
Synthesis of (2S,3S)-3-Aroyl Pyroglutamic Acid Amides †


Abstract
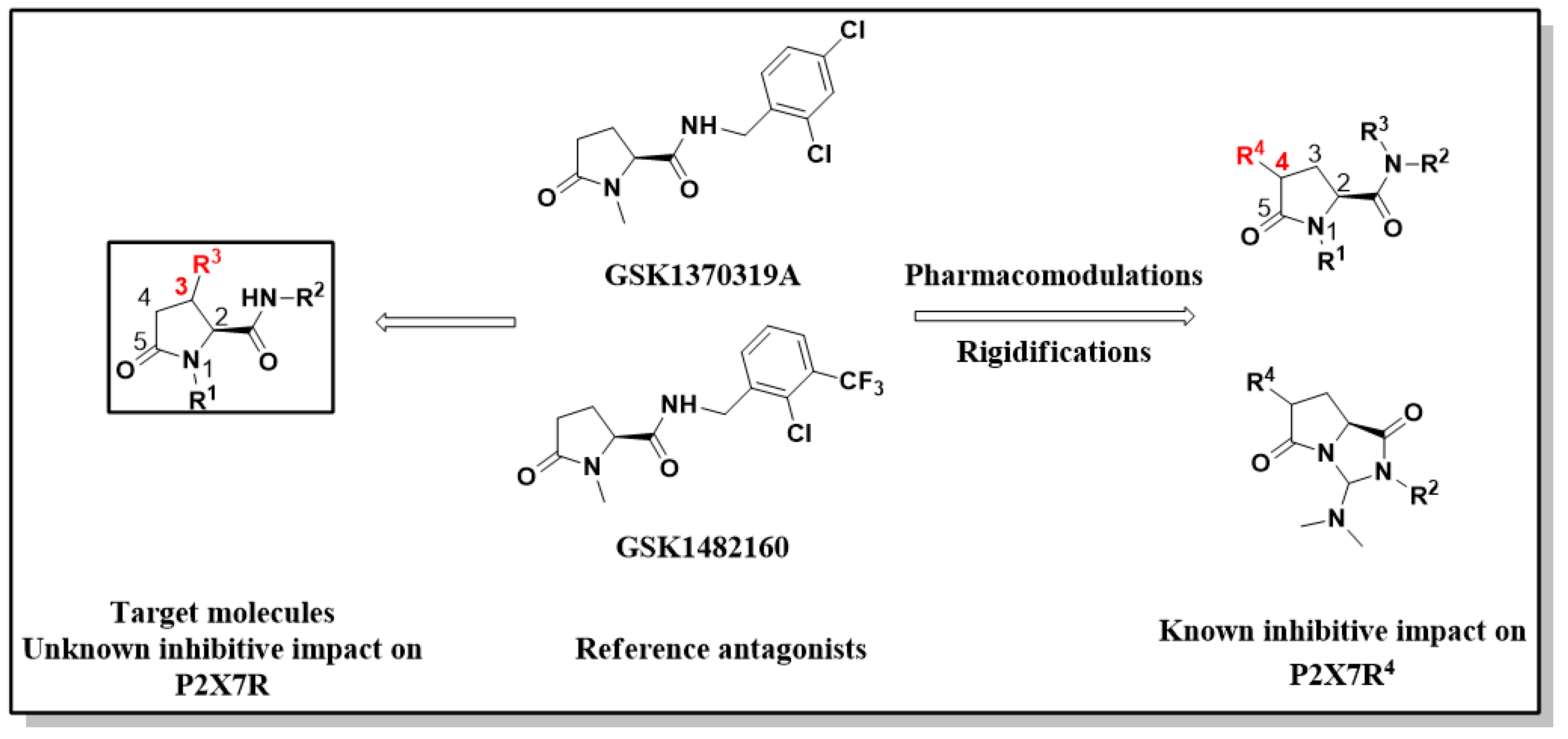
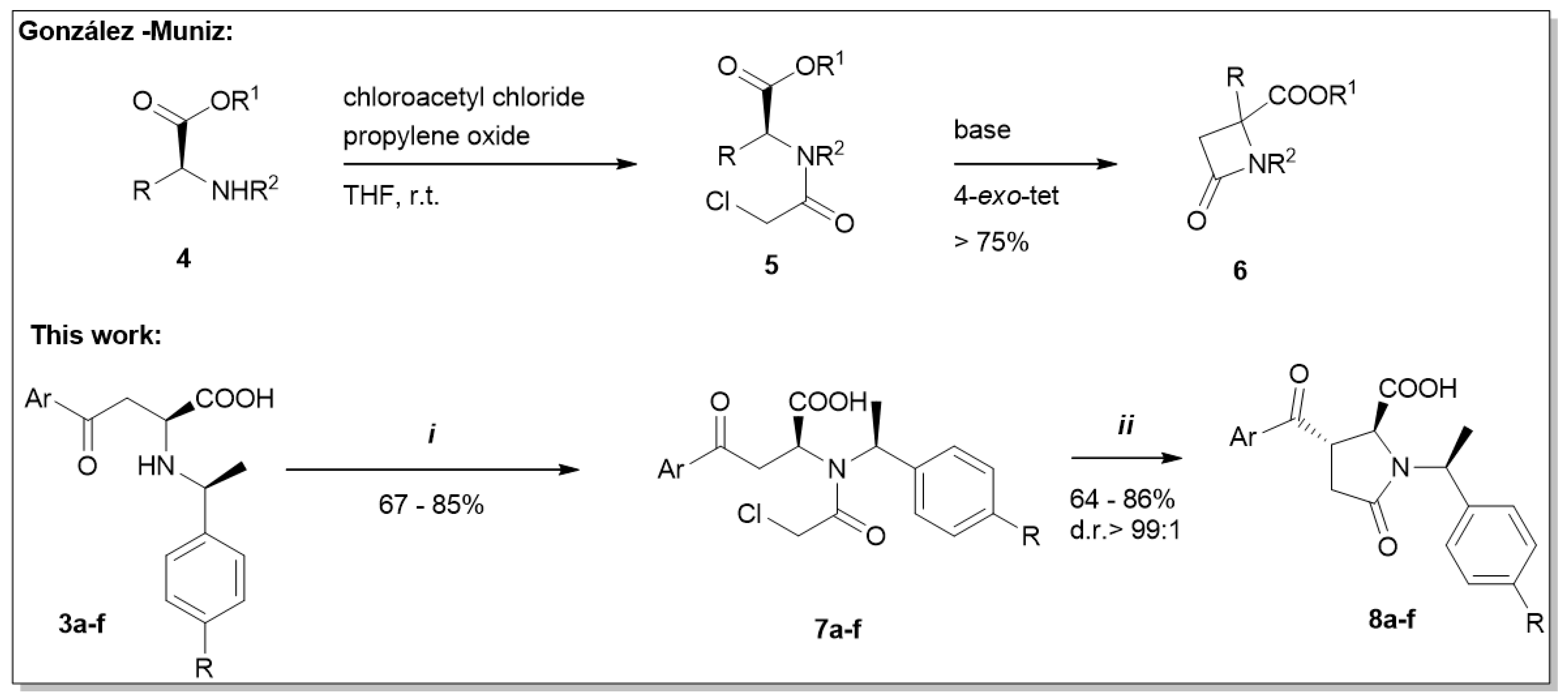
:1. Introduction
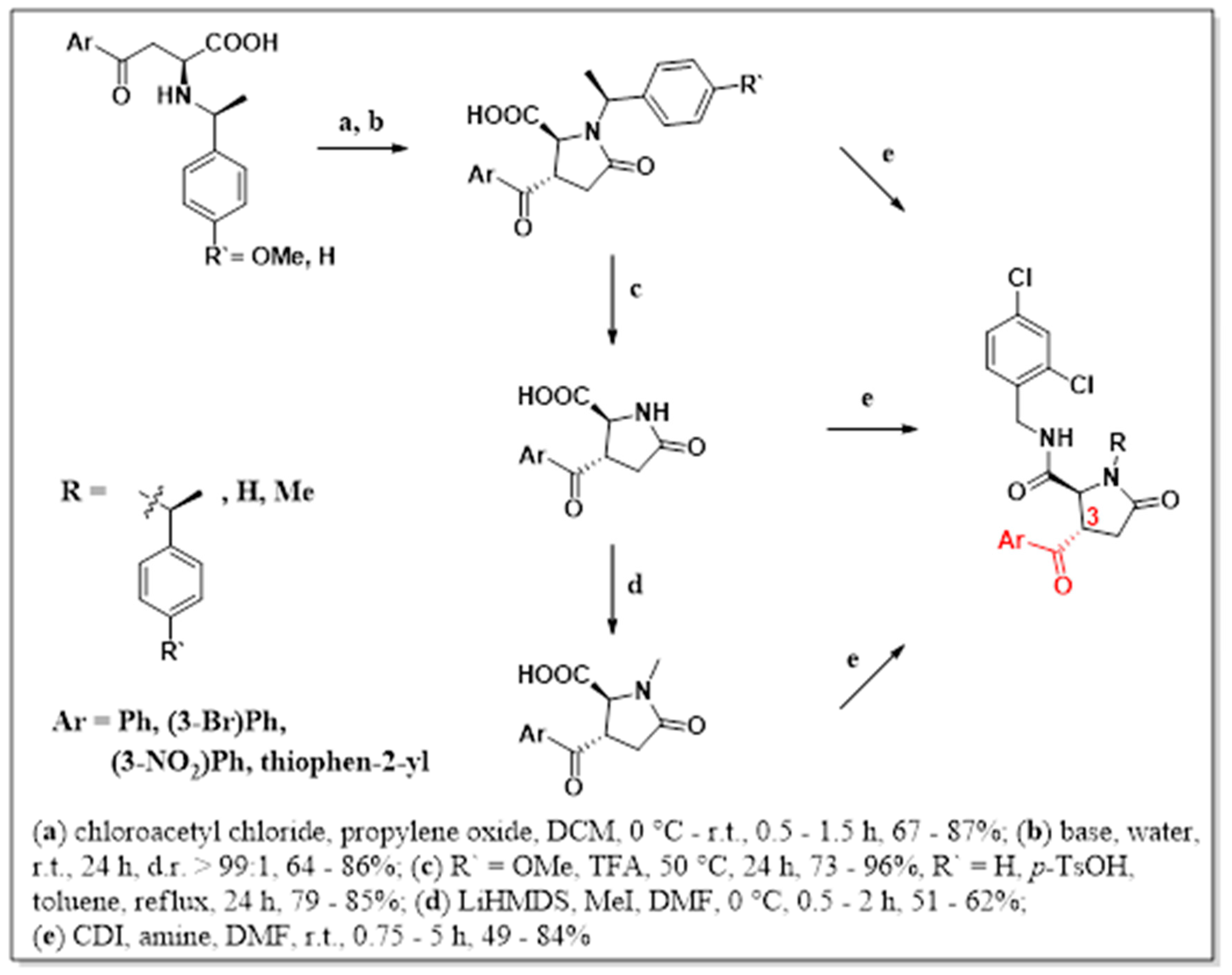
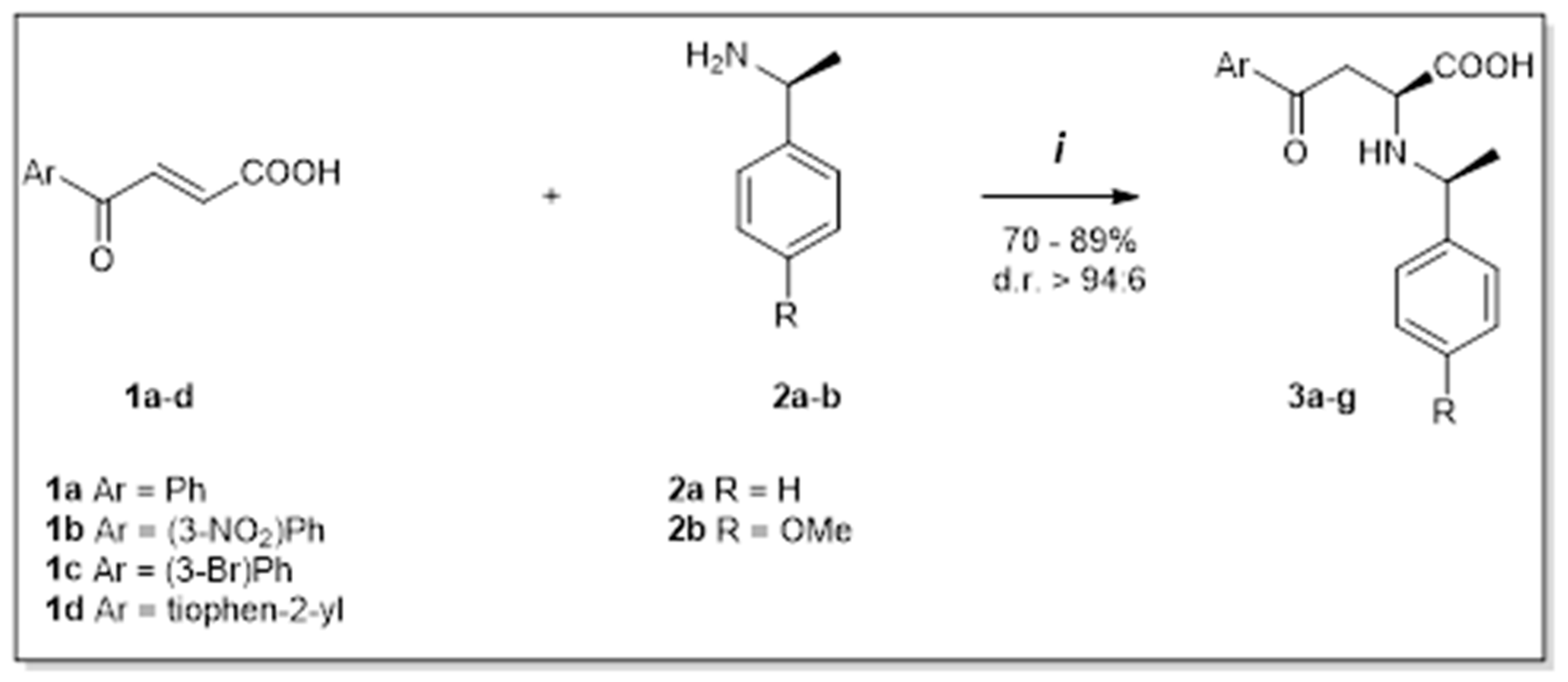
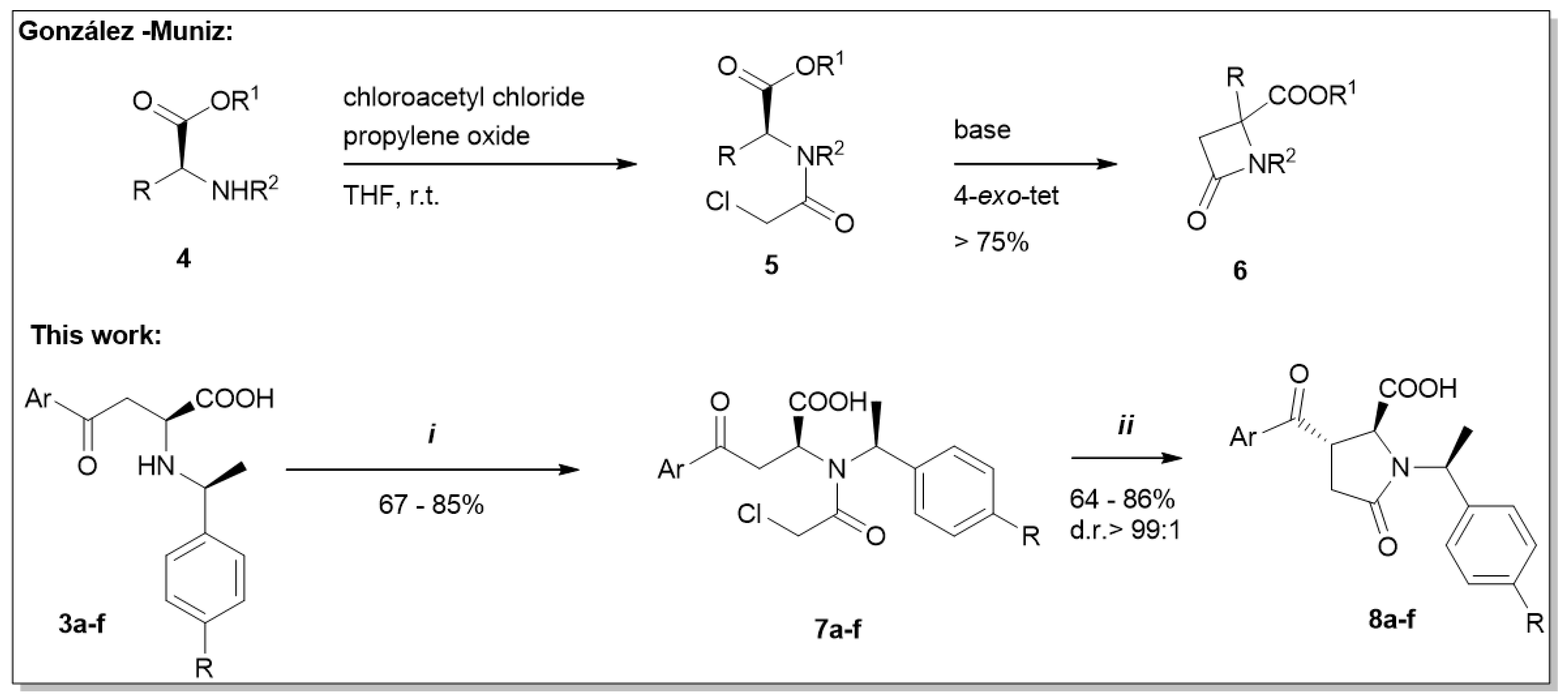
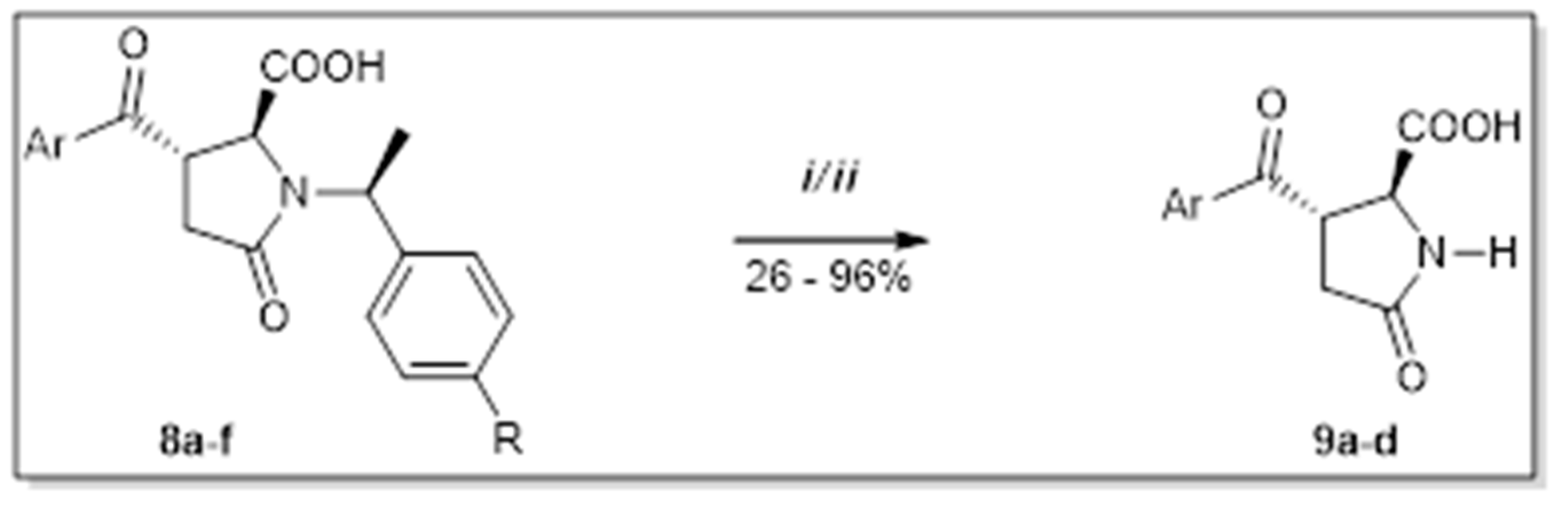
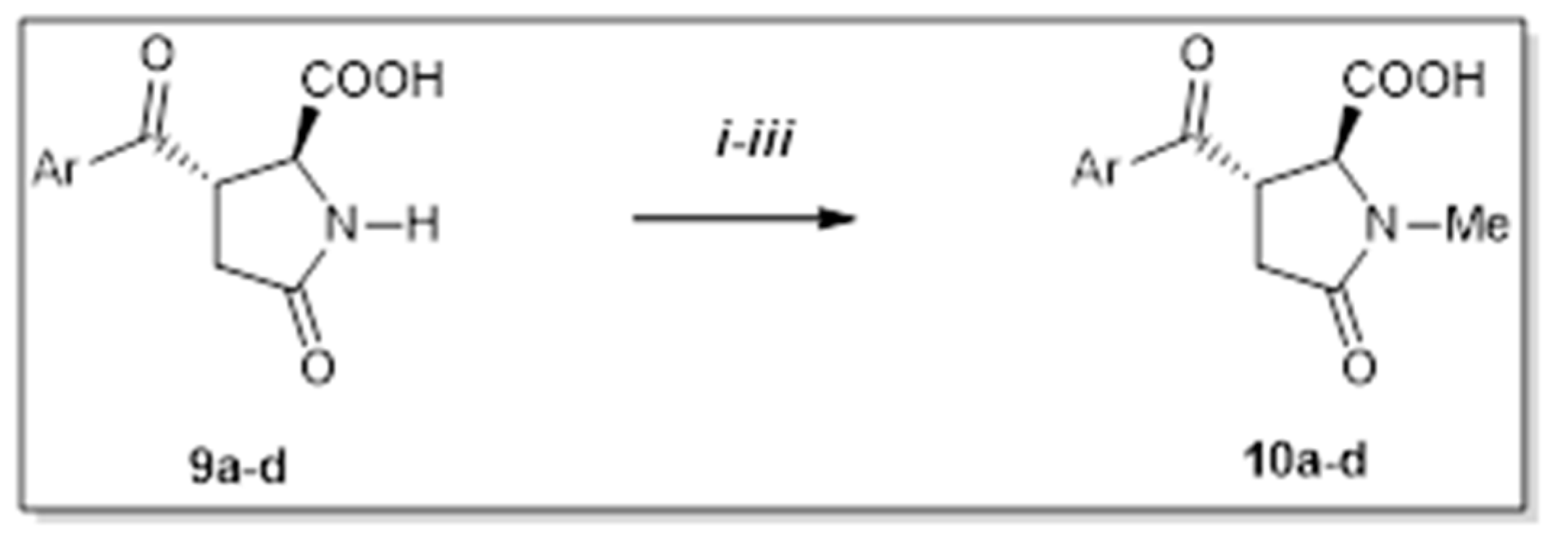
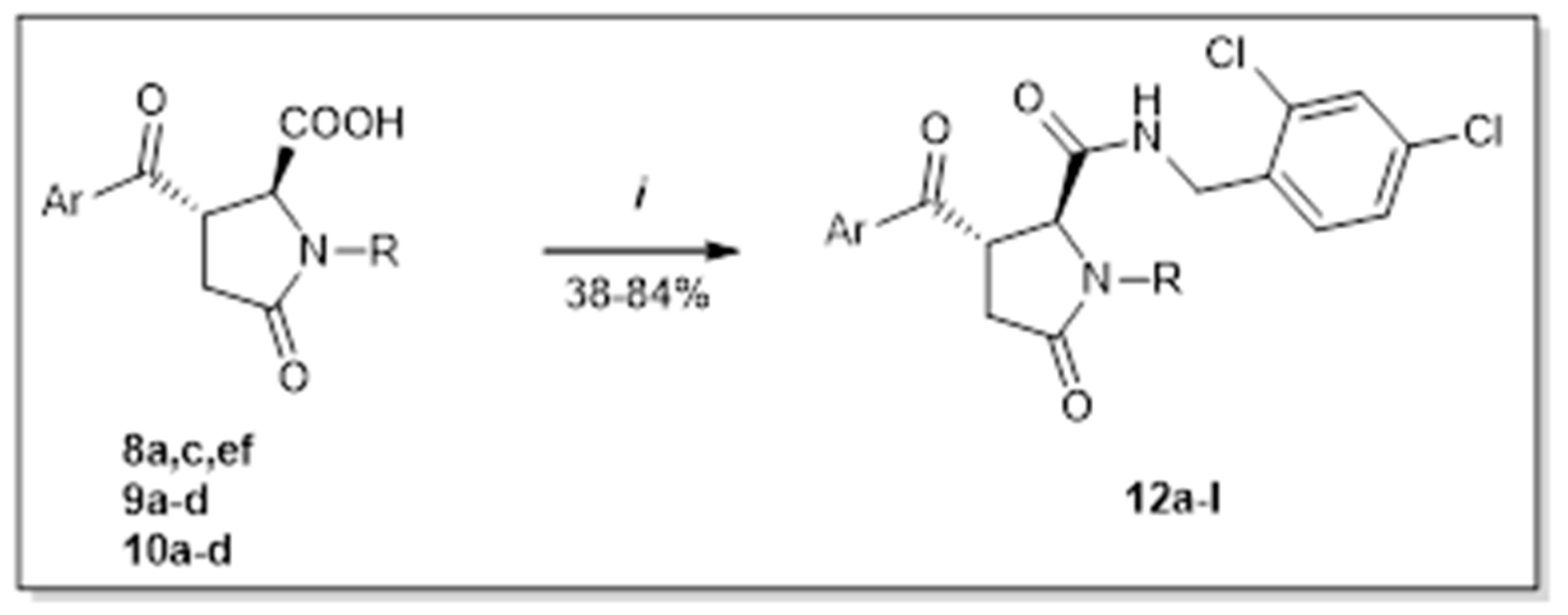
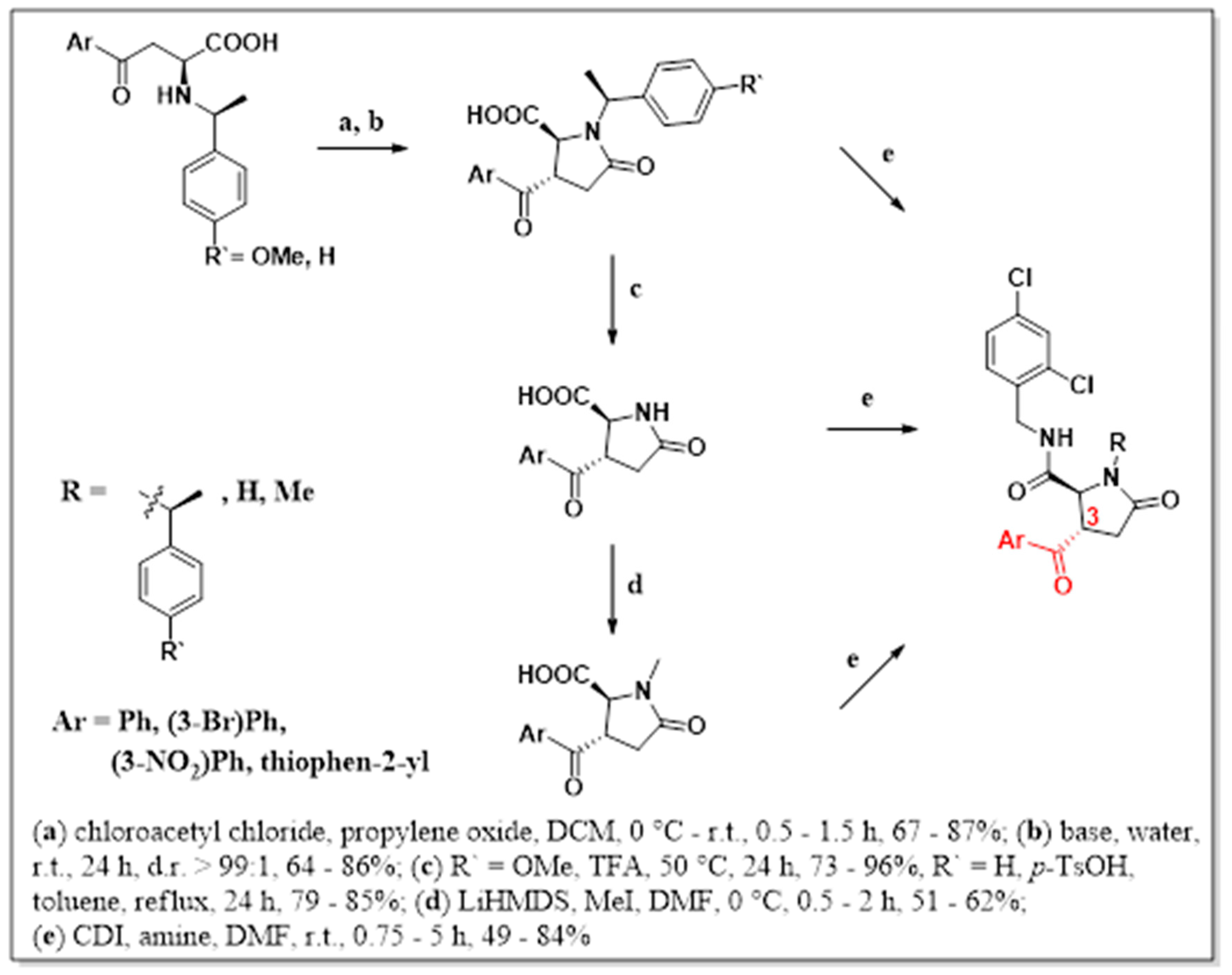
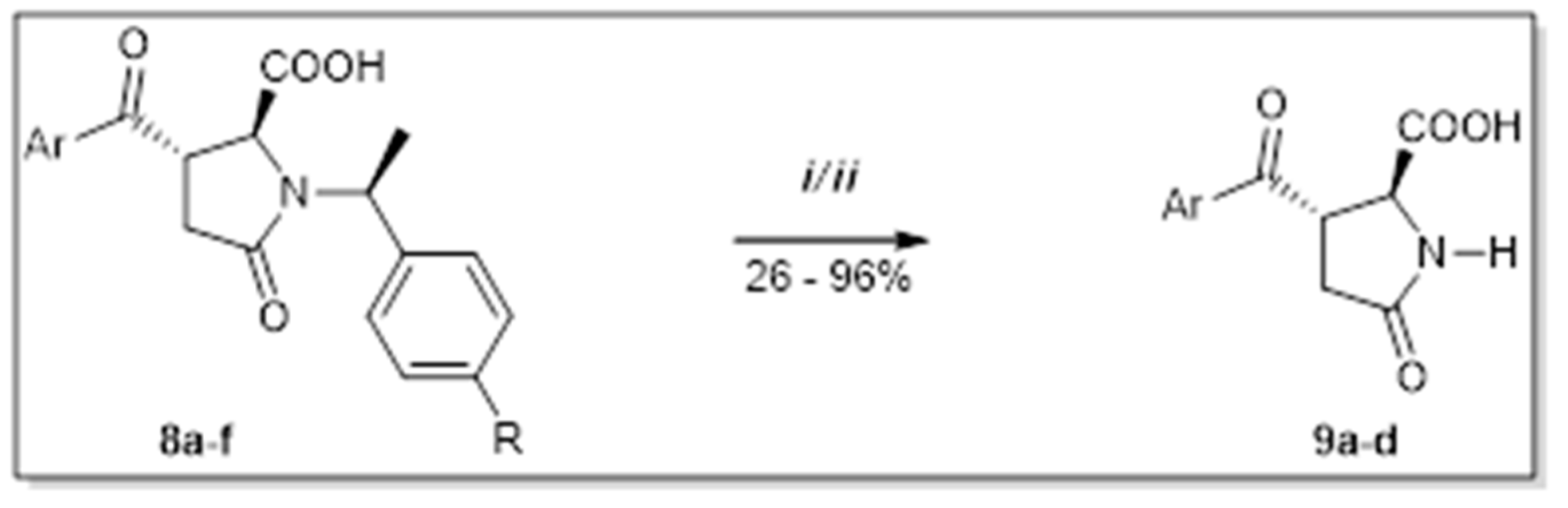
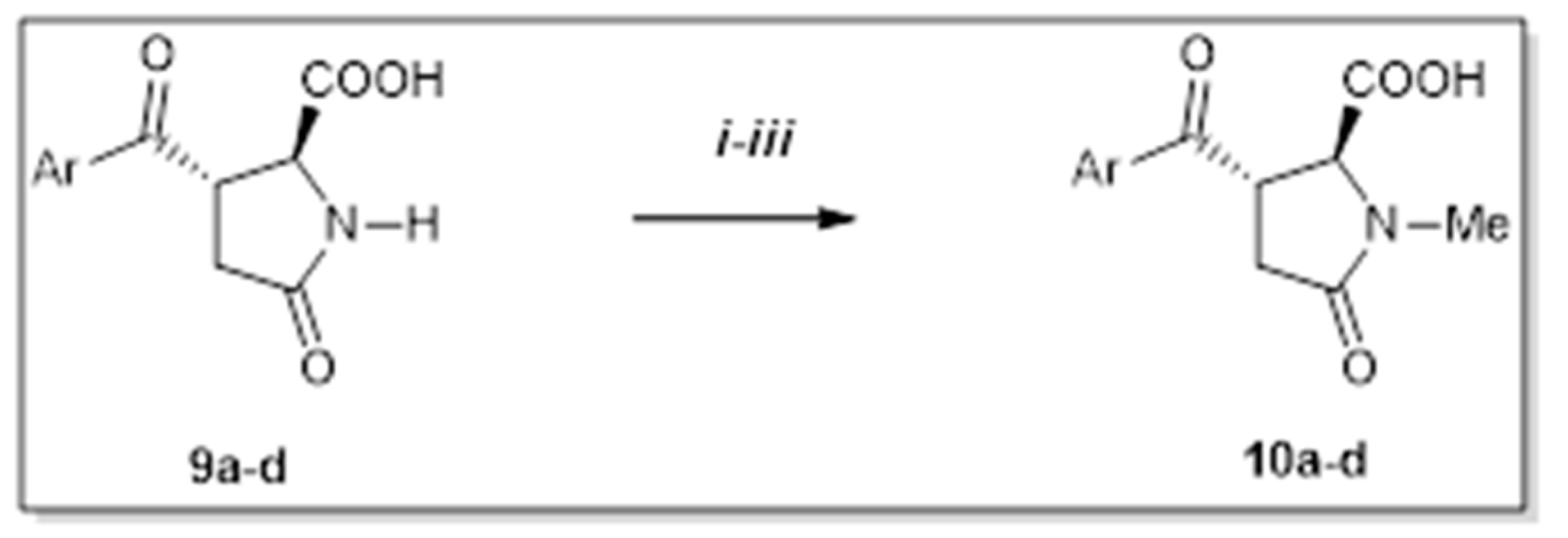
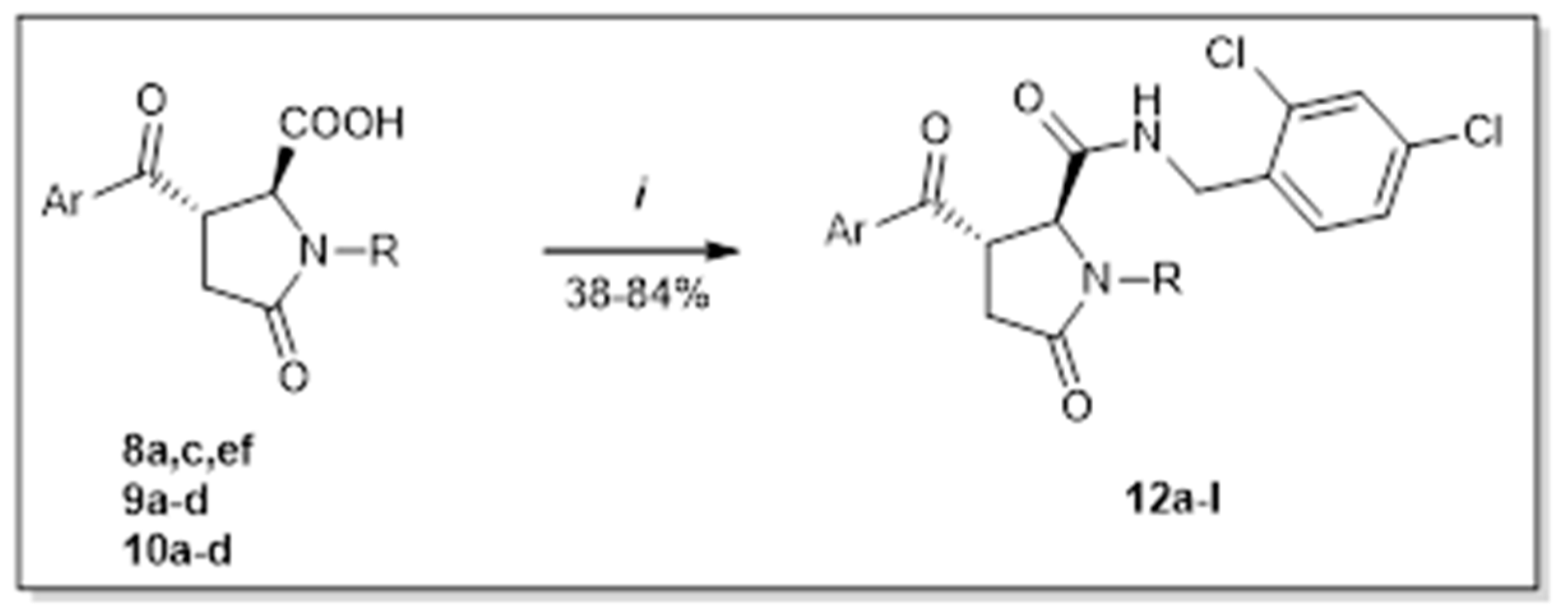
2. Results and Discussion
3. General Methods
4. Experimental
4.1. Procedure A
4.2. Procedure B
4.3. Procedure C
4.4. Procedure D
4.5. Procedure E
4.6. Procedure F
Author Contributions
Funding
Conflicts of Interest
References
- Khopade, T.M.; Warghude, P.K.; Sonawane, A.D.; Bhat, R.G. Multicomponent synthesis of pyroglutamic acid derivatives via Knoevenagel–Michael-hydrolysis-lactamization-decarboxylation (KMHL-D) sequence. Org. Biomol. Chem. 2019, 17, 561. [Google Scholar] [CrossRef] [PubMed]
- Sperlagh, B.; Illes, P. P2X7 receptor: An emerging target in central nervous system diseases. Trends. Pharmacol. Sci. 2014, 35, 537. [Google Scholar] [CrossRef] [PubMed]
- Abdi, M.H.; Beswick, P.J.; Billinton, A.; Chambers, L.J.; Charlton, A.; Collins, S.D.; Collis, K.L.; Dean, D.K.; Fonfria, E.; Gleave, R.J.; et al. Discovery and structure-activity relationships of a series of pyroglutamic acid amide antagonists of the P2X7 receptor. Bioorg. Med. Chem. Lett 2010, 20, 5080. [Google Scholar] [CrossRef] [PubMed]
- Homerin, G.; Jawhara, S.; Dezitter, X.; Baudelet, D.; Dufrénoy, P.; Rigo, B.; Millet, R.; Furman, C.; Ragé, G.; Lipka, E.; et al. Pyroglutamide-Based P2X7 Receptor Antagonists Targeting Inflammatory Bowel Disease. J. Med. Chem. 2019, 63, 2074–2094. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yang, X.; Li, S.; Zhu, Y.; Li, Y.; Zhang, Y. Visible light-induced aerobic oxidative cross-coupling of glycine esters with α-angelicalactone: A facile pathway to γ-lactams. Org. Biomol. Chem. 2018, 16, 6728. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.G. Developing processes for crystallization-induced asymmetric transformation. Org. Process. Res. Dev. 2005, 9, 800. [Google Scholar] [CrossRef]
- Berkeš, D.; Jakubec, P.; Winklerová, D.; Považanec, F.; Daich, A. CIAT with simultaneous epimerization at two stereocenters. Synthesis of substituted β-methyl-α-homophenylalanines. Org. Biomol. Chem. 2007, 5, 121. [Google Scholar] [CrossRef] [PubMed]
- Jakubec, P.; Berkeš, D. Crystallisation-induced asymmetric transformation (CIAT) for the synthesis of dipeptides containing homophenylalanine. Tetrahedron Asymmetry 2010, 21, 2807. [Google Scholar] [CrossRef]
- Brands, K.M.J.; Davies, A.J. Crystallization-induced diastereomer transformations. Chem. Rev. 2006, 106, 2711–2733. [Google Scholar] [CrossRef] [PubMed]
- Markus, J.; Puchľová, E.; Pinčeková, L.; Moncol, J.; Doháňošová, J.; Berkeš, D.; Caletková, O. Synthesis and Derivatization of 3-Aroyl Pyroglutamic Acids. ChemistrySelect 2020, 5, 2115. [Google Scholar] [CrossRef]
- Gerona-Navarro, G.; Bonache, M.A.; Herranz, R.; García-López, M.T.; González-Muniz, R. Entry to new conformationally constrained amino acids. First synthesis of 3-unsubstituted 4-alkyl-4-carboxy-2-azetidinone derivatives via an intramolecular N α-C α-cyclization strategy. J. Org. Chem. 2001, 66, 3538. [Google Scholar] [CrossRef]
- Chern, C.-Y.; Huang, Y.-P.; Kan, W.M. Selective N-debenzylation of amides with p-TsOH. Tetrahedron Lett. 2003, 44, 1039. [Google Scholar] [CrossRef]
- Vaidyanathan, R.; Kalthod, V.G.; Ngo, D.P.; Manley, J.M.; Lapekas, S.P. Amidations using N, N′-carbonyldiimidazole: Remarkable rate enhancement by carbon dioxide. J. Org. Chem. 2004, 69, 2565. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-W.; Liu, Y.-K. Enantio-and diastereoselective synthesis of tetrahydrofuro [2, 3-b] furan-2 (3 H)-one derivatives and related oxygen heterocycles via an asymmetric organocatalytic cascade process. Org. Chem. Front. 2017, 4, 2358. [Google Scholar] [CrossRef]
- Zhao, S.; Lin, J.-B.; Zhao, Y.-Y.; Liang, Y.-M.; Xu, P.-F. Hydrogen-bond-directed formal [5+ 1] annulations of oxindoles with ester-linked bisenones: Facile access to chiral spirooxindole δ-lactones. Org. Lett. 2014, 16, 1802. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Xu, J.; Zhang, G.; Xu, C.; Zhao, L.; You, X.; Wang, Y.; Lu, Y.; Yu, L.; Wang, J. Design, synthesis, and bioevaluation of a novel class of (E)-4-oxo-crotonamide derivatives as potent antituberculosis agents. Bioorg. Med. Chem. Lett. 2019, 29, 539. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product 3a–g | Ar | R | Yield 3 (%) | d.r. | m.p. (°C) |
|---|---|---|---|---|---|
| 3a | Ph | OMe | 84 | 98:2 | 174–177 |
| 3b | (3-NO2)Ph | H | 89 | 99:1 | 172–173 |
| 3c | OMe | 86 | 99:1 | 179–181 | |
| 3d | (3-Br)Ph | H | 70 | 98:2 | 172–175 |
| 3e | OMe | 73 | 97:3 | 173–178 | |
| 3f | Thiophen-2-yl | H | 80 | 94:6 | 196–198 |
| 3g | OMe | - | - | - |
| Substrate 3a–f | Ar | R | Yield 7 (%) | m.p. (°C) | Yield 8 (%) | m.p. (°C) |
|---|---|---|---|---|---|---|
| 3a | Ph | OMe | 75 | 164–165 | 74 | 220–222 |
| 3b | (3-NO2)Ph | H | 85 | 144–146 | 64 | 232–234 |
| 3c | OMe | 67 | 173–175 | 86 | 182–183 | |
| 3d | (3-Br)Ph | H | 85 | 129–132 | 73 | 216–217 |
| 3e | OMe | 78 | 158–160 | 73 | 162–167 | |
| 3f | Thiophen-2-yl | H | 79 | 204–205 | 72 | 227–229 |
| Substrate 8a–g | Ar | R | Conditions | Yield 9 (%) | m.p. (°C) |
|---|---|---|---|---|---|
| 8a | Ph | OMe | i | 96 | 166–167 |
| 8b | (3-NO2)Ph | H | ii | 85 | 221–223 |
| 8c | OMe | i | 94 | 221–222 | |
| 8d | (3-Br)Ph | H | ii | 79 | 212–215 |
| 8e | OMe | i | 73 | 215–216 | |
| 8f | Thiophen-2-yl | H | ii | 26 | - |
| Substrate 9a–d | Ar | Overall Yield 10 (3 Steps) (%) |
|---|---|---|
| 9a | Ph | 44 |
| 9b | (3-NO2)Ph | 36 |
| 9c | (3-Br)Ph | 42 |
| 9d | Thiophen-2-yl | - |
| Substrate | Ar | R | Yield 12 (%) | m.p. (°C) |
|---|---|---|---|---|
| 8a | Ph | (S)-1-((4-OMe)Ph)Et | 70 | 136–137 |
| 9a | H | 65 | 134–136 | |
| 10a | Me | 84 | - | |
| 8c | (3-NO2)Ph | (S)-1-((4-OMe)Ph)Et | 49 | 191–192 |
| 9b | H | 36 | 223–225 | |
| 10b | Me | 56 | - | |
| 8e | (3-Br)Ph | (S)-1-((4-OMe)Ph)Et | 69 | - |
| 9c | H | 49 | 116–118 | |
| 10c | Me | 57 | - | |
| 8f | Thiophen-2-yl | (S)-1-(Ph)Et – | 77 | 154–156 |
| 9d | H | 82 | 157–159 | |
| 10d | Me | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pincekova, L.; Berkes, D. Synthesis of (2S,3S)-3-Aroyl Pyroglutamic Acid Amides. Chem. Proc. 2021, 3, 86. https://doi.org/10.3390/ecsoc-24-08377
Pincekova L, Berkes D. Synthesis of (2S,3S)-3-Aroyl Pyroglutamic Acid Amides. Chemistry Proceedings. 2021; 3(1):86. https://doi.org/10.3390/ecsoc-24-08377
Chicago/Turabian StylePincekova, Lucia, and Dusan Berkes. 2021. "Synthesis of (2S,3S)-3-Aroyl Pyroglutamic Acid Amides" Chemistry Proceedings 3, no. 1: 86. https://doi.org/10.3390/ecsoc-24-08377
APA StylePincekova, L., & Berkes, D. (2021). Synthesis of (2S,3S)-3-Aroyl Pyroglutamic Acid Amides. Chemistry Proceedings, 3(1), 86. https://doi.org/10.3390/ecsoc-24-08377