2. Materials and Methods
Monitoring the progress of caution and determining the composition of basic mixtures and the individual types of isolated products and their identification were carried out using TLC methods, including 1H NMR spectroscopy. TLC analysis was conducted on Silufol UV-254 plates, eluting ent ethyl acetate/hexane/chloroform (2: 2: 1) (developer—iodine vapor). Chromatographic column/sorbent –silica gel 60, eluent –ethyl acetate/hexane/chloroform (2:2:1). FT-IR spectrum was recorded on a Nicolet 6700 spectrometer (Thermo Scientific, Waltham, MA, USA) using KBr pellet (wavenumber range of 4000–400 cm−1) with a spectral resolution of 4 cm-1. The absorption spectra of the solutions under study were recorded on a SHIMADZU-1800 spectrophotometer in 1 cm cuvettes with a scanning step of 1 nm. Solvents and reagents were of special grade. Working solutions were prepared by dissolving exactly weighed samples of the compounds (C = 5·10−5 M) in an appropriate solvent. A Varian 400 spectrometer was used to record 1H NMR, 13C, and HSQC spectra at 20–25 °C. The operating frequency for the 1H NMR spectrum was 400 MHz, and 100 MHz for the 13C NMR spectrum. The internal standard was TMS, and the solvents were CDCl3, dimethyl sulfoxide-d6.
2.1. Synthesis of 6-(4-methoxyphenyl)-3,9-dimethyl-12-phenyl-1H-dipyrano[4,3-d:3′,4′-g][1,3,2]dioxaphosphocine-1,11(12H)-dione-6-sulfide (2a), and 3,7-dimethyl-10-phenyl-1H-thiopyrano [3,2-c:5,6-c’]dipyran-1,9(10H)-dione (4a)
A mixture of 6-(4-methoxyphenyl)-3,9-dimethyl-12-phenyl-1H-dipyrano[4,3-d:3′,4′-g][1,3,2]dioxaphosphocine-1,11(12H)-dione-6-sulfide (2a) and 3,7-dimethyl-10-phenyl-1H-thiopyrano[3,2-c:5,6-c’]dipyran-1,9(10H)-dione (4a) was synthesized by the reaction of phenylbis(4-hydroxy-6-methyl-2-oxo-2H-pyran-3-yl)methane (1a) and 0.12 g (0.29 mmol) of Lawesson’s reagent with stirring in toluene for 6 h at 120 °C. The yield was dioxaphosphocinedionesulfide 2a—0.014 g (12%), thiopyran 4a—0.036 g (36.7%) m.p. 196–197 °C; FTIR, cm−1 3522, 3407, 3058, 3032, 2923, 1716, 1601, 1547, 1518, 1452, 1347, 1307, 1262, 1222, 1230, 1172, 1112, 838, 798 753, 688, 619, 519. 1H NMR (400 MHz, CDCl3, δ): 2.06 (s, 3Н, СН3), 2.21 (s, 18Н, СН3), 2.33 (s, 3Н, СН3), 3.84 (s, 3Н, ОСН3), 6.08 (s, 1Н, СН), 6.27 (s, 2H, CHvinyl), 5.50 (s, 3Н, СН), 5.92 (s, 6H, CHvinyl), 6.98–7.80 (m, 24Н, Ar).
2.2. Synthesis of 3,7-dimethyl-10-(3-nitrophenyl)-1H-dipyrano[4,3-b:3′,4′-e]pyran-1,9(10H)-dione (3b)
We obtained 3,7-Dimethyl-10-(3-nitrophenyl)-1H-dipyrano[4,3-b:3′,4′-e]pyran-1,9(10H)-dione (3b) in a similar manner using 0.10 g (0.26 mmol) of nitrophenylbis(4-hydroxy-6-methyl-2-oxo-2H-pyran-3-yl)methane (1b), and 0.10 g (0.26 mmol) of Lawesson’s reagent in 10 mL of toluene. The yield was 0.04 g (50%) m.p. 251–252 °C; FTIR, cm−1 3550, 3420, 3032, 2953, 1720, 1608, 1525, 1518, 1452, 1347, 1307, 1262, 1222, 1172, 1112, 838, 798 753, 688, 619, 519. 1H NMR (400 MHz, CDCl3, δ): 2.26 (s, 6Н, СН3), 6.27 (s, 1Н, СН), 5.96 (s, 2H, CHvinyl), 6.85–8.26 (m, 4Н, Ar).
2.3. Synthesis of 3,7-dimethyl-10-(4-chlorophenyl)-1H-dipyrano[4,3-b:3′,4′-e]pyran-1,9(10H)-dione (3c) and 3,7-dimethyl-10-(4-chlorophenyl)-1H-thiopyrano[3,2-c:5,6-c’]dipyran-1,9(10H)-dione (4c)
The mixture of 3,7-dimethyl-10-(4-chlorophenyl)-1H-dipyrano[4,3-b:3′,4′-e]pyran-1,9(10H)-dione (3c) and 3,7-dimethyl-10-(4-chlorophenyl)-1H-thiopyrano[3,2-c:5,6-c’]dipyran-1,9(10H)-dione (4c) was obtained in a similar manner using 0.11 g (0.27 mmol) of chlorophenylbis(4-hydroxy-6-methyl-2-oxo-2H-pyran-3-yl)methane (1c), and 0.11 g (0.27 mmol) of Lawesson’s reagent in 10 mL of toluene. The yield was pyran 3c—0.026 g (27%), thiopyran 4c—0.014 g (14%) m.p. 198–200 °C; FTIR, cm−1 3589, 3398, 3103, 3042, 1725, 1610, 1520, 1498, 1409, 1378, 1305, 1287, 1220, 1189, 1110, 958, 878 773, 675, 587. 1H NMR (400 MHz, CDCl3, δ):2.21–2.38 (s, 18Н, СН3), 5.70 (s, 1Н, СН), 5.99–6.07 (m, 4H, CHvinyl), 5.92 (s, 2Н, СНvinyl), 6.21 (m, 1H, CH), 6.88–7.56 (m, 12Н, Ar).
2.4. Decomposition of UV–Vis Spectra
The computer program of the MILCA algorithm and examples of solving practical tasks are available as independent executables with a MATLAB interface (MathWorks, Natick, MA, USA).
The essence of automodel curve splitting is in the decomposition of the M × N spectrum matrix X of a multicomponent system (N is the number of records by wavelength, M is the number of spectra of the mixture) into the K × N spectrum matrix S of the individual components and the M × K matrix of their relative concentrations A (K being the number of components in the system) (Equation (1)).
3. Results and Discussion
It is known that the interaction with Lawesson’s reagent can lead to a variety of products depending on the reaction conditions and the structure of the starting substrates. The direction of the transformation is determined, in particular, by the tautomeric form that is stabilized under the specific reaction conditions. Compounds containing a (benzo)pyranone fragment with an oxo function at the C-4 position and a mobile proton at C-3 tend to enolize, and the quantitative ratio of tautomers is determined by the polarity of the medium.
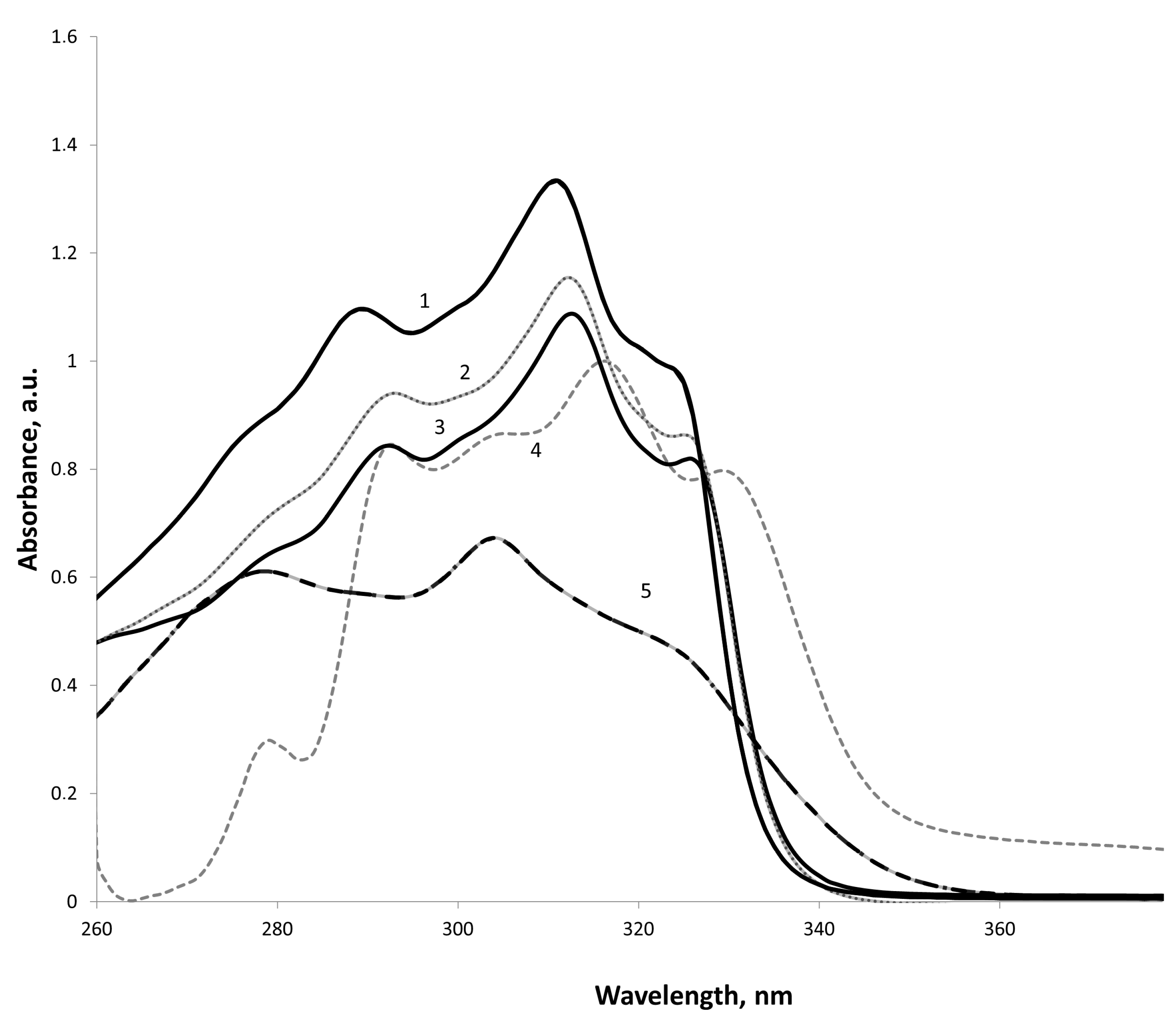
Three intense bands are observed in the UV spectra of the bis-substrate (
Figure 1). The bands with λ
мах 254–293 nm (lg ε 4.00–4.33) and λ
мах 304–313 nm (lg ε 3.94–4.42) correspond to symmetry-forbidden π-π* transitions of aromatic rings. The presence of a lactonic fragment results in a bathochromic shift and essential strengthening of the latter one, which obtains one of the most intense bands of the ketonic form.
The range of λмах 325–326 nm (lg ε 4.20–4.29) corresponding to the π-π* and n-π* transitions of the enecarbonyl fragment is most characteristic of the enolic form.
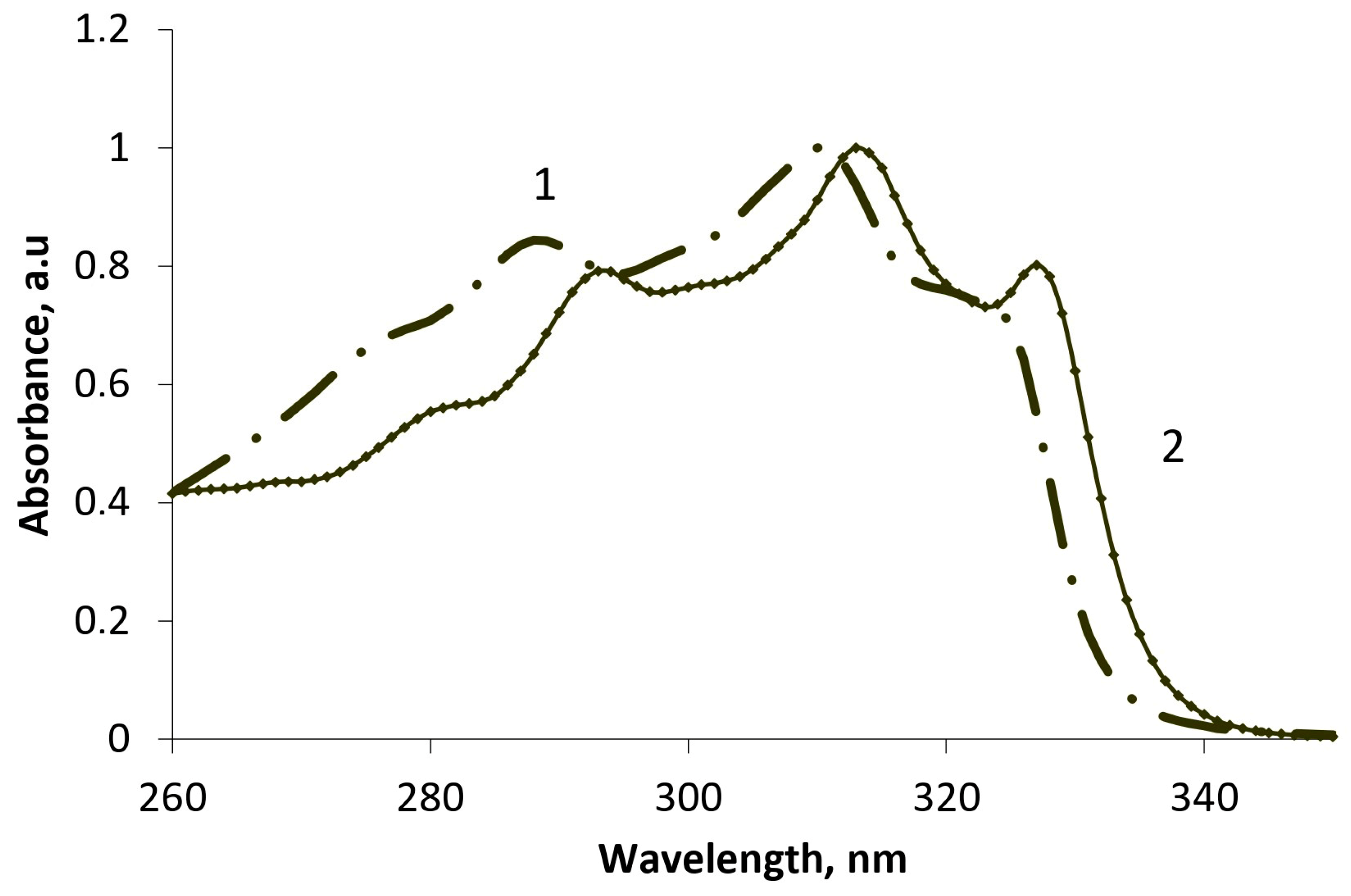
In benzannulated analogs, we have shown a trend of influence of the nature (donor or acceptor) and position of the substituent at the C-3 of the benzopyranone ring. Using a chemometric method (independent component analysis) implemented in the MILCA algorithm [
2], we estimated the quantitative ratio of components in the system and isolated the spectral contours of individual components (
Figure 2), in particular, the ketone and enol forms in solvents of different polarity.
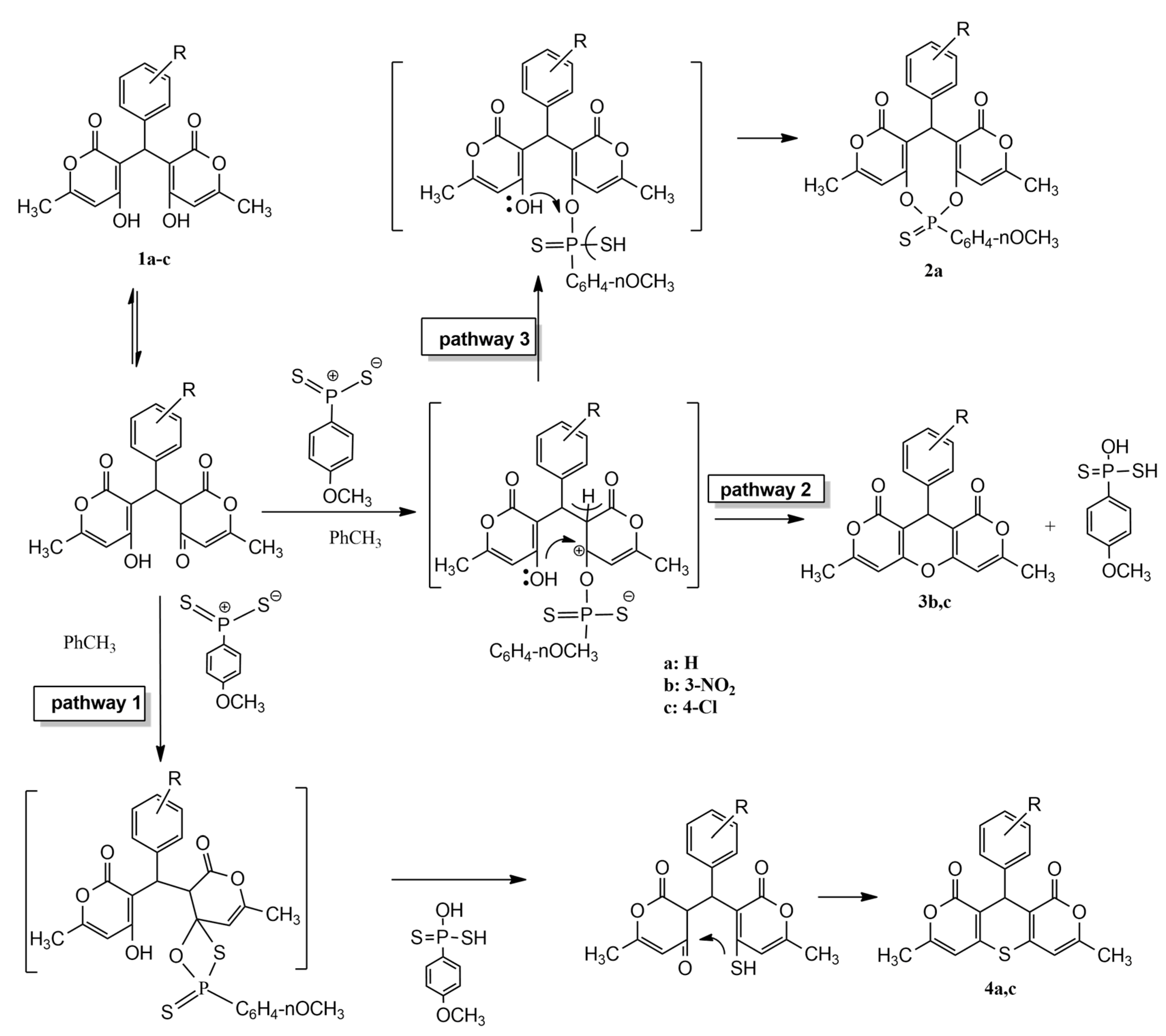
According to the classical scheme of the interaction of Lawesson’s reagent with the studied substrate, the reaction can proceed via a mechanism of thionation of the keto group at C-4 through a four-membered cyclic transition state followed by S-heterocyclization (
Scheme 1) (pathway 1). Probably due to the reaction being carried out in non-polar toluene (the solvent choice was based on the solubility of the substrate and reagent), the process culminates in intramolecular heterocyclization rather than stopping at the stage of thione formation. It is also known that Lawesson’s reagent, acting as a Lewis acid, activates the oxo function and promotes the ketalization of the substrate, resulting in intramolecular O-heterocyclization (pathway 2). A competing process in the latter case is the intramolecular nucleophilic attack of the oxygen of the enol hydroxyl on the phosphorus atom, which, due to its greater affinity for oxygen, ultimately forms a phosphorus-sulfur-organic compound (pathway 3). The yields of all types of the obtained products are presented in
Table 1.
Based on the obtained results of decomposition using the MILCA algorithm for the 3-nitro- and 4-chlorophenyl derivatives, the dynamic equilibrium is shifted towards the enol form (
Table 2), which explains their tendency towards O-heterocyclization. Moreover, the stronger the electron-withdrawing effect, the higher the yield of the 4H-pyranobispyranone system. For the unsubstituted substrate, the proportion of the ketone form is higher, resulting in a predominance of products of nucleophilic attack on the carbonyl function in the reaction mixture.




{kind=link}
{kind=link}
{kind=link}