
In Silico Pharmacological Prediction of Capitavine, Buchenavianine and Related Flavonoid Alkaloids †


Abstract
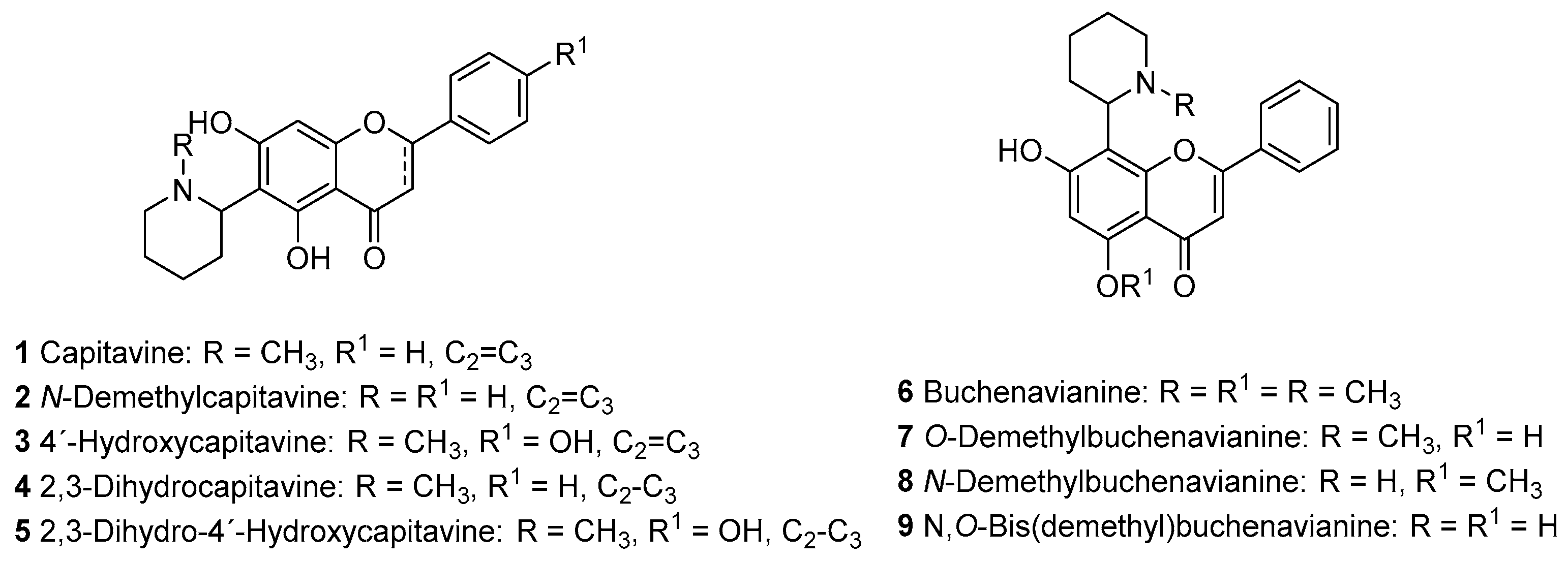
1. Introduction
2. Material and Methods
3. Results and Discussion
3.1. Molinspiration
3.2. SwissADME
3.3. Osiris
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khadem, S.; Marles, R.J. Chromone and Flavonoid Alkaloids: Occurrence and Bioactivity. Molecules 2012, 17, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Ilkei, V.; Hazai, L.; Antus, S.; Bölcskei, H. Flavonoid Alkaloids: Isolation, Bioactivity, and Synthesis. Stud. Nat. Prod. Chem. 2018, 56, 247–285. [Google Scholar] [CrossRef]
- Ahond, A.; Fournet, A.; Moretti, C.; Philogene, E.; Poupat, C.; Thoison, O.; Potier, P. Premiers alcaloïdes vrais isolés de Combretacées: Buchenavia macrophylla Eichl. et Buchenavia capitata Eichl. Bull. Soc. Chim. Fr. Partie I 1984, 1–2, 41–45. [Google Scholar]
- Beutler, J.A.; Cardellina II, J.H.; McMahon, J.B.; Boyd, M.R.; Cragg, G.M. Anti-HIV and Cytotoxic Alkaloids from Buchenavia capitata. J. Nat. Prod. 1992, 55, 207–213. [Google Scholar] [CrossRef]
- Maliar, T.; Maliarová, M.; Purdešová, A.; Jankech, T.; Gerhardtová, I.; Beňovič, P.; Dvořáček, V.; Jágr, M.; Viskupičová, J. The Adapted POM Analysis of Avenanthramides in Silico. Pharmaceuticals 2023, 16, 717. [Google Scholar] [CrossRef] [PubMed]
- Apan, A.; Casoni, D.; Leonte, D.; Pop, C.; Iaru, I.; Mogosan, C.; Zaharia, V. Heterocycles 52: The Drug-Likeness Analysis of Anti-Inflammatory Thiazolo[3,2-b][1,2,4]triazole and Imidazo[2,1-b][1,3,4]thiadiazole Derivatives. Pharmaceuticals 2024, 17, 295. [Google Scholar] [CrossRef]
- Molinspiration Chemoinformatics Software. Available online: https://www.molinspiration.com/ (accessed on 1 June 2024).
- SwissADME Software. Available online: http://www.swissadme.ch/ (accessed on 10 June 2024).
- OSIRIS Property Explorer; Actelion Pharmaceuticals Ltd.: Allschwil, Switzerland, 2010; Available online: http://www.organic-chemistry.org/prog/peo/ (accessed on 5 June 2024).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Duong, V.A.; Maeng, H.J. Pharmaceutical Formulations with P-Glycoprotein Inhibitory Effect as Promising Approaches for Enhancing Oral Drug Absorption and Bioavailability. Pharmaceutics 2021, 13, 1103. [Google Scholar] [CrossRef]
- Šestić, T.L.; Ajduković, J.J.; Marinović, M.A.; Petri, E.T.; Savić, M.P. In silico ADMET analysis of the A-, B- and D-modified androstane derivatives with potential anticancer effects. Steroids 2023, 189, 109147. [Google Scholar] [CrossRef] [PubMed]
- Teague, S.J.; Davis, A.M.; Leeson, P.D.; Oprea, T. The Design of Leadlike Combinatorial Libraries. Angew. Chem. Int. Ed. 1999, 38, 3743–3748. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| No. | logP | TPSA | MW | nA/nD | rot | GPCR | ICM | KI | NRL | PI | EI |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 4.23 | 73.91 | 351.40 | 5/2 | 2 | 0.12 | 0.02 | 0.06 | 0.07 | −0.05 | 0.17 |
| 2 | 3.09 | 82.69 | 337.38 | 5/3 | 2 | 0.09 | −0.01 | 0.06 | 0.12 | −0.04 | 0.21 |
| 3 | 3.75 | 94.13 | 367.40 | 6/2 | 2 | 0.13 | 0.03 | 0.07 | 0.12 | −0.05 | 0.18 |
| 4 | 3.68 | 70.00 | 353.42 | 5/2 | 2 | 0.20 | −0.10 | −0.27 | 0.23 | 0.07 | 0.20 |
| 5 | 3.20 | 90.23 | 369.42 | 6/3 | 2 | 0.20 | −0.09 | −0.24 | 0.28 | 0.07 | 0.20 |
| 6 | 4.51 | 62.91 | 365.43 | 5/1 | 3 | 0.15 | −0.10 | 0.12 | 0.06 | −0.10 | 0.11 |
| 7 | 4.23 | 73.91 | 351.40 | 5/1 | 2 | 0.18 | −0.05 | 0.14 | 0.12 | −0.05 | 0.18 |
| 8 | 3.36 | 71.70 | 351.40 | 5/2 | 3 | 0.12 | −0.14 | 0.12 | 0.11 | −0.10 | 0.14 |
| 9 | 3.09 | 82.69 | 337.38 | 5/3 | 2 | 0.16 | −0.08 | 0.15 | 0.17 | −0.04 | 0.22 |
| No | GIA | BBB | P-gpS | CYP | Lipinski | Ghose | Veber | Egan | Muegge | PAINS | Brenk | LL | SA | LogKp | BA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | H | Y | Y | Y,Y,Y,Y,Y | Y | Y | Y | Y | Y | 1 | 0 | N | 3.69 | −5.79 | 0.55 |
| 2 | H | N | Y | Y,Y,Y,Y,Y | Y | Y | Y | Y | Y | 1 | 0 | N | 3.58 | −5.68 | 0.55 |
| 3 | H | N | N | N,N,Y,Y,Y | Y | Y | Y | Y | Y | 1 | 0 | N | 3.72 | −5.89 | 0.55 |
| 4 | H | Y | Y | N,N,N,Y,Y | Y | Y | Y | Y | Y | 1 | 0 | N | 3.73 | −6.00 | 0.55 |
| 5 | H | N | Y | N,N,N,Y,Y | Y | Y | Y | Y | Y | 1 | 0 | N | 3.81 | −6.25 | 0.55 |
| 6 | H | Y | Y | N,N,N,Y,Y | Y | Y | Y | Y | Y | 1 | 0 | N | 3.95 | −6.24 | 0.55 |
| 7 | H | Y | Y | N,N,N,Y,Y | Y | Y | Y | Y | Y | 1 | 0 | N | 3.83 | −6.00 | 0.55 |
| 8 | H | Y | Y | N,Y,N,Y,Y | Y | Y | Y | Y | Y | 1 | 0 | N | 3.84 | −6.49 | 0.55 |
| 9 | H | N | Y | N,N,N,Y,N | Y | Y | Y | Y | Y | 1 | 0 | N | 3.73 | −6.25 | 0.55 |
| No | MUT | TUM | IRR | REP | DL | DS |
|---|---|---|---|---|---|---|
| 1 | ++ | ++ | ++ | ++ | 3.52 | 0.70 |
| 2 | ++ | ++ | ++ | ++ | 0.52 | 0.63 |
| 3 | ++ | ++ | ++ | ++ | 3.68 | 0.79 |
| 4 | ++ | ++ | ++ | ++ | 4.12 | 0.79 |
| 5 | ++ | ++ | ++ | ++ | 4.06 | 0.81 |
| 6 | ++ | ++ | ++ | ++ | 3.57 | 0.71 |
| 7 | - | ++ | ++ | ++ | 3.52 | 0.46 |
| 8 | ++ | ++ | ++ | ++ | 0.61 | 0.59 |
| 9 | - | ++ | ++ | ++ | 0.52 | 0.38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gašparová, R.; Kabaňová, N. In Silico Pharmacological Prediction of Capitavine, Buchenavianine and Related Flavonoid Alkaloids. Chem. Proc. 2024, 16, 55. https://doi.org/10.3390/ecsoc-28-20222
Gašparová R, Kabaňová N. In Silico Pharmacological Prediction of Capitavine, Buchenavianine and Related Flavonoid Alkaloids. Chemistry Proceedings. 2024; 16(1):55. https://doi.org/10.3390/ecsoc-28-20222
Chicago/Turabian StyleGašparová, Renata, and Natália Kabaňová. 2024. "In Silico Pharmacological Prediction of Capitavine, Buchenavianine and Related Flavonoid Alkaloids" Chemistry Proceedings 16, no. 1: 55. https://doi.org/10.3390/ecsoc-28-20222
APA StyleGašparová, R., & Kabaňová, N. (2024). In Silico Pharmacological Prediction of Capitavine, Buchenavianine and Related Flavonoid Alkaloids. Chemistry Proceedings, 16(1), 55. https://doi.org/10.3390/ecsoc-28-20222