1. Introduction
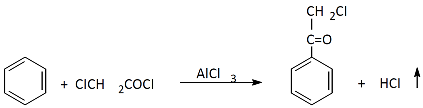
The chloroacetylation reaction of an aromatic ring was first studied by Ch Friedel and J. M. Crafts. They carried out the reaction between benzene and chloroacetyl chloride in the presence of an equimolecular amount of AlCI3 in a carbon sulfide solution, and, as a result, obtained the appropriate chloroketone with a yield of 66% [
1]:
O. Gore used monochloroacetic anhydride as an acylating agent in the chloroacetylation reaction of benzene and obtained phenacyl chloride in 88% yield.
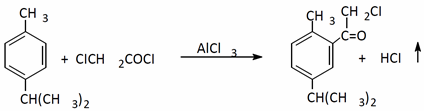
They studied the interaction of p-Tsymol and chloroacetyl chloride. 2-methyl-5-isopropylphenacyl chloride was obtained with a 30% yield using AlCl3 as a catalyst [
2]:
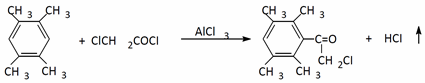
Buy-Khoi and co-workers [
3] carried out the chloroacetylation reaction of durol in the presence of AlCI3. They were able to obtain chloroketone with a high yield (78%):
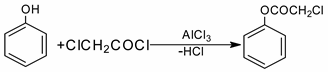
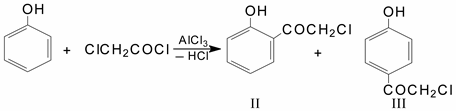
The reaction of chloroacetylation of phenol was first performed by F. Kunkel and F. Johannson, who studied phenol:chloroacetyl chloride:AlCl3 in a ratio of 1:1:0.7 (CS2, 0 °C); they determined that phenylchloroacetate is formed as a result of the reaction [
4]:
N. M. Cullinan and F. R. Edwards carried out the same reaction at 10–20 °C and in an excess of phenol, resulting in an S-acylation reaction took place, and 2- and 4-hydroxyphenacyl chlorides were formed [
5]:
K. Fries and A. Fink practically proved that in the reaction of chloroacetylation of phenols, O—acylation reaction takes place first, after which complex ether is formed, and then regrouping occurs to form the S—acylation product, ketone; later on, this reaction was named “Fries rearrangement” [
6,
7].
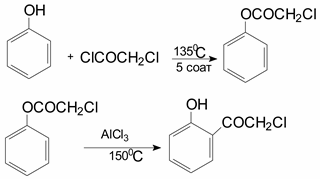
Later K. Fries and V. Pfaffendorfs synthesized phenylchloroacetate by heating a mixture of phenol and chloroacetyl chloride at 135 °C for 5 h and regrouping it at 150 °C in the presence of AlCl3; they found that only 2-hydroxyphenacyl chloride was formed as a result of the reaction:
G. N. Dorofenko and E. N. Sodikov [
8] also studied the reaction of the chloroacetylation of phenol. Unlike the above authors, they heated phenol and chloroacetyl chloride in equimolecular amounts without a catalyst (14 h), which resulted in the formation of the O-acylation product phenylchloroacetate with 79% yield.
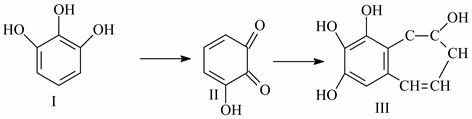
Considering the above information, the o-chloroacetylation reaction of triatomic phenols was studied. Pyuragallol (1,2,3-trioxybenzene), oxyhydroquinone, and fluoroglucin (1,3,5-tri oxybenzene) are examples of trihydric phenol compounds:
Pyrogallol has a special property among three-atom phenols. Depending on the condition, it easily undergoes an oxidation reaction and turns into a tarry black liquid. Research has revealed that when pyrogallol is oxidized, the following process occurs: pyrogallol is first transformed to orthquinone, and then at the conclusion of the process, it is converted to purpurogallin [
9,
10,
11].
The chemical activity of pyrogallol was increased by a chloroacetylation process. It is well recognized that the chloroacetylation reaction’s byproducts are extensively employed in the chemical industry, in medicine, and in several agricultural industries. The national economy, therefore, places a high value on phenol chloroacetylation goods and items made from their base. Finding practical and affordable ways to synthesize new chloroacetyl products and their derivatives, as well as studying their biological properties, is one of the urgent tasks of organic chemistry.
Information on the 0-chloroacetylation reactions of more phenols and their uses is also provided in our earlier works [
12,
13].
2. Experimental Procedure
To ascertain the composition of the reaction products, Silufol-254 plates were subjected to thin-layer chromatography (TLC). In the mobile phase system of benzene and ethyl ether of acetic acid (95:5), TLC was used to assess the reaction’s development and the purity of the compounds that were produced during the process. Aluminum plates with a silica gel coating (silica gel 60 F254) purchased from MERCK in India were used for the TLC stationary phase. UV light was used to see how compounds were distributed on TLC plates. Column chromatography was used to clean the reaction mixture, and the yield of the chemical reaction that resulted from isolation was determined. After separation by column chromatography, the reaction mixture was confirmed by TLC using benzene and ethyl acetate (95:5) as the mobile phase. The reaction was subsequently completed, and the liquid was poured into ice-cold water. The solid material that had crystallized was filtered and dried. The starting material was cleaned using column chromatography, petroleum ether, and ethyl ester of acetic acid. The FT-IR spectra of the goods were obtained on a Carl Sies (Hamburg, Germany) Specord IR-71 spectrophotometer using the KBr pellet method. TMS was used as the internal standard for the 1H NMR recordings, and chemical shift values were expressed in ppm scale using a Bruker (Bremen, Germany) 400 MHz NMR apparatus. Using Mvtec melting point equipment and the open capillary approach, the uncorrected melting points of the produced compounds were determined [
14,
15,
16,
17].
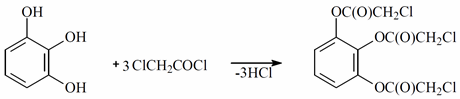
Synthesis of trichloroacetylpyragallol. The pyrogallol chloroacetylation process was tried. In a 100 mL round flask with a refrigerant (1:3), 6.3 g (ops mol) of pyrogallol and 16.95 g (p-1.4 g/mL, 12.1 mL; 0.15 mL) of chloroacetyl chloride were combined. A total of 35 cc of chloroform (along with other solvents) was added before being poured over them. The combination was heated, and it was anticipated that the process would begin when the solvent reached its boiling point. The radiation lasted for 22 h. In reality, the separation of the HCl (as confirmed by wet litmus paper) is evidence of the progress of the process. The solvent was released when the process had finished. Tests revealed that the leftover mixture was TLC (0.644).
3. Reaction Results and Discussion of Results
When the O-chloroacetylation process was carried out in a chloroform solution, the degree of generality was at its highest. Pyrogallol was chloroacetylated to produce 95% O-chloroacetyl pyrogallol in a chloroform solution.
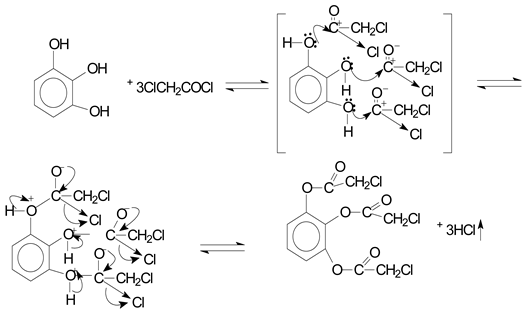
The sorbed electron density from the chloroacetyl chloride molecule provided an oxygen molecule in the reaction between pyrogallol and chloroacetyl chloride, which had a partially negative charge. The carbon atom interacted with the double electrons of the hydroxyl group in the pyrogallol molecule to form complex I, and the chlorine and oxygen atoms’ electrons gave carbon a partially positive charge. Complex II, from which the byproduct of the reaction with hydrogen chloride was removed, was generated during the process when an oxygen-to-carbon valence bond was formed.
The organic chlorine compounds used in the reaction process were more efficient. However, in other solvents (benzene, hexane, and heptane), the reaction was more difficult, and in the process, the product was mixed with the oxidation product of pyrogallol. This made the process more complicated.
It was investigated how pyrogallol reacted with trichloroacetypirogallol and chloroacetylchloride.
The procedure continued till the conclusion of HCl elevation as the reaction followed the type of nucleophilic exchange (S
N1). The yield of the product in chloroform was high, and the procedure was carried out in apraton solvents.
Table 1 shows the yields of the reaction in relation to the reducers.
Reaction mechanisms were as follows:
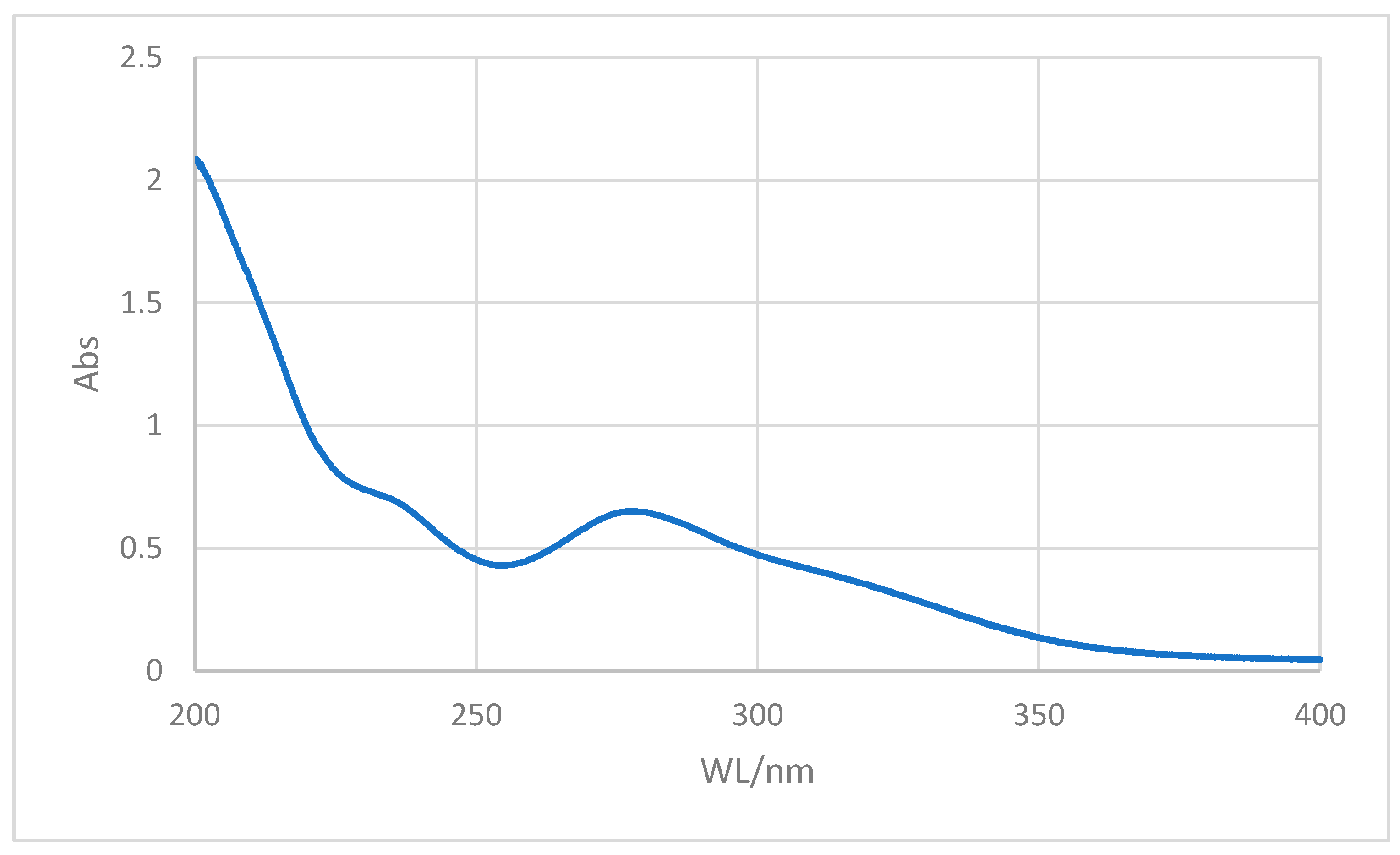
UV-spectral data of trichloroacetylpyrogallol: The aromatic ring for pyrogallol exhibited UV absorption spectra in the region of 228 nm (π→π*) and 265 nm (n→π*) of the OH group linked to the aromatic ring. These absorption regions, however, were not preserved in the molecule we created throughout the reaction. The development of the estimated product was further supported by the fact that the aromatic ring’s absorption area relocated to 250 nm, and a new absorption region between 295 and 310 nm was created as a result of the synthesis of the carbonyl group (C=O) (
Figure 1).
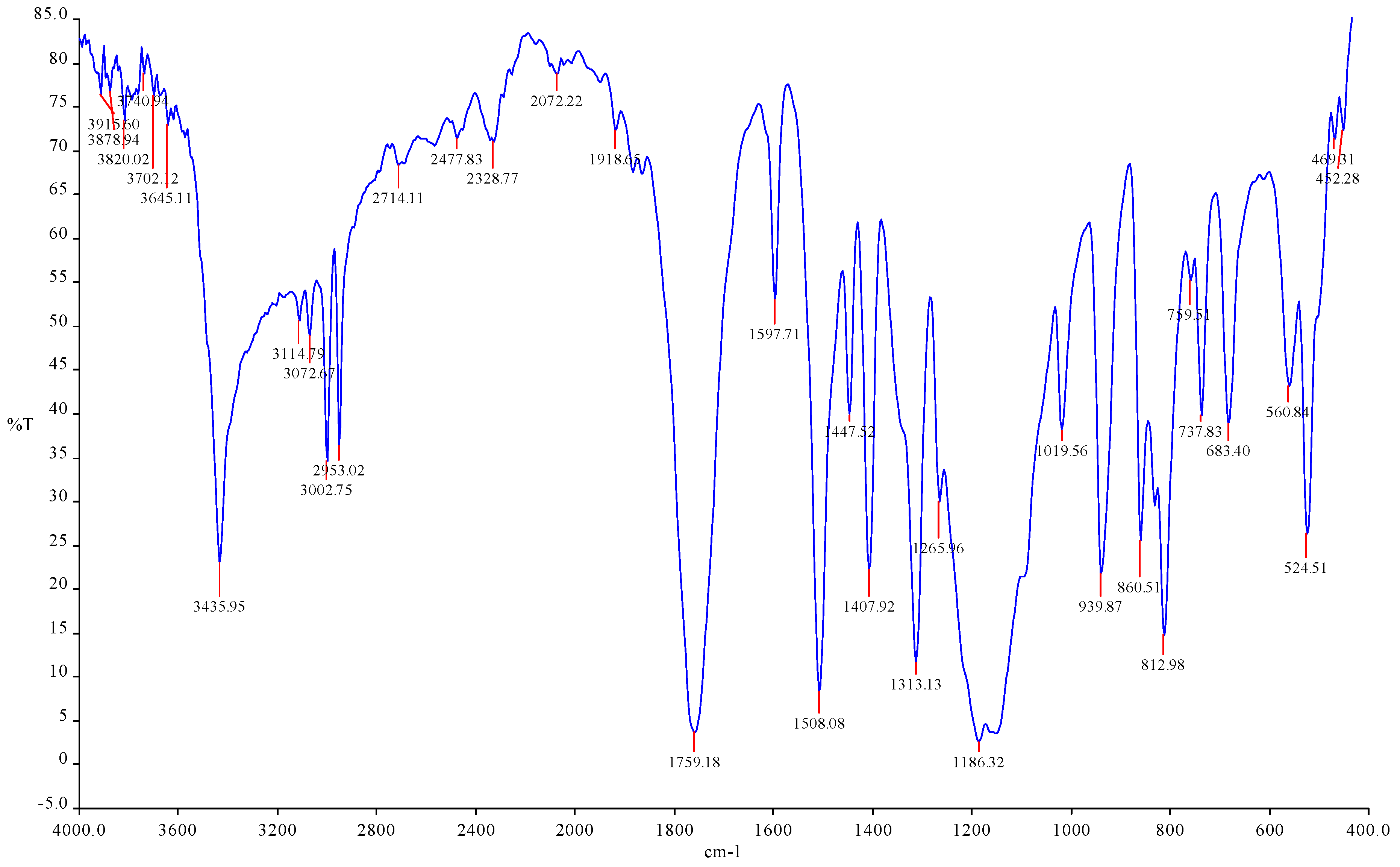
IR spectral data of trichloroacetylpyrogallol (Figure 2): (Ar)C-C bond in the 1500–1400 sm
−1 range; (Ar)C-H bond in the low absorption region of 3100–3000 sm
−1; carbonyl group valence oscillation (ν
C=O) with 1790–1720 sm
−1 intensity, which is an extremely strong field characterized by the carbonyl group C-O bond with 1280–1270 sm
−1 area, and (Ar) =C-O- bond with 1270–1180 sm
−1 broad intense absorption field in the 800–600 sm
−1 region of absorption, where intense valence bond oscillations of the -CH
2-Cl bond were seen. In the resulting product, the following absorptions were seen: strong, medium-intensity acetyl group valence oscillations in the region of absorption at 3452.17 sm
−1; average weak-intensity absorption valence oscillations specific to group C-H in the aromatic ring were 311.27, 307.25, and 3011.65 sm
−1; average weak-intensity absorption valence oscillations specific to group C=C in the aromatic ring were 1598.03, 1509.24, 1455.96, 1465.40, 1441.33, and 1410.82 sm
−1; average weak-intensity absorption deformation oscillations specific to group C-H in the aromatic ring were 947.85, 922.51, 826.57, 769.65, 769.83, 709.45, 734.19, and 654.45 sm
−1; average C=O group specific deformation vibration was 1759.51 sm
−1; average valence oscillations specific to group C-O in the area of weak absorption was 1054.80; oscillations unique to the C-Cl group were avoided in the region with an average absorption of 522.68 sm
−1.
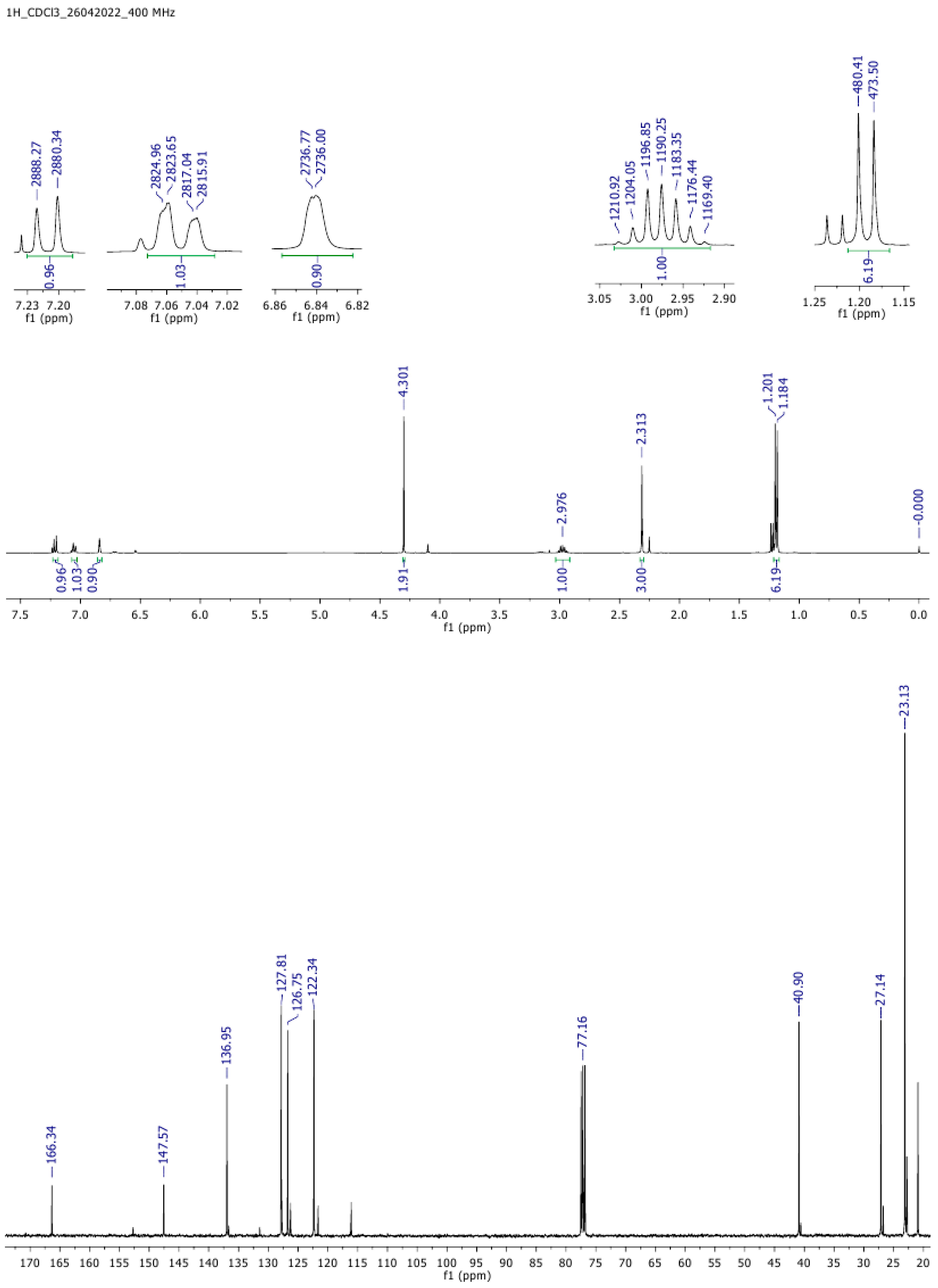
1H NMR spectral data of trichloroacetylpyrogallol: The
1H NMR (400 MHz, CDCl
3)spectra of O-trichloroacetyl pyrogallol are presented in
Figure 3 and characterize the absorption lines of the hydrogen atoms in the molecule as follows: δ 6.7 (m, 1H, ArH), 7.07 (d,
I = 8.5 Hz, 1H, ArH), 7.25 (m, 1H, ArH), and 12.23 (s, 1H, -OH).
13C-NMR (400 MHz, CDCl
3): δ 163.8, 153.0, 136.1, 131.7, 126.2, 121.9, 116.0, 77.5, 26.7, and 22.8.






{kind=link}
{kind=link}
{kind=link}