
In Silico Assessment of Enaminone–Sulfanilamides as Potential Carbonic Anhydrase II Inhibitors: Molecular Docking and ADMET Prediction †


Abstract
1. Introduction
2. Materials and Methods
2.1. Molecular Docking
2.2. ADMET Prediction
3. Results and Discussion
3.1. Molecular Docking
3.2. ADMET Prediction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Occhipinti, R.; Boron, W.F. Role of Carbonic Anhydrases and Inhibitors in Acid-Base Physiology: Insights from Mathematical Modeling. Int. J. Mol. Sci. 2019, 20, 3841. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Roumeliotis, A.; Dounousi, E.; Eleftheriadis, T.; Liakopoulos, V. Biomarkers of vascular calcification in serum. Adv. Clin. Chem. 2020, 98, 91–147. [Google Scholar] [PubMed]
- Alsharidi, A.; Al-Hamed, M.; Alsuwaida, A. Carbonic anhydrase II deficiency: Report of a novel mutation. CEN Case Rep. 2016, 5, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Supuran, C.T.; Scozzafava, A. Sulfonamides and Their Isosters As Carbonic Anhydrase Inhibitors. Future Med. Chem. 2014, 6, 1149–1165. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.swissadme.ch/ (accessed on 2 September 2024).
- Available online: https://www.molsoft.com/ (accessed on 2 September 2024).
- Available online: https://tox-new.charite.de/protox_II/ (accessed on 2 September 2024).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
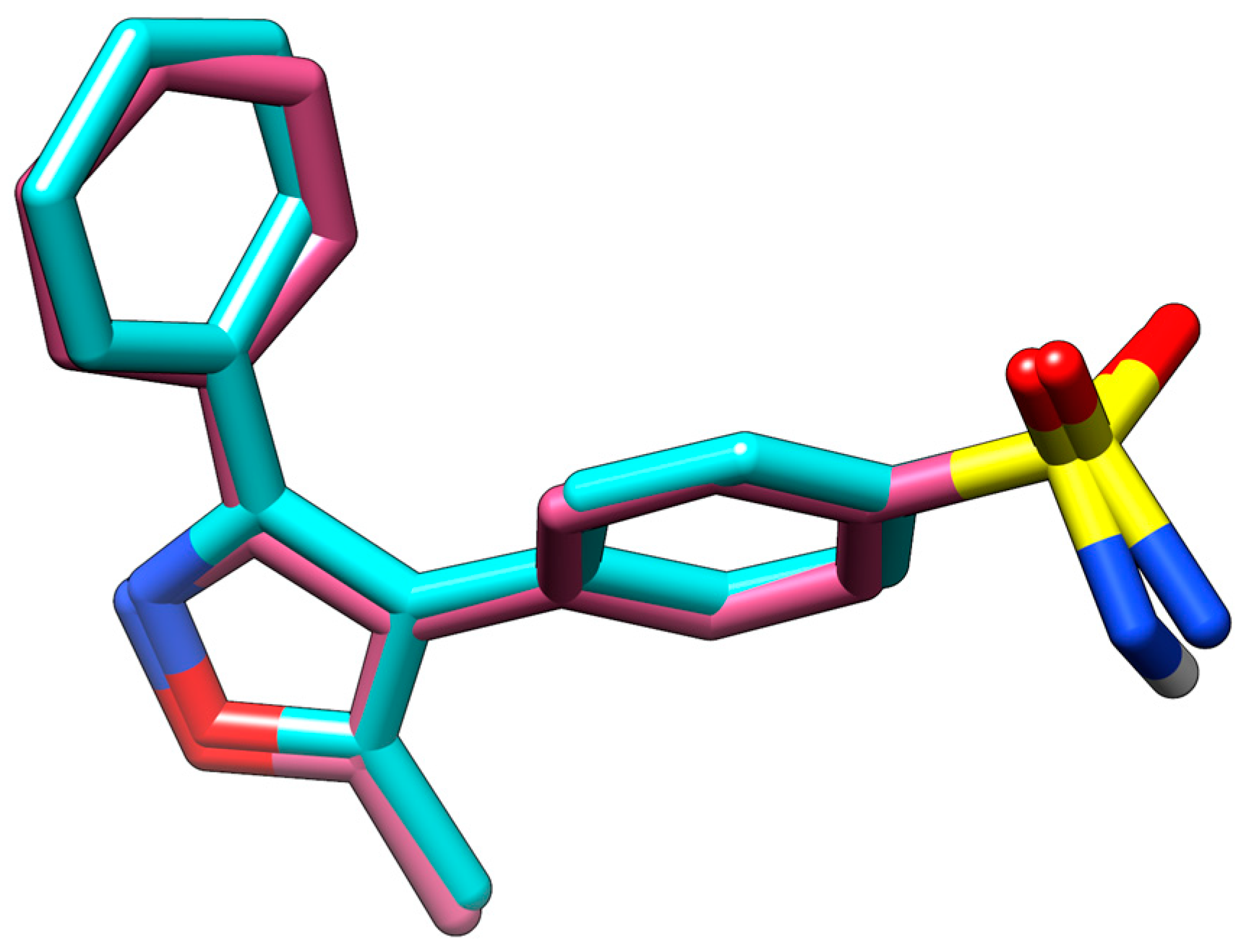


{kind=link}
{kind=link}
{kind=link}
{kind=link}
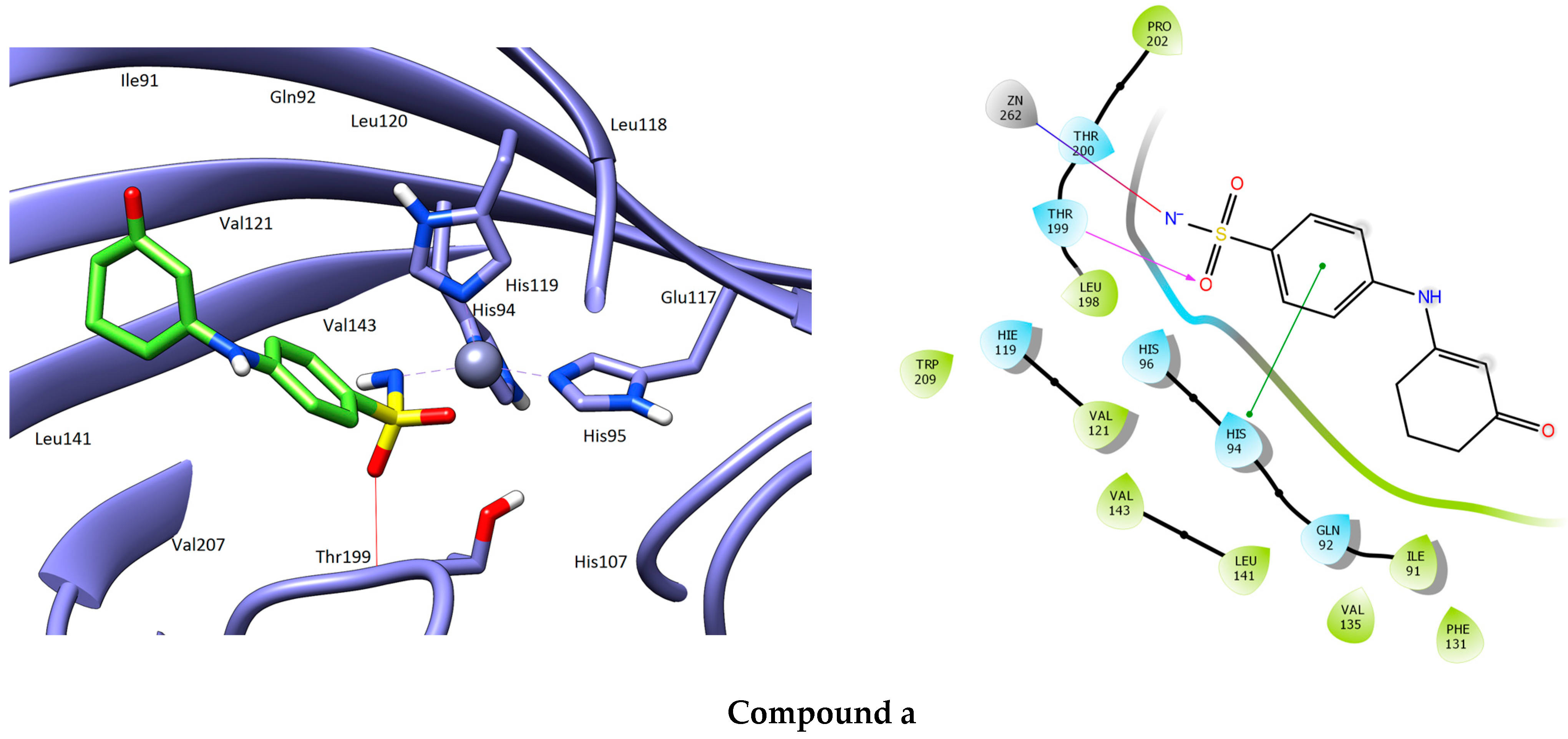
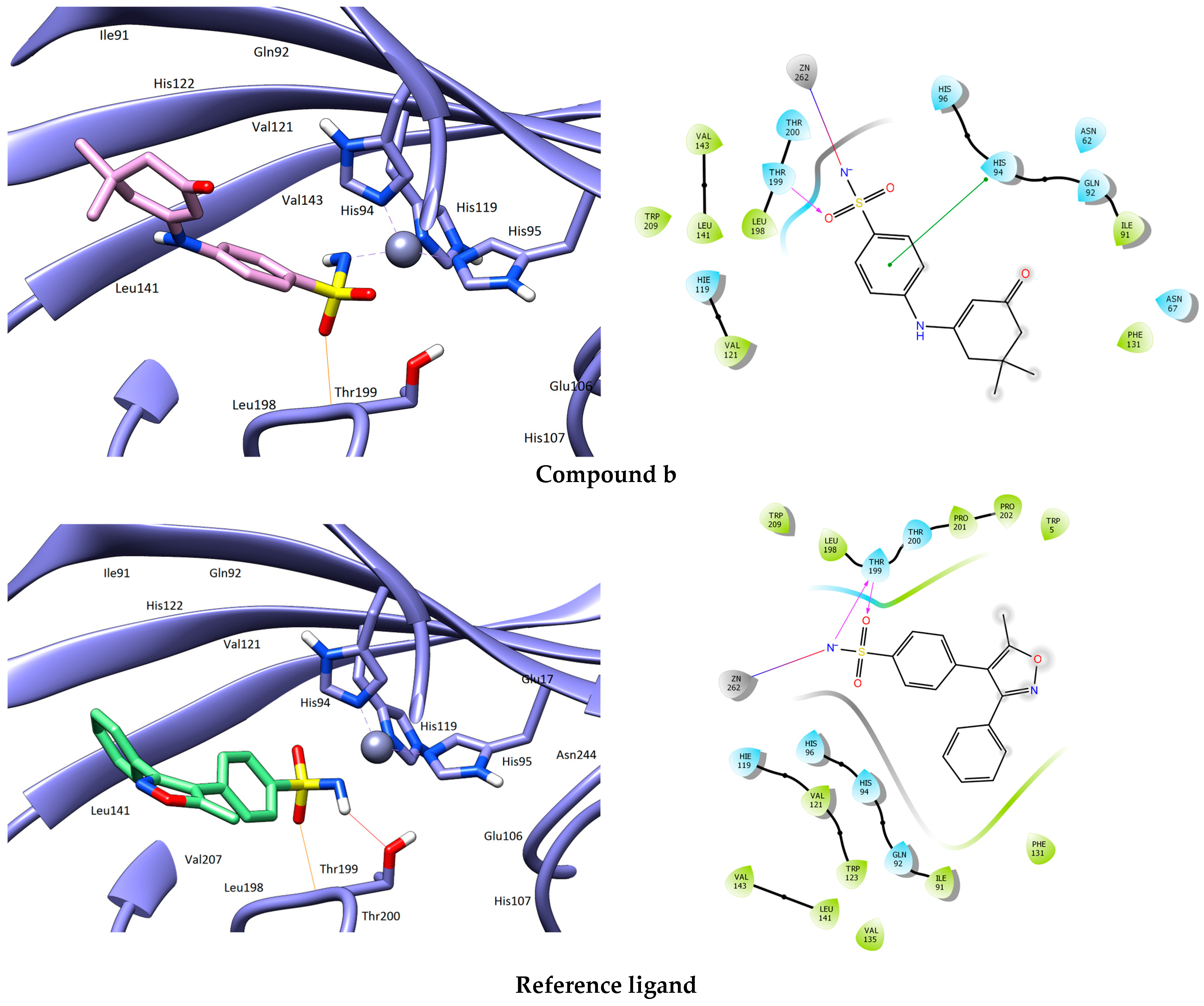
| Compound | Structure | H-Bonds | Hydrophobic Interactions | Docking Score (kcal.mol−1) |
|---|---|---|---|---|
| a | 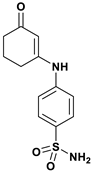 | Thr199 | Pro202, Leu198, Val121, Trp209, Val143, Leu141, Val135, Ile91, Phe131 | −8.099 |
| b | 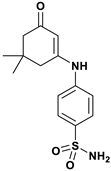 | Thr199 | Leu198, Val121, Trp209, Val143, Leu141, Ile91, Phe131 | −7.053 |
| Reference ligand | 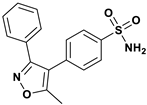 | Thr199 | Pro201, Pro202, Leu198, Val121, Trp209, Val143, Leu141, Val135, Ile91, Phe131, Trp5, Trp123 | −9.001 |
| Properties | Compound a | Compound b |
|---|---|---|
| Molecular weight (g per mole) | 266.32 | 294.37 |
| Rotatable bonds | 3 | 3 |
| H-bond donor | 2 | 2 |
| H-bond acceptor | 4 | 4 |
| Violations | 0 | 0 |
| Log Po/w iLOGP | 1.40 | 1.71 |
| Log S ESOL | −1.99 | −2.67 |
| GI | High | High |
| BBB | No | No |
| Log Kp (cm/s) | −7.41 | −7.00 |
| Bioavailability score | 0.55 | 0.55 |
| TPSA (Å2) | 97.64 | 97.64 |
| DLS | 0.01 | −0.07 |
| Predicted LD50 (mg/kg) | 5000 | 5000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouone, Y.O.; Bouzina, A.; Mansouri, R.; Aouf, N.-E. In Silico Assessment of Enaminone–Sulfanilamides as Potential Carbonic Anhydrase II Inhibitors: Molecular Docking and ADMET Prediction. Chem. Proc. 2024, 16, 117. https://doi.org/10.3390/ecsoc-28-20211
Bouone YO, Bouzina A, Mansouri R, Aouf N-E. In Silico Assessment of Enaminone–Sulfanilamides as Potential Carbonic Anhydrase II Inhibitors: Molecular Docking and ADMET Prediction. Chemistry Proceedings. 2024; 16(1):117. https://doi.org/10.3390/ecsoc-28-20211
Chicago/Turabian StyleBouone, Yousra Ouafa, Abdeslem Bouzina, Rachida Mansouri, and Nour-Eddine Aouf. 2024. "In Silico Assessment of Enaminone–Sulfanilamides as Potential Carbonic Anhydrase II Inhibitors: Molecular Docking and ADMET Prediction" Chemistry Proceedings 16, no. 1: 117. https://doi.org/10.3390/ecsoc-28-20211
APA StyleBouone, Y. O., Bouzina, A., Mansouri, R., & Aouf, N.-E. (2024). In Silico Assessment of Enaminone–Sulfanilamides as Potential Carbonic Anhydrase II Inhibitors: Molecular Docking and ADMET Prediction. Chemistry Proceedings, 16(1), 117. https://doi.org/10.3390/ecsoc-28-20211