
In Silico Study of FDA-Approved Drugs on Leishmania infantum CYP51, a Drug Repositioning Approach in Visceral Leishmaniasis †



Abstract
1. Introduction
2. Materials and Methods
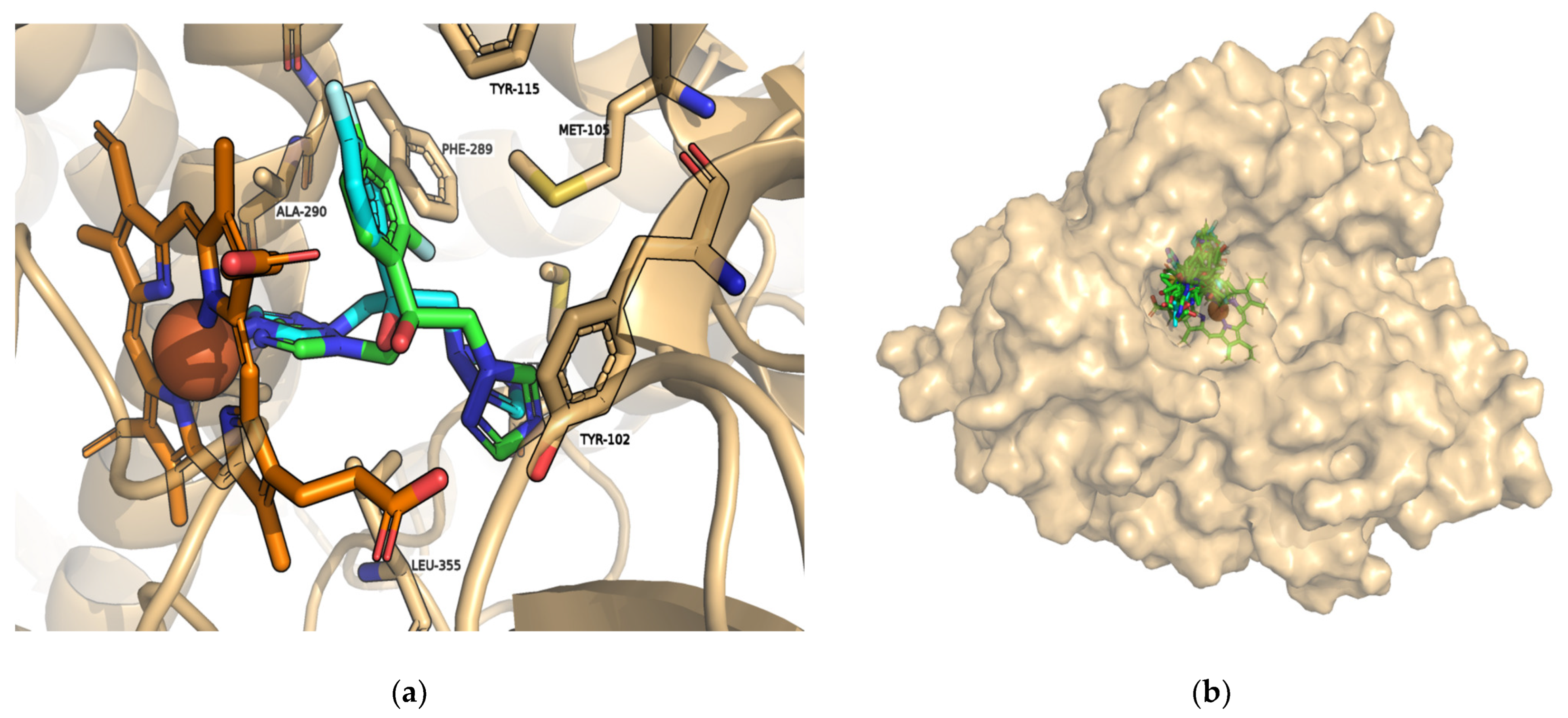
3. Results and Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef] [PubMed]
- Leite, J.C.; Gonçalves, A.A.M.; de Oliveira, D.S.; Resende, L.A.; Boas, D.F.V.; Ribeiro, H.S.; Pereira, D.F.; Da Silva, A.V.; Mariano, R.M.; Reis, P.C.; et al. Transmission-Blocking Vaccines for Canine Visceral Leishmaniasis: New Progress and Yet New Challenges. Vaccines 2023, 11, 1565. [Google Scholar] [CrossRef] [PubMed]
- Organización Panamericana de la Salud. Available online: https://iris.paho.org/handle/10665.2/59156 (accessed on 12 August 2024).
- Shmueli, M.; Ben-Shimol, S. Review of Leishmaniasis Treatment: Can We See the Forest through the Trees? Pharmacy 2024, 12, 30. [Google Scholar] [CrossRef] [PubMed]
- Khurana, G.; Rohilla, A.; Deep, A. Drug Development Process and Novel Drugs Approved by FDA for 2017–18. Appl. Clin. Res. Clin. Trials Regul. Aff. 2018, 5, 80–98. [Google Scholar] [CrossRef]
- Novel Drug Approvals at FDA. Available online: https://www.fda.gov/drugs/development-approval-process-drugs/novel-drug-approvals-fda (accessed on 24 May 2024).
- Lepesheva, G.I.; Friggeri, L.; Waterman, M.R. CYP51 as Drug Targets for Fungi and Protozoan Parasites: Past, Present and Future. Parasitology 2018, 145, 1820–1836. [Google Scholar] [CrossRef] [PubMed]
- Van Rhijn, N.; Bromley, M.; Richardson, M.; Bowyer, P. CYP51 Paralogue Structure Is Associated with Intrinsic Azole Resistance in Fungi. MBio 2021, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Wawrzak, Z.; Liu, J.; Nes, W.D.; Waterman, M.R.; Lepesheva, G.I. Substrate Preferences and Catalytic Parameters Determined by Structural Characteristics of Sterol 14α-Demethylase (CYP51) from Leishmania infantum. J. Biol. Chem. 2011, 286, 26838–26848. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 Update. Nucleic Acids Res. 2022, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High-Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED and HYBRID Docking Performance on Standardized Datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Kelley, B.P.; Brown, S.P.; Warren, G.L.; Muchmore, S.W. POSIT: Flexible Shape-Guided Docking for Pose Prediction. J. Chem. Inf. Model. 2015, 55, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef] [PubMed]
- Guarimata, J.D.; Alcívar, C.; Lavecchia, M.; Poveda, A. Molecular Docking for the Development of Alternative Therapies against Leishmaniasis. Chem. Proc. 2023, 14, 82. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 2.5.0 Schrödinger, LLC. Available online: https://storage.googleapis.com/pymol-storage/installers/index.html (accessed on 24 May 2024).
- BIOVIA Discovery Studio-Dassault Systèmes. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/ (accessed on 20 June 2024).
- Riebel, M.; Brunner, L.-M.; Nothdurfter, C.; Wein, S.; Schwarzbach, J.; Liere, P.; Schumacher, M.; Rupprecht, R. Neurosteroids and translocator protein 18 kDa (TSPO) ligands as novel treatment options in depression. In European Archives of Psychiatry and Clinical Neuroscience; Springer: Berlin/Heidelberg, Germany, 2024; Volume 2024, pp. 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Xu, H.; Ye, L.; Li, C.; Zhu, H.; Jiang, W.; Wang, W.; Yang, H.; Yang, Y.; Wang, Y.; et al. Synthesis and evaluation of neuroactive steroids with novel pharmacophore at C-21 let identify a compound with advantageous PK profile and higher EC(50) and E(max) as PAM on GABAA receptor. Eur. J. Med. Chem. 2024, 276, 116602. [Google Scholar] [CrossRef] [PubMed]
- Battipaglia, C.; Feliciello, L.; Genazzani, A.D.; Facchinetti, F.; Grandi, G. Combined Oral Contraceptive with Estetrol Plus Drospirenone: From Pharmacokinetics to Clinical Applications. Expert Opin. Drug Metab. Toxicol. 2023, 19, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Gérard, C.; Foidart, J.M. Estetrol: From Preclinical to Clinical Pharmacology and Advances in the Understanding of the Molecular Mechanism of Action. Drugs R D 2023, 23, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Tsagogiorgas, C.; Anger, F.; Beck, G.; Breedijk, A.; Yard, B.; Hoeger, S. Impact of Different Emulsifiers on Biocompatibility and Inflammatory Potential of Perfluorohexyloctane (F6H8) Emulsions for New Intravenous Drug Delivery Systems. Drug Des. Dev. Ther. 2019, 13, 2097–2110. [Google Scholar] [CrossRef] [PubMed]
- Habbe, K.J.; Frings, A.; Saad, A.; Geerling, G. The Influence of a Mineral Oil Cationic Nanoemulsion or Perfluorohexyloctane on the Tear Film Lipid Layer and Higher Order Aberrations. PLoS ONE 2023, 18, e0279977. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Molecule | FRED Chemgauss4 Score | Interactions with CYP51 |
|---|---|---|
| Sofpironium cation | −17.7642 | Y102, F104, M105, F109, Y115, P209, A286, F289, A290, T294, L355, M357, M459, heme group. |
| Perfluorohexyloctane | −16.2173 | Y102, F104, M105, F109, Y115, P209, V212, A286, F289, A290, T294, L355, M357, M359, M459, heme group. |
| Estetrol | −16.0491 | TYR102, M105, TYR115, F289, A290, T294, L355, M357, L358, M459, heme group. |
| Triclabendazole | −15.3390 | Y102, M105, F109, Y115, F289, A290, L355, V356, M357, M459, heme group. |
| Drospirenone | −15.1884 | Y102, F104, M105, Y115, A286, F289, A290, L355, M357, M359, M459, heme group. |
| Capivasertib | −15.1749 | Y102, F104, M105, F109, A114, Y115, L126, P209, A286, F289, A290, L355, V356, M357, M459, heme group. |
| Zuranolone | −15.1568 | Y102, F104, M105, F109, A114, Y115, L126, M283, A286, F289, A290, L355, M357, L358, M359, M459, heme group. |
| Ganaxolone | −15.0057 | Y102, F104, M105, F109, Y115, A286, F289, A290, L355, M357, L358, M459, heme group. |
| Brexanolone | −14.8929 | Y102, F104, M105, F109, Y115, A286, F289, A290, L355, M357, L358, M459, heme group. |
| Sotagliflozin | −14.7156 | Y102, F104, M105, F109, Y115, P209, V212, A286, F289, A290, L355, M357, L358, M359, M459, heme group. |
| Vonoprazan | −14.6532 | Y102, M105, F109, Y115, L126, A286, F289, A290, T294, L355, M459, heme group. |
| Clascoterone | −14.6196 | Y102, F104, M105, F109, A114, Y115, L126, M283, A286, F289, A290, L355, V356, M357, C422, M459, heme group. |
| Oliceridine | −14.4961 | Y102, F104, M105, F109, Y115, L126, A286, F289, A290, T294, L355, M357, M459, V460, heme group. |
| Voxelotor | −14.4840 | Y102, M105, F109, Y115, F289, A290, T294, L355, M357, L358, M459, heme group. |
| Remimazolam | −14.4406 | Y102, F109, Y115, A286, F289, A290, T294, L355, M357, L358, C422, M459, V460, heme group. |
| Umbralisib | −14.3285 | Y102, F104, M105, F109, A114, Y115, L126, M283, A286, F289, A290, T294, L355, V356, M357, M459, V460, heme group. |
| Fluconazole | −14.2743 | A290, L355, A286, M357, F289, Y102, M105, F109, T294, Y115, heme group. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guarimata, J.D.; Lavecchia, M. In Silico Study of FDA-Approved Drugs on Leishmania infantum CYP51, a Drug Repositioning Approach in Visceral Leishmaniasis. Chem. Proc. 2024, 16, 11. https://doi.org/10.3390/ecsoc-28-20201
Guarimata JD, Lavecchia M. In Silico Study of FDA-Approved Drugs on Leishmania infantum CYP51, a Drug Repositioning Approach in Visceral Leishmaniasis. Chemistry Proceedings. 2024; 16(1):11. https://doi.org/10.3390/ecsoc-28-20201
Chicago/Turabian StyleGuarimata, Juan Diego, and Martin Lavecchia. 2024. "In Silico Study of FDA-Approved Drugs on Leishmania infantum CYP51, a Drug Repositioning Approach in Visceral Leishmaniasis" Chemistry Proceedings 16, no. 1: 11. https://doi.org/10.3390/ecsoc-28-20201
APA StyleGuarimata, J. D., & Lavecchia, M. (2024). In Silico Study of FDA-Approved Drugs on Leishmania infantum CYP51, a Drug Repositioning Approach in Visceral Leishmaniasis. Chemistry Proceedings, 16(1), 11. https://doi.org/10.3390/ecsoc-28-20201