
Molecular Docking Study: Application to the Epidermal Growth Factor Receptor †


Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
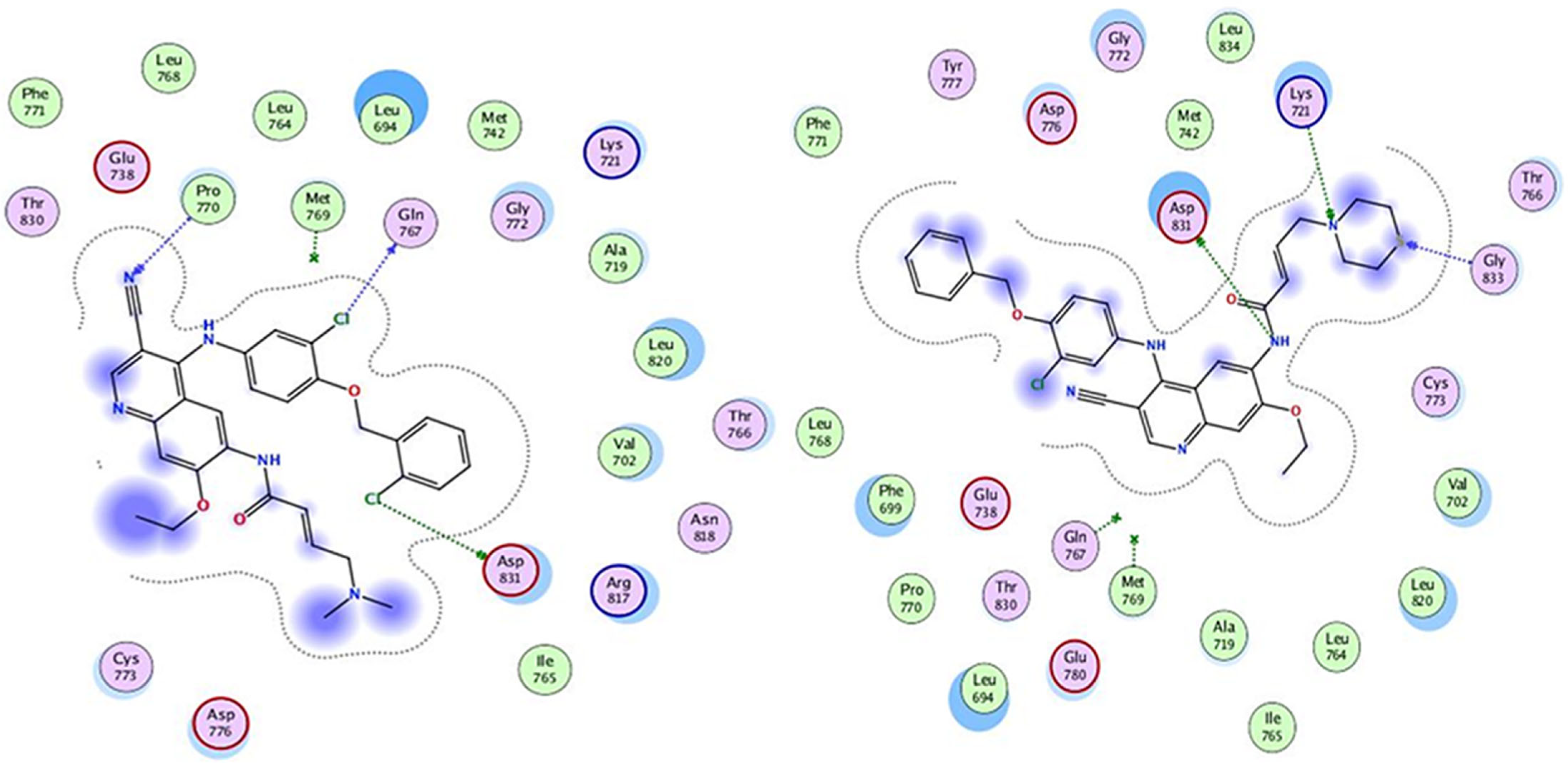
3.1. Molecular Docking
3.2. Evaluation ADME-TOX
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rojas, K.D.; Perez, M.E.; Marchetti, M.A.; Nichols, A.J.; Penedo, F.J.; Jaimes, N. Skin cancer: Primary, secondary, and tertiary prevention. Part II. J. Am. Acad. Dermatol. 2022, 87, 271–288. [Google Scholar] [CrossRef] [PubMed]
- Dildar, M.; Akram, S.; Irfan, M.; Khan, H.U.; Ramzan, M.; Mahmood, A.R.; Alsaiari, S.A.; Saeed, A.H.M.; Alraddadi, M.O.; Mahnashi, M.H. Skin Cancer Detection: A Review Using Deep Learning Techniques. Int. J. Environ. Res. Public Health 2021, 18, 5479. [Google Scholar] [CrossRef] [PubMed]
- Asli, F. Étude de Docking Moleculaire: Application au Recepteur du Facteur de Croissances Epidermique. Master’s Thesis, Université Mohamed Khider de Biskra, Biskra, Algeria, 2020. Available online: http://archives.univ-biskra.dz:80/handle/123456789/15601 (accessed on 22 January 2025).
- Shang, X.-F.; Morris-Natschke, S.L.; Liu, Y.-Q.; Li, X.-H.; Zhang, J.-Y.; Lee, K.-H. Chapter One—Biology of quinoline and quinazoline alkaloids. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Academic Press: Cambridge, MA, USA, 2022; Volume 88, pp. 1–47. [Google Scholar] [CrossRef]
- Evren, A.E.; Özkan, B.N.S.; Akalin-Çiftçi, G.; Yurttaş, L. Pharmacophore-Based Modeling, Synthesis, and Biological Evaluation of Novel Quinazoline/Quinoline Derivatives: Discovery of EGFR Inhibitors with Low Nanomolar Activity. Adv. Theory Simul. 2025, 8, 2400811. [Google Scholar] [CrossRef]
- Kiso-Farnè, K.; Tsuruyama, T. Epidermal growth factor receptor cascade prioritizes the maximization of signal transduction. Sci. Rep. 2022, 12, 16950. Available online: https://www.nature.com/articles/s41598-022-20663-0 (accessed on 14 January 2025). [CrossRef] [PubMed]
- RCSB PDB. 1M17: Epidermal Growth Factor Receptor Tyrosine Kinase Domain with 4-Anilinoquinazoline Inhibitor Erlotinib. 2022. Available online: https://www.rcsb.org/structure/1m17 (accessed on 14 January 2025).
- Wissner, A.; Berger, D.M.; Boschelli, D.H.; Floyd, M.B.; Greenberger, L.M.; Gruber, B.C.; Johnson, B.D.; Mamuya, N.; Nilakantan, R.; Reich, M.F.; et al. 4-Anilino-6,7-dialkoxyquinoline-3-carbonitrile inhibitors of epidermal growth factor receptor kinase and their bioisosteric relationship to the 4-anilino-6,7-dialkoxyquinazoline inhibitors. J. Med. Chem. 2000, 43, 3244–3256. [Google Scholar] [CrossRef] [PubMed]
- Tsou, H.-R.; Mamuya, N.; Johnson, B.D.; Reich, M.F.; Gruber, B.C.; Ye, F.; Nilakantan, R.; Shen, R.; Discafani, C.; DeBlanc, R.; et al. 6-Substituted-4-(3-bromophenylamino)quinazolines as putative irreversible inhibitors of the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor (HER-2) tyrosine kinases with enhanced antitumor activity. J. Med. Chem. 2001, 44, 2719–2734. [Google Scholar] [CrossRef] [PubMed]
- Wissner, A.; Floyd, M.B.; Rabindran, S.K.; Nilakantan, R.; Greenberger, L.M.; Shen, R.; Wang, Y.-F.; Tsou, H.-R. Syntheses and EGFR and HER-2 kinase inhibitory activities of 4-anilinoquinoline-3-carbonitriles: Analogues of three important 4-anilinoquinazolines currently undergoing clinical evaluation as therapeutic antitumor agents. Bioorg. Med. Chem. Lett. 2002, 12, 2893–2897. [Google Scholar] [CrossRef] [PubMed]
- Tsou, H.-R.; Overbeek-Klumpers, E.G.; Hallett, W.A.; Reich, M.F.; Floyd, M.B.; Johnson, B.D.; Michalak, R.S.; Nilakantan, R.; Discafani, C.; Golas, J.; et al. Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J. Med. Chem. 2005, 48, 1107–1131. [Google Scholar] [CrossRef] [PubMed]
- Wissner, A.; Overbeek, E.; Reich, M.F.; Floyd, M.B.; Johnson, B.D.; Mamuya, N.; Rosfjord, E.C.; Discafani, C.; Davis, R.; Shi, X.; et al. Synthesis and structure-activity relationships of 6,7-disubstituted 4-anilinoquinoline-3-carbonitriles. The design of an orally active, irreversible inhibitor of the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor-2 (HER-2). J. Med. Chem. 2003, 46, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group (CCG). Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2014; Available online: https://www.chemcomp.com/ (accessed on 7 July 2024).

{kind=link}
| No of Ligands | IC50 (µM) | S-Score (kcal mol) | RMSD (Å) | Bonds Between the Compounds Atoms and the Active Site Residues | |||
|---|---|---|---|---|---|---|---|
| Compound Atoms | Receptor Atoms | Interaction Type | Distance (Å) | ||||
| L1 | 0.016 | −9.0419 | 1.9563 | N10 | OD2 | H-donor | 2.98 |
| S18 | CA | H-acceptor | 3.83 | ||||
| N25 | NZ | H-acceptor | 3.71 | ||||
| L2 | 0.011 | −9.0158 | 1.2483 | CL69 | OD2 | H-donor | 3.33 |
| CL70 | O | H-donor | 3.05 | ||||
| N43 | CA | H-acceptor | 3.63 | ||||
| L3 | 0.015 | −9.0054 | 1.6614 | N12 | NZ | H-acceptor | 3.19 |
| 6-ring | CD | pi-H | 3.90 | ||||
| 6-ring | CD | pi-H | 4.33 | ||||
| 6-ring | CA | pi-H | 3.54 | ||||
| L4 | 0.030 | −8.8247 | 1.4567 | CL67 | OD1 | H-donor | 3.09 |
| N12 | OG1 | H-acceptor | 3.28 | ||||
| L5 | 0.031 | −8.7421 | 1.5109 | 6-ring | CG1 | pi-H | 3.82 |
| 6-ring | CD | pi-H | 3.94 | ||||
| 6-ring | N | pi-H | 4.00 | ||||
| L6 | 0.020 | −8.6277 | 2.0115 | N10 | OD1 | H-donor | 3.51 |
| O23 | OD2 | H-donor | 3.16 | ||||
| N47 | N | H-acceptor | 3.47 | ||||
| 6-ring | CD1 | pi-H | 4.25 | ||||
| L7 | 0.024 | −8.3984 | 1.9097 | CL55 | O | H-donor | 3.05 |
| L8 | 0.026 | −8.3727 | 1.6572 | N 24 | N | H-acceptor | 3.42 |
| 6-ring | CB | pi-H | 3.71 | ||||
| L9 | 0.007 | −8.1837 | 1.7923 | 6-ring | CB | pi-H | 3.74 |
| 6-ring | CB | pi-H | 4.65 | ||||
| L10 | 0.034 | −8.1624 | 1.4415 | 6-ring | N | pi-H | 4.05 |
| L_Ref | 0.020 | −8.0480 | 1.4130 | N44 | N | H-acceptor | 3.13 |
| Category | L_148 | L_177 | L_198 | L_140 | L_143 | L_Ref | ||
|---|---|---|---|---|---|---|---|---|
| Physicochemical properties | Molecular weight (MW g/mol) < 500 | 614.16 | 590.50 | 494.38 | 465.30 | 467.92 | 393.4 | |
| Heavy atoms | 43 | 41 | 32 | 30 | 33 | 29 | ||
| Rotatable bonds | 12 | 12 | 9 | 7 | 9 | 10 | ||
| H-bond acceptors < 10 | 6 | 6 | 5 | 5 | 6 | 6 | ||
| H-bond donors < 5 | 2 | 2 | 2 | 2 | 2 | 1 | ||
| TPSA 140 (Å2) | 124.81 | 99.51 | 90.28 | 96.27 | 90.28 | 74.73 | ||
| Drug-likeness | Lipinski violation | Yes | Yes | Yes | Yes | Yes | Yes | |
| Veber violation | No | No | Yes | No | Yes | Yes | ||
| Ghose violation | No | No | No | Yes | Yes | Yes | ||
| Muegge violations | No | No | Yes | No | Yes | Yes | ||
| Egan violations | No | No | Yes | Yes | Yes | Yes | ||
| Bioavailability | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | ||
| Consensus log Po/w < 5 | 5.26 | 5.48 | 3.86 | 3.40 | 4.10 | 3.20 | ||
| Log S (ESOL) | −7.22 | −7.22 | −5.52 | −5.24 | −5.37 | −4.11 | ||
| Pharmacokinetics | GI absorption | Low | Low | High | High | High | High | |
| BBB permeability | No | No | No | No | No | Yes | ||
| P-gp substrate | Yes | Yes | No | No | No | No | ||
| CYP inhibitor | 1A2 | No | No | No | Yes | No | Yes | |
| 2C19 | Yes | Yes | Yes | Yes | Yes | Yes | ||
| 2C9 | Yes | No | Yes | Yes | Yes | Yes | ||
| 2D6 | No | Yes | Yes | Yes | Yes | Yes | ||
| 3A4 | No | No | Yes | Yes | Yes | Yes | ||
| Log Kp (cm/s) | −5.55 | −5.27 | −6.12 | −6.23 | −5.93 | −6.35 | ||
| Toxicity | Oral rat acute toxicity LD50 (mol/kg) | 2.426 | 3.033 | 2.531 | 2.386 | 2.641 | 2 368 | |
| Hepatotoxicity | Inactive | Inactive | Inactive | Inactive | Inactive | active | ||
| Carcinogenicity | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | ||
| Cytotoxicity | Inactive | Inactive | Inactive | Inactive | Inactive | active | ||
| Mutagenicity | Active | Inactive | Inactive | Inactive | Inactive | active | ||
| Immunotoxicity | Inactive | Inactive | active | Active | active | active | ||
| Ames toxicity | No | No | No | No | No | No | ||
| Skin sensitization | No | No | No | No | No | No | ||
| hERG I inhibition | No | No | No | No | No | No | ||
| hERG II inhibition | Yes | Yes | Yes | Yes | Yes | Yes | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asli, F.; Bensahbane, I. Molecular Docking Study: Application to the Epidermal Growth Factor Receptor. Chem. Proc. 2024, 16, 82. https://doi.org/10.3390/ecsoc-28-20219
Asli F, Bensahbane I. Molecular Docking Study: Application to the Epidermal Growth Factor Receptor. Chemistry Proceedings. 2024; 16(1):82. https://doi.org/10.3390/ecsoc-28-20219
Chicago/Turabian StyleAsli, Faiza, and Imane Bensahbane. 2024. "Molecular Docking Study: Application to the Epidermal Growth Factor Receptor" Chemistry Proceedings 16, no. 1: 82. https://doi.org/10.3390/ecsoc-28-20219
APA StyleAsli, F., & Bensahbane, I. (2024). Molecular Docking Study: Application to the Epidermal Growth Factor Receptor. Chemistry Proceedings, 16(1), 82. https://doi.org/10.3390/ecsoc-28-20219