Study to Identify Some Potential Inhibitors of SARS-CoV-2 Main Protease from Biological Active Quinolones. Chemistry Proceedings. 2022; 7(1):10. https://doi.org/10.3390/chemproc2022007010
Chicago/Turabian StylePintilie, Lucia. 2022. "A Computational Study to Identify Some Potential Inhibitors of SARS-CoV-2 Main Protease from Biological Active Quinolones" Chemistry Proceedings 7, no. 1: 10. https://doi.org/10.3390/chemproc2022007010
APA StylePintilie, L. (2022). A Computational Study to Identify Some Potential Inhibitors of SARS-CoV-2 Main Protease from Biological Active Quinolones. Chemistry Proceedings, 7(1), 10. https://doi.org/10.3390/chemproc2022007010