
Anti-Helicobacter Pylori Activity of Phytochemicals from Artocarpus spp.: In Silico Analysis †

Abstract
1. Introduction
2. Materials and Methods
2.1. Molecular Docking Simulations
2.2. In Silico Drug-Likeness and ADMET Analysis
2.3. Boiled Egg Model Analysis
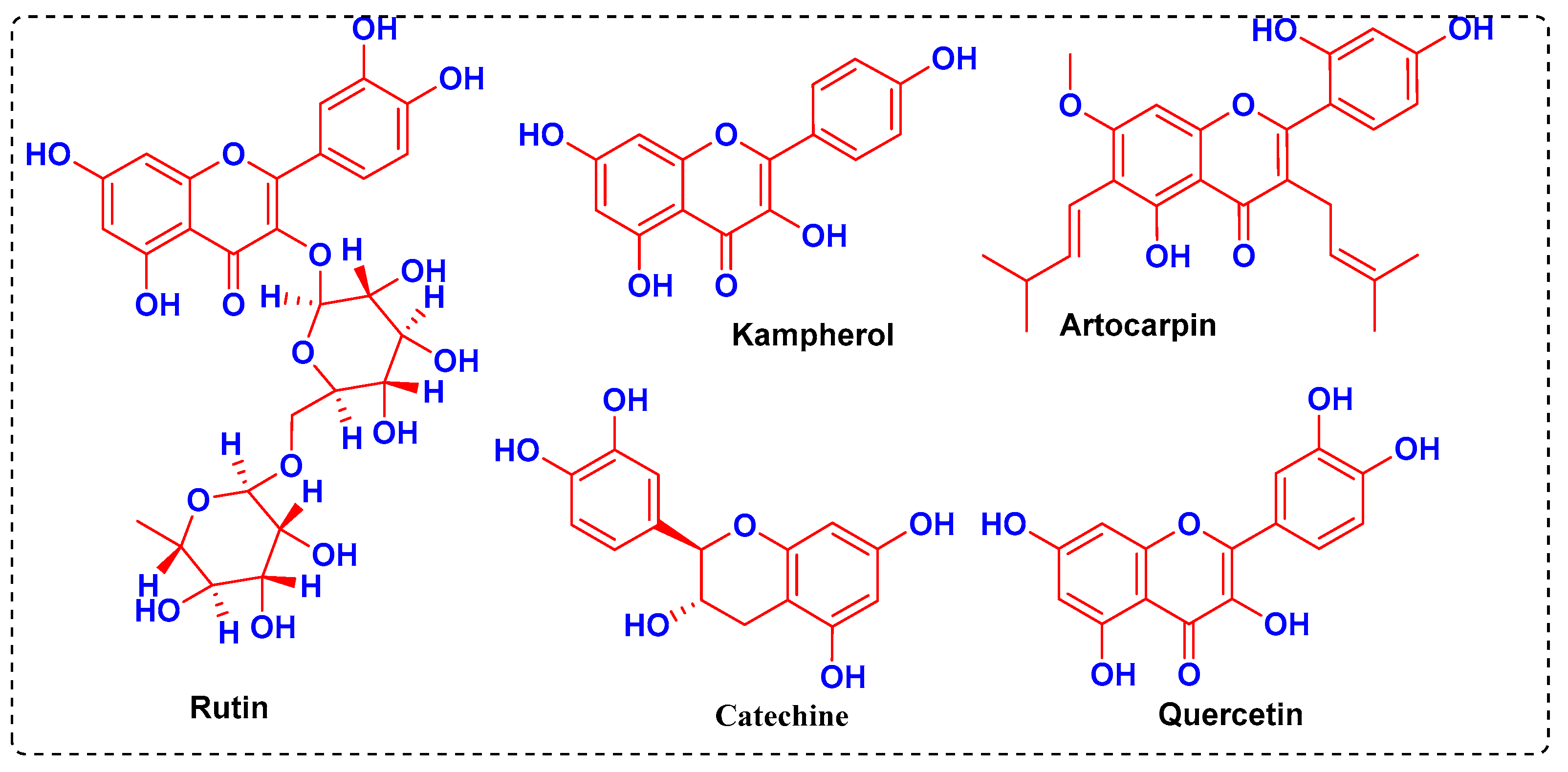
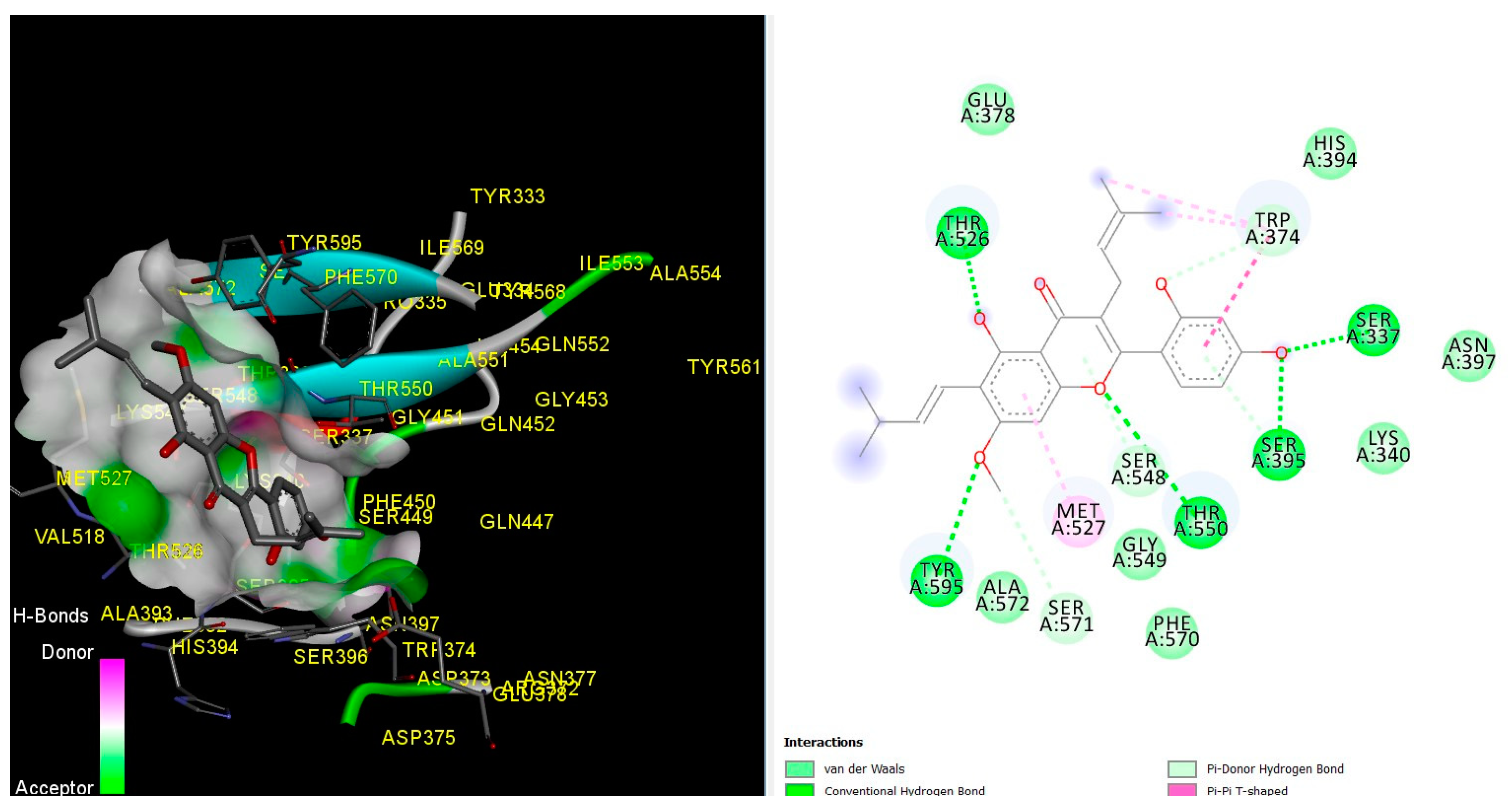
3. Results and Discussion
3.1. Molecular Docking Simulations
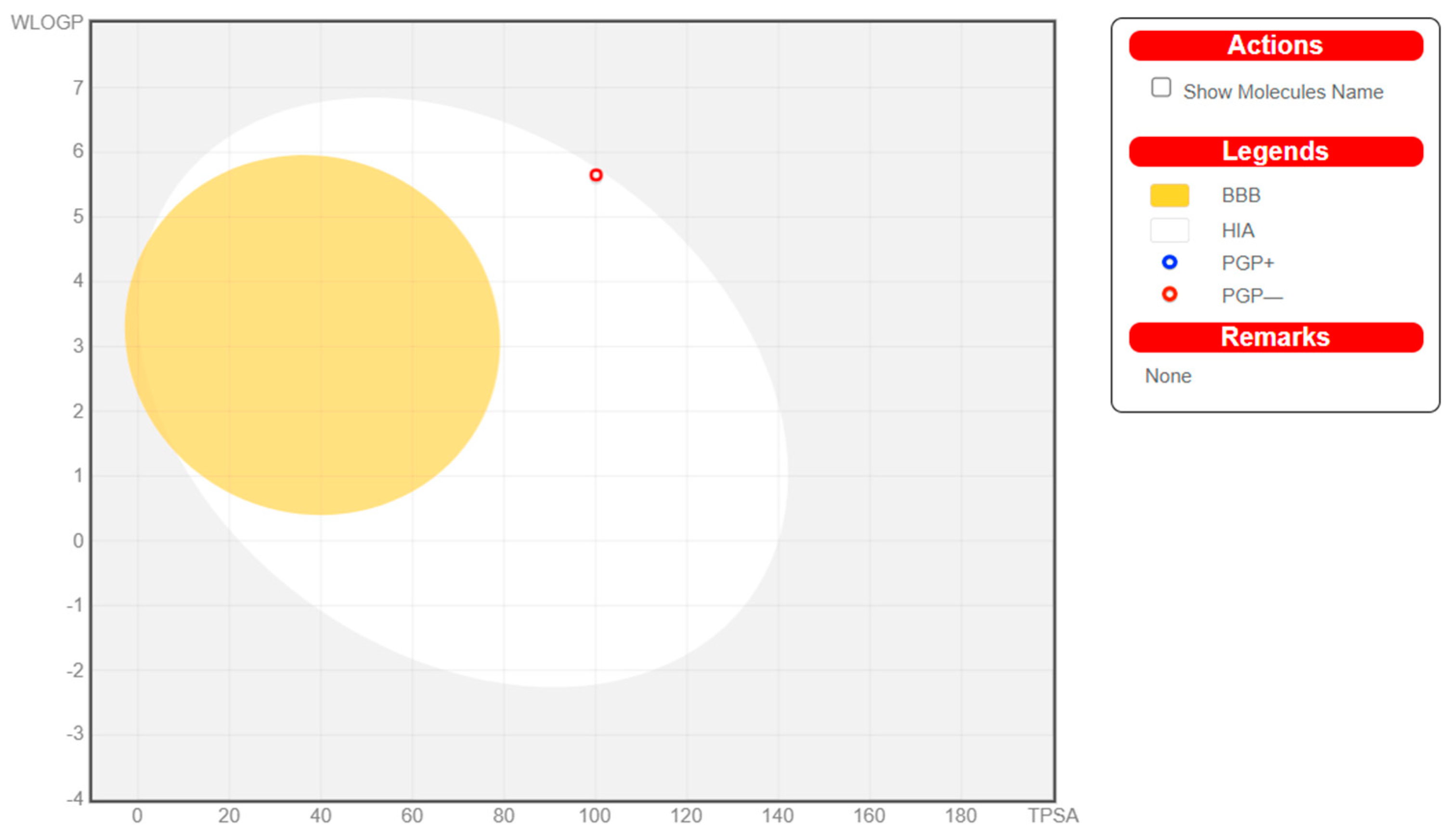
3.2. In Silico ADMET Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yadav, S.; Pandey, A.; Mali, S.N. From Lab to Nature: Recent Advancements in the Journey of Gastroprotective Agents from Medicinal Chemistry to Phytotherapy. Eur. J. Med. Chem. 2024, 272, 116436. [Google Scholar] [CrossRef]
- Sugano, K.; Tack, J.; Kuipers, E.J.; Graham, D.Y.; El-Omar, E.M.; Miura, S.; Haruma, K.; Asaka, M.; Uemura, N.; Malfertheiner, P. Kyoto global consensus report on Helicobacter pylori gastritis. Gut 2015, 64, 1353–1367. [Google Scholar] [CrossRef]
- Dadgostar, P. Antimicrobial resistance: Implications and costs. Infect. Drug Resist. 2019, 12, 3903–3910. [Google Scholar] [CrossRef]
- Guerra-Valle, M.; Orellana-Palma, P.; Petzold, G. Plant-based polyphenols: Anti-Helicobacter pylori effect and improvement of gut microbiota. Antioxidants 2022, 11, 109. [Google Scholar] [CrossRef]
- Gerrits, M.M.; Schuijffel, D.; Van Zwet, A.A.; Kuipers, E.J.; Vandenbroucke-Grauls, C.M.J.E.; Kusters, J.G. Alterations in penicillin-binding protein 1A confer resistance to β-lactam antibiotics in Helicobacter pylori. Antimicrob. Agents Chemother. 2002, 46, 2229–2233. [Google Scholar] [CrossRef]
- Hsu, K.C.; Chen, Y.F.; Lin, S.R.; Yang, J.M. iGEMDOCK: A graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinform. 2011, 12, S33. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Mishra, V.R.; Ghanavatkar, C.W.; Mali, S.N.; Qureshi, S.I.; Chaudhari, H.K.; Sekar, N. Design, synthesis, antimicrobial activity and computational studies of novel azo linked substituted benzimidazole, benzoxazole and benzothiazole derivatives. Comput. Biol. Chem. 2019, 78, 330–337. [Google Scholar] [CrossRef]
- Kshatriya, R.; Kambale, D.; Mali, S.; Jejurkar, V.P.; Lokhande, P.; Chaudhari, H.K.; Saha, S.S. Brønsted acid catalyzed domino synthesis of functionalized 4H-chromens and their ADMET, molecular docking and antibacterial studies. ChemistrySelect 2019, 4, 7943–7948. [Google Scholar] [CrossRef]
- Jadhav, B.S.; Yamgar, R.S.; Kenny, R.S.; Mali, S.N.; Chaudhari, H.K.; Mandewale, M.C. Synthesis, In silico and Biological Studies of Thiazolyl-2h-chromen-2-one Derivatives as Potent Antitubercular Agents. Curr. Comput.-Aided Drug Des. 2020, 16, 511–522. [Google Scholar] [CrossRef]
- Desale, V.J.; Mali, S.N.; Chaudhari, H.K.; Mali, M.C.; Thorat, B.R.; Yamgar, R.S. Synthesis and Anti-mycobacterium Study on Halo-substituted 2-aryl oxyacetohydrazones. Curr. Comput.-Aided Drug Des. 2020, 16, 618–628. [Google Scholar] [CrossRef]
- Shelke, P.B.; Mali, S.N.; Chaudhari, H.K.; Pratap, A.P. Chitosan hydrochloride mediated efficient, green catalysis for the synthesis of perimidine derivatives. J. Heterocycl. Chem. 2019, 56, 3048–3054. [Google Scholar] [CrossRef]
- Thorat, B.R.; Rani, D.; Yamgar, R.S.; Mali, S.N. Synthesis, Spectroscopic, In-vitro and Computational Analysis of Hydrazones as Potential Antituberculosis Agents:(Part-I). Comb. Chem. High Throughput Screen. 2020, 23, 392–401. [Google Scholar] [CrossRef]
- Thorat, B.R.; Mali, S.N.; Rani, D.; Yamgar, R.S. Synthesis, In silico and In vitro Analysis of Hydrazones as Potential Antituberculosis Agents. Curr. Comput.-Aided Drug Des. 2021, 17, 294–306. [Google Scholar] [CrossRef]
- The Protein Database Bank, PDB Database. Available online: www.rcsb.org (accessed on 10 October 2021).
- Mali, S.N.; Pandey, A. Balanced QSAR and Molecular Modeling to Identify Structural Requirements of Imidazopyridine Analogues as Anti-infective Agents against Trypanosomiases. J. Comput. Biophys. Chem. 2021, 21, 83–114. [Google Scholar] [CrossRef]
- Mishra, V.R.; Ghanavatkar, C.W.; Mali, S.N.; Chaudhari, H.K.; Sekar, N. Schiff base clubbed benzothiazole: Synthesis, potent antimicrobial and MCF-7 anticancer activity, DNA cleavage and computational study. J. Biomol. Struct. Dyn. 2019, 38, 1772–1785. [Google Scholar] [CrossRef]
- Jejurkar, V.P.; Mali, S.N.; Kshatriya, R.; Chaudhari, H.K.; Saha, S. Synthesis, antimicrobial screening and in silico appraisal of iminocarbazole derivatives. ChemistrySelect 2019, 4, 9470–9475. [Google Scholar] [CrossRef]
- Anuse, D.G.; Mali, S.N.; Thorat, B.R.; Yamgar, R.S.; Chaudhari, H.K. Synthesis, SAR, in silico appraisal and anti-microbial study of substituted 2-aminobenzothiazoles derivatives. Curr. Comput.-Aided Drug Des. 2020, 16, 802–813. [Google Scholar] [CrossRef]
- Kapale, S.S.; Mali, S.N.; Chaudhari, H.K. Molecular modelling studies for 4-oxo-1,4-dihydroquinoline-3-carboxamide derivatives as anticancer agents. Med. Drug Discov. 2019, 2, 100008. [Google Scholar] [CrossRef]
- Desale, V.J.; Mali, S.N.; Thorat, B.R.; Yamgar, R.S. Synthesis, admetSAR Predictions, DPPH Radical Scavenging Activity, and Potent Anti-mycobacterial Studies of Hydrazones of Substituted 4-(anilino methyl) benzohydrazides (Part 2). Curr. Comput.-Aided Drug Des. 2021, 17, 493–503. [Google Scholar] [CrossRef]
- Kshatriya, R.; Shelke, P.; Mali, S.; Yashwantrao, G.; Pratap, A.; Saha, S. Synthesis and evaluation of anticancer activity of pyrazolone appended triarylmethanes (TRAMs). ChemistrySelect 2021, 6, 6230–6239. [Google Scholar] [CrossRef]
- Mali, S.N.; Thorat, B.R.; Gupta, D.R.; Pandey, A. Mini-Review of the Importance of Hydrazides and Their Derivatives—Synthesis and Biological Activity. Eng. Proc. 2021, 11, 21. [Google Scholar] [CrossRef]
- Nagre, D.T.; Mali, S.N.; Thorat, B.R.; Thorat, S.A.; Chopade, A.R.; Farooqui, M.; Agrawal, B. Synthesis, In-Silico Potential Enzymatic Target Predictions, Pharmacokinetics, Toxicity, Anti-Microbial and Anti-Inflammatory Studies of Bis-(2-Methylindolyl) Methane Derivatives. Curr. Enzym. Inhib. 2021, 17, 127–143. [Google Scholar] [CrossRef]
- Ghosh, S.; Mali, S.N.; Bhowmick, D.N.; Pratap, A.P. Neem oil as natural pesticide: Pseudo ternary diagram and computational study. J. Indian Chem. Soc. 2021, 98, 100088. [Google Scholar] [CrossRef]
- Mali, S.N.; Tambe, S.; Pratap, A.P.; Cruz, J.N. Molecular Modeling Approaches to Investigate Essential Oils (Volatile Compounds) Interacting with Molecular Targets. In Essential Oils; Santana de Oliveira, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2022; Volume 1, pp. 417–442. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules | -iGemDock Interaction Energy | Molecules | -iGemDock Interaction Energy |
|---|---|---|---|
| Quercetin | −111.11 | Artonin A | −117.1 |
| Ascorbates | −84.29 | Artocarpanone | −98.62 |
| Catechine | −94.99 | Caffeic acid | −78.51 |
| Lupeol acetate | −90.55 | Cycloheterophyllin | −125.23 |
| Bita -sitosterol | −97.62 | Cyclocommunol | −106.54 |
| Kaempferol | −107.95 | Isobavachalcone | −121.11 |
| Gallic acid | −90.35 | Artonin E | −128.21 |
| Engeletin 5 | −134.89 | Heterophyllin | −131.91 |
| Oxyresveratrol | −72.08 | Cyclomorusin | −120.05 |
| Artocarpin | −48.24 | Cryptoxanthin | −92.1799 |
| Cycloartocarpin | −111.69 | Myricetin | −82.5936 |
| Cudraflavone | −111.35 | Ascorbic acid | −85.54 |
| Vanillic acid | −79.88 | Cinnamic acid | −69.96 |
| Isorhamnetin | −113.3 | Ferulic acid | −79.6 |
| Psoralenoside | -107.8 | Betulinic acid | −95.9 |
| Epigallocatechin Gallate | −140 | Resorcinol | −59.9 |
| Morin | −106.1 | Norartocarpetin | −105 |
| Ursolic acid | −99.6 | Pyrogallol | −69.1 |
| Artocarpesin | −112.9 | Rutin | −148.07 |
| Albanin A | −107.9 | Arbutin | −98.2 |
| Engeletin | −116.79 | Diethyl phthalate | −83.86 |
| α-amyrin acetate | −92.9 | β-amyrin acetate | −95.27 |
| Cycloartenol | −92.66 | ||
| Amoxicillin * | −109.20 |
| Sr. No. | Molecules | Residues with Contribution Energy |
|---|---|---|
| 1. | Amoxicillin | THR 526, TRP 374, Ser 337, Ser 395, Thr 550, Met 527, and Tyr 595 |
| 2. | Artocarpin (Best docked) | THR 526, TRP 374, Ser 337, Ser 395, Thr 550, Met 527, and Tyr 595 |
| Properties | Artocarpin * | Rutin | Engeletin 5 |
|---|---|---|---|
| GI absorption | High | High | High |
| BBB permeant | Low | Low | Low |
| P-gp substrate | NO | NO | NO |
| CYP1A2 inhibitor | NO | NO | NO |
| CYP2C19 inhibitor | Yes | Yes | Yes |
| CYP2C9 inhibitor | NO | NO | NO |
| CYP2D6 inhibitor | NO | NO | NO |
| CYP3A4 inhibitor | NO | NO | NO |
| Lipinski | Yes | Yes | Yes |
| Ghose | NO | NO | NO |
| Veber | Yes | Yes | Yes |
| Egan | Yes | Yes | Yes |
| Blood–Brain Barrier | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yadav, S.; Pandey, A. Anti-Helicobacter Pylori Activity of Phytochemicals from Artocarpus spp.: In Silico Analysis. Chem. Proc. 2024, 16, 17. https://doi.org/10.3390/ecsoc-28-20200
Yadav S, Pandey A. Anti-Helicobacter Pylori Activity of Phytochemicals from Artocarpus spp.: In Silico Analysis. Chemistry Proceedings. 2024; 16(1):17. https://doi.org/10.3390/ecsoc-28-20200
Chicago/Turabian StyleYadav, Susmita, and Anima Pandey. 2024. "Anti-Helicobacter Pylori Activity of Phytochemicals from Artocarpus spp.: In Silico Analysis" Chemistry Proceedings 16, no. 1: 17. https://doi.org/10.3390/ecsoc-28-20200
APA StyleYadav, S., & Pandey, A. (2024). Anti-Helicobacter Pylori Activity of Phytochemicals from Artocarpus spp.: In Silico Analysis. Chemistry Proceedings, 16(1), 17. https://doi.org/10.3390/ecsoc-28-20200