
Aromatic Iodides: Synthesis and Conversion to Heterocycles †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
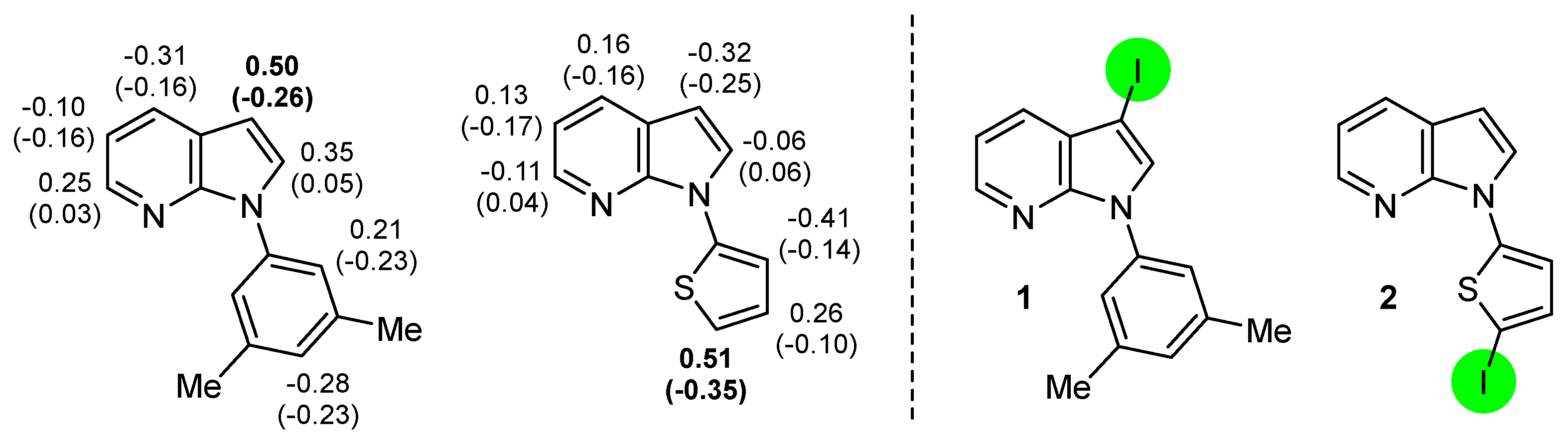
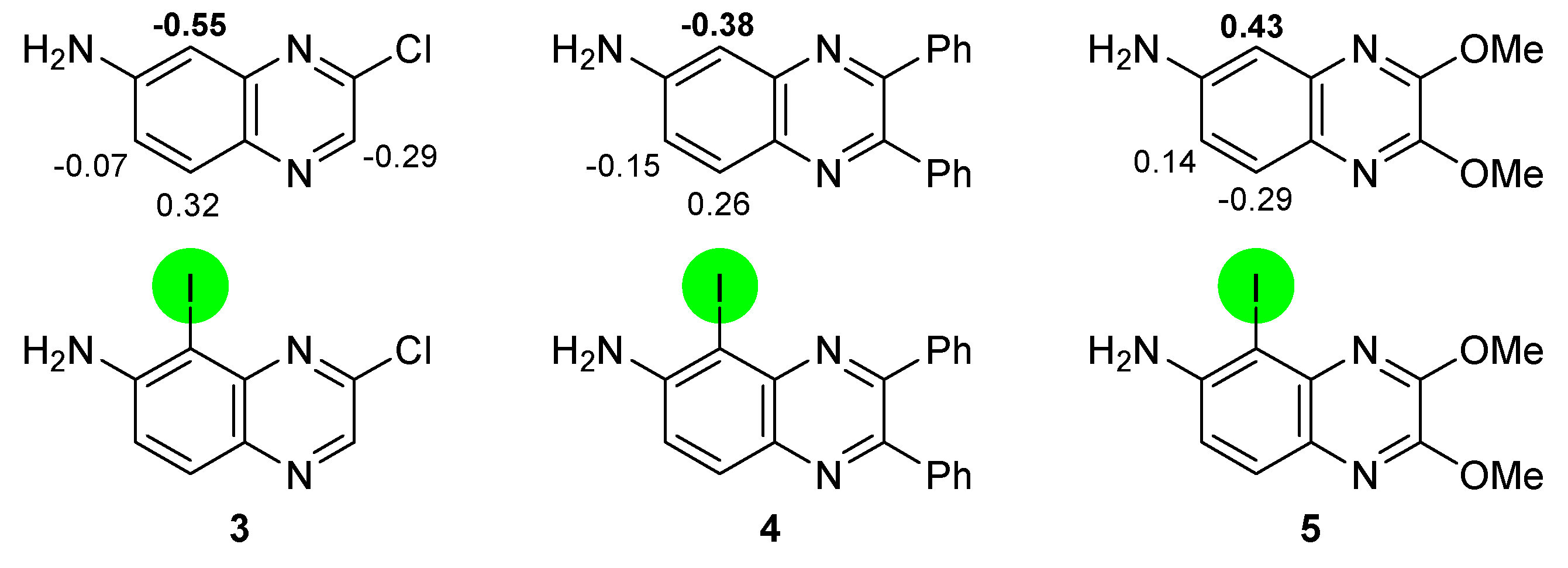
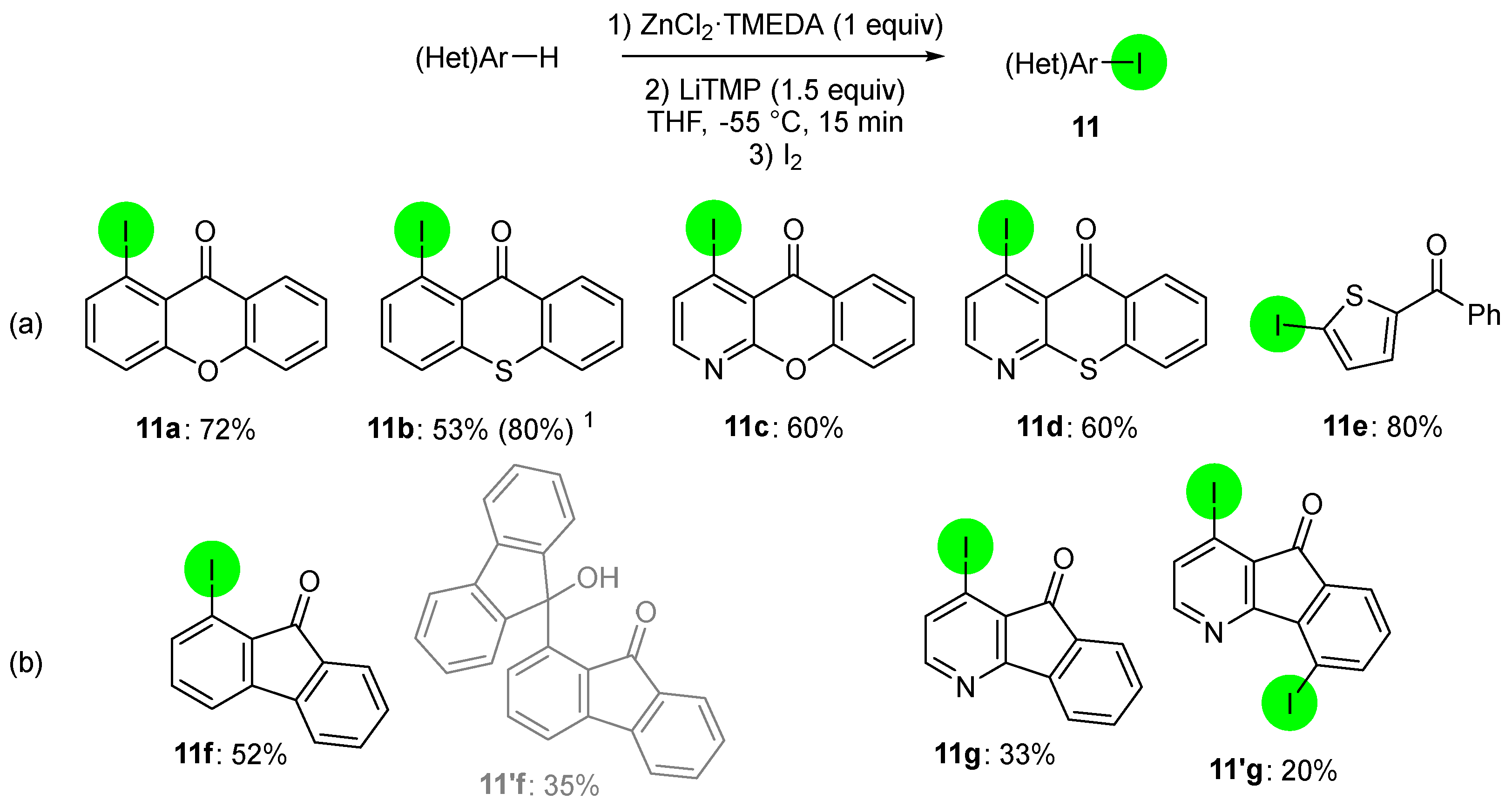
2. Selective Introduction of Iodine onto Aromatic Compounds
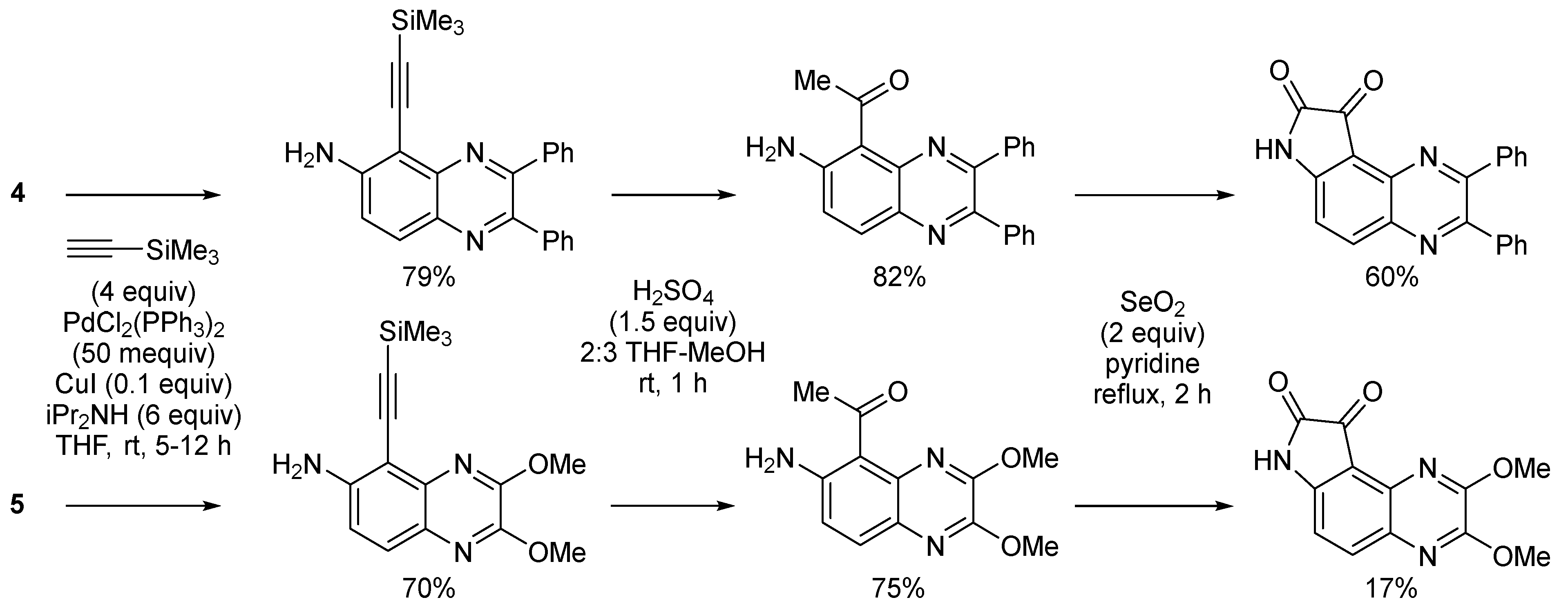
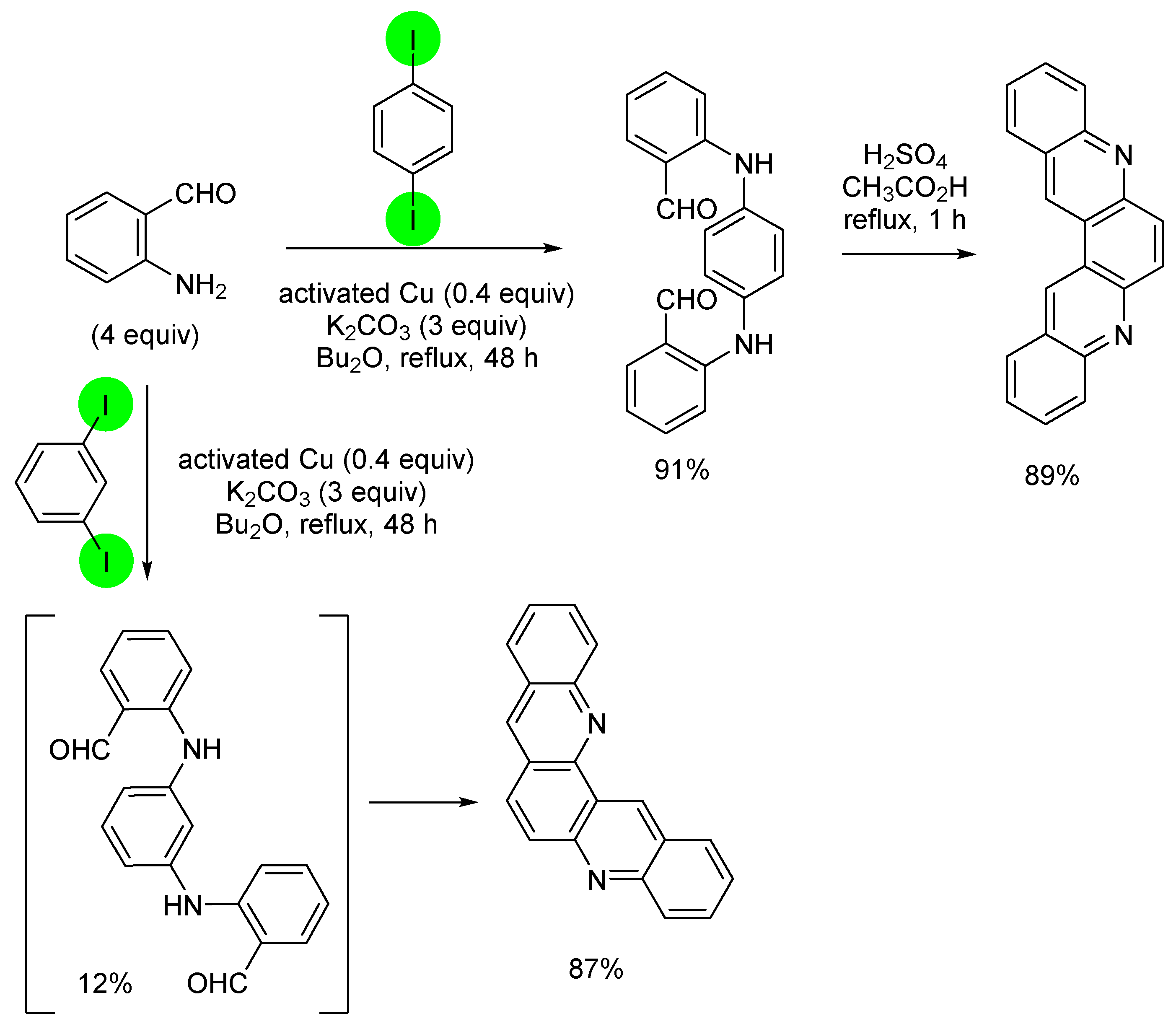
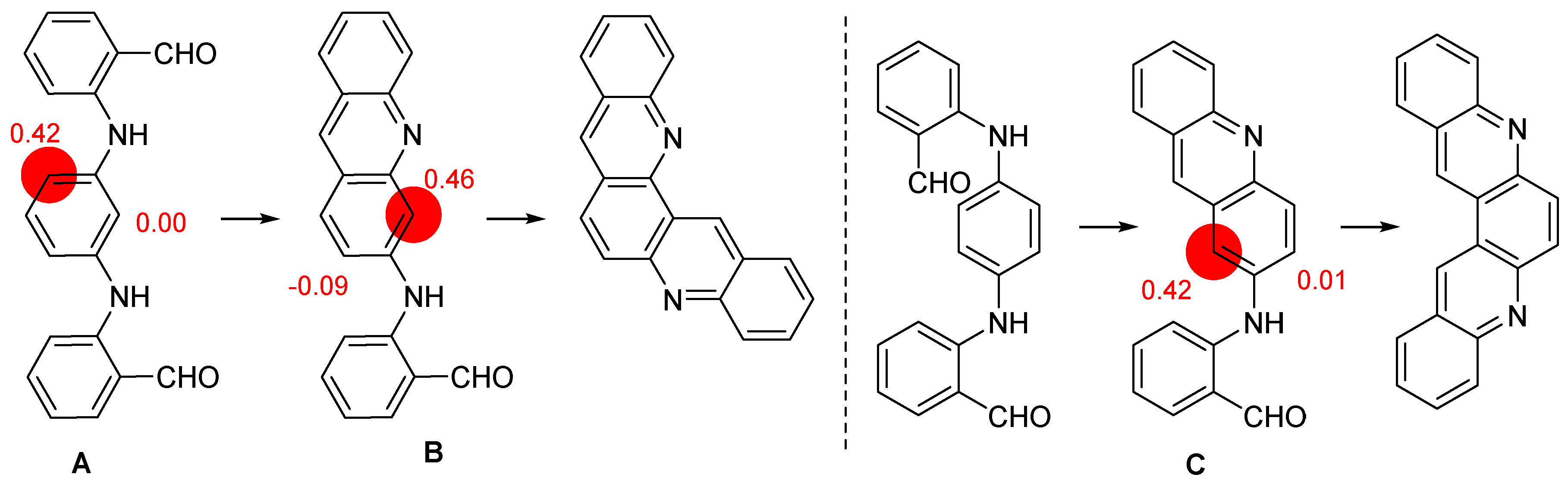
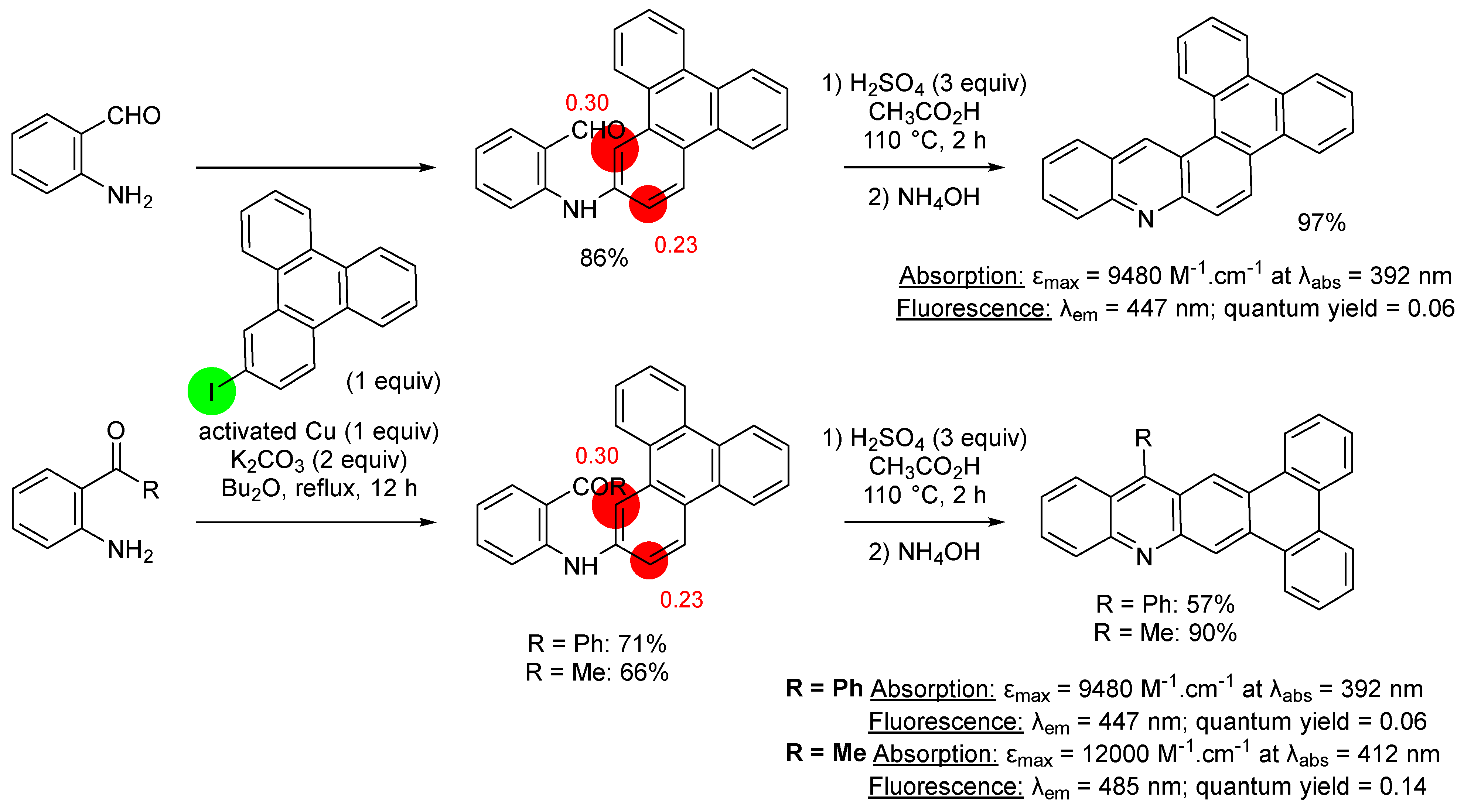
2.1. Direct Iodination and Application to the Synthesis of Heterocycles of Interest
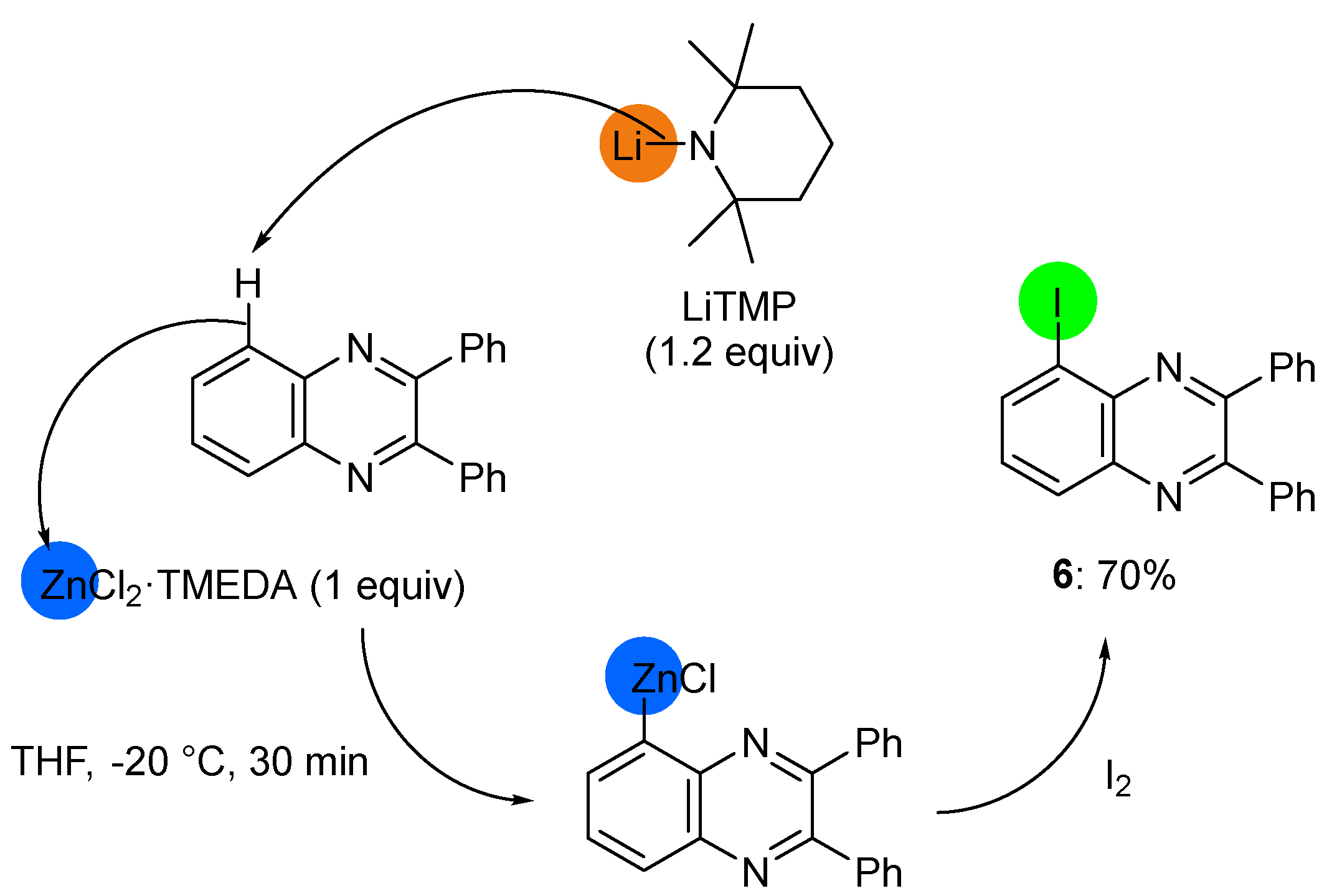
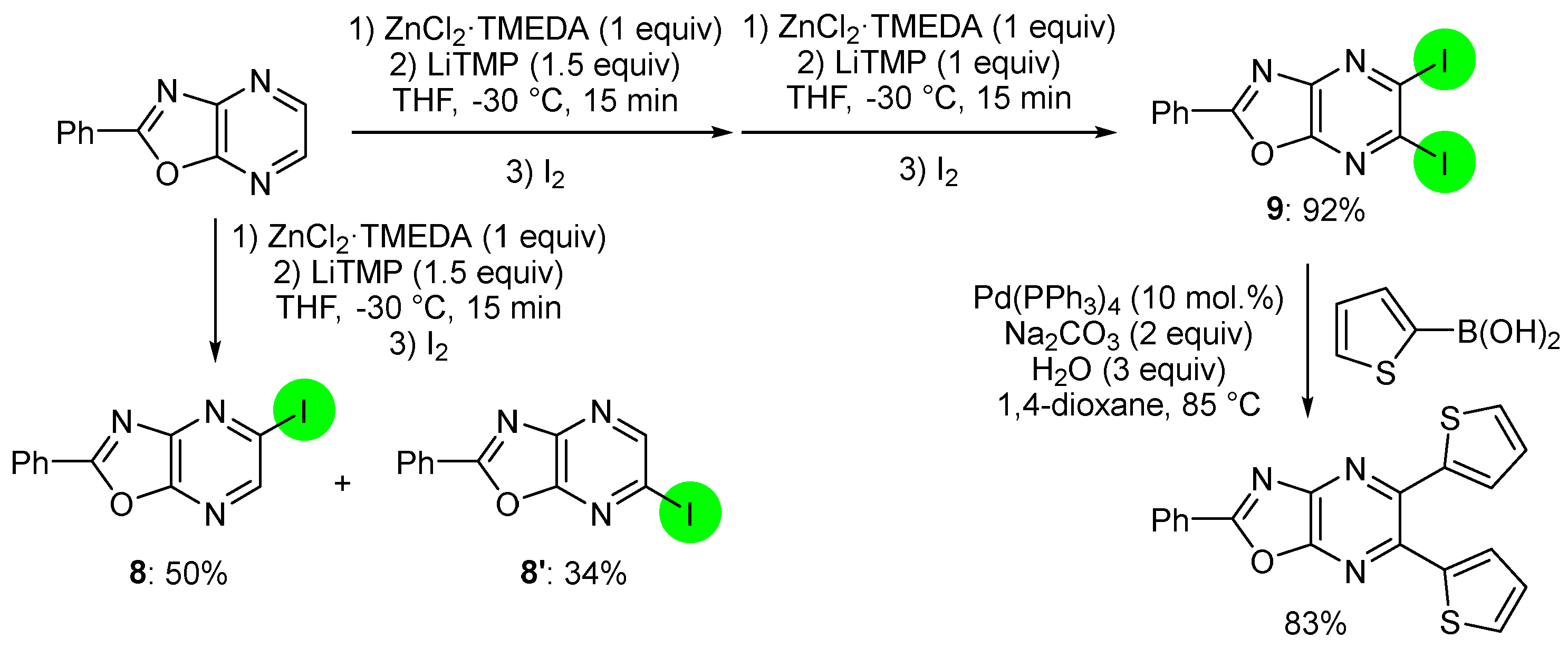
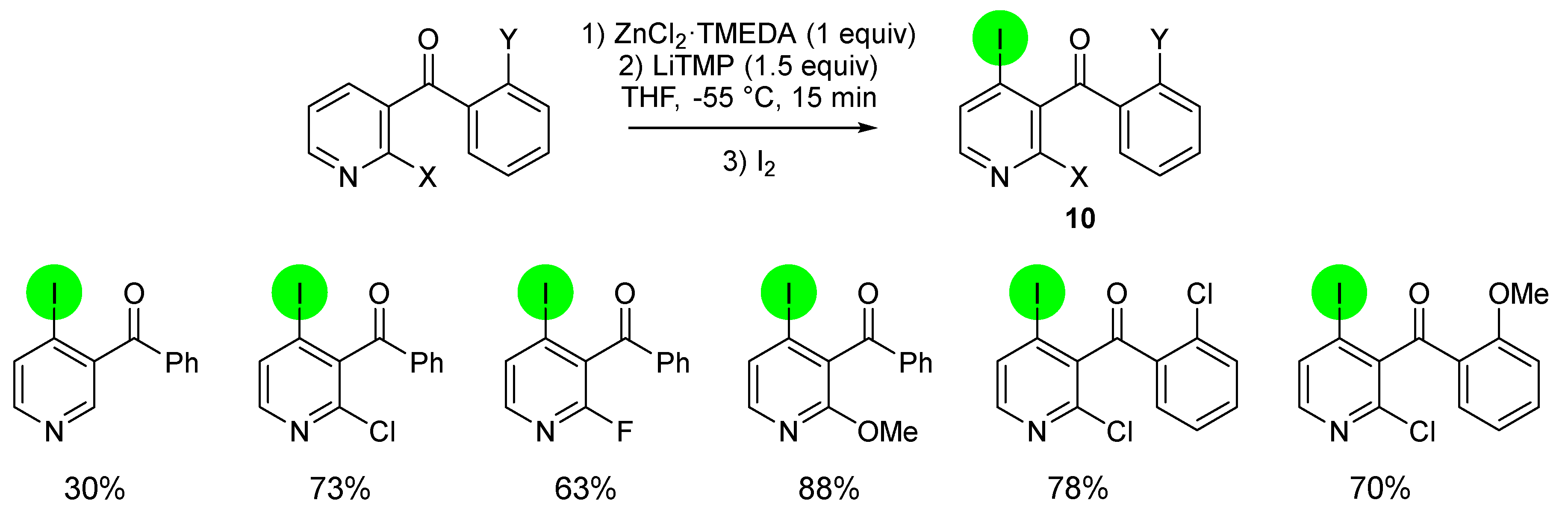
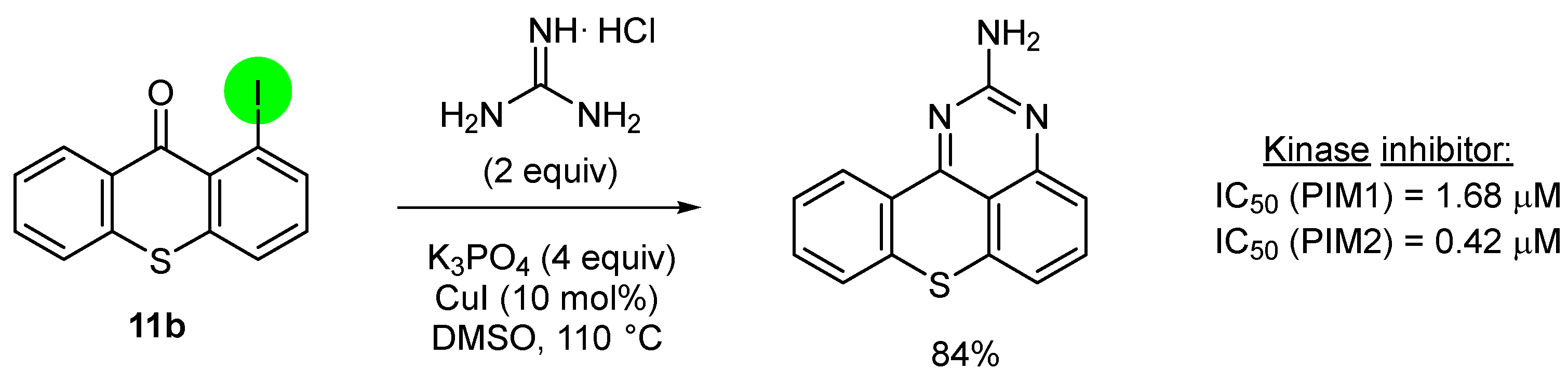
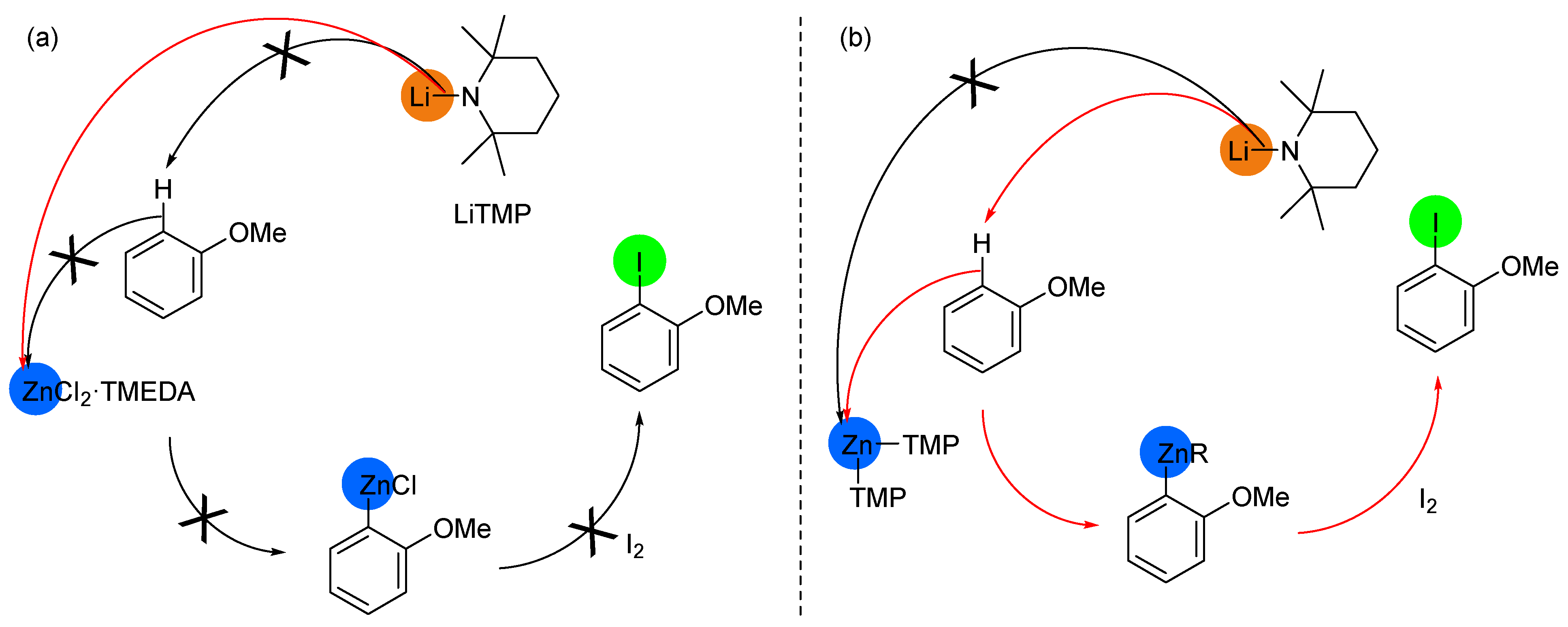
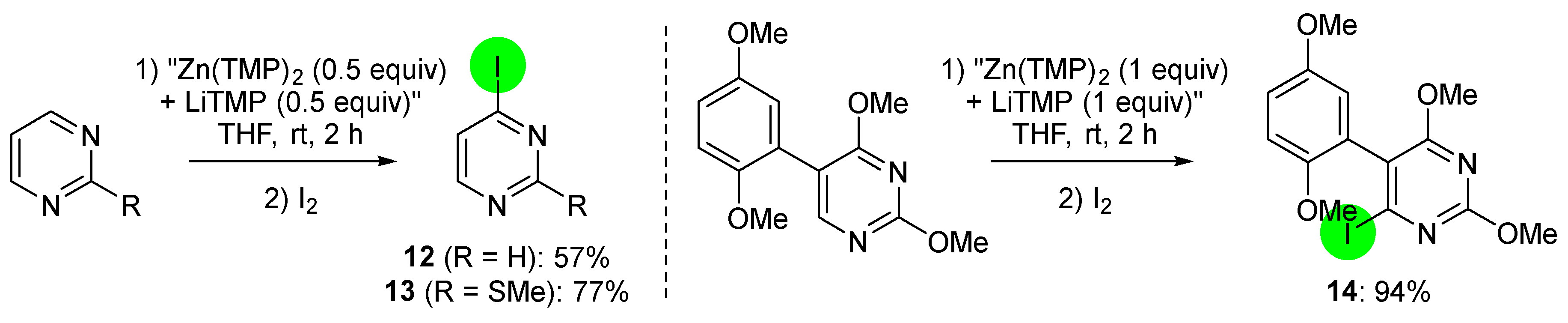
2.2. Deprotometalation with In Situ Trapping–Iodolysis as an Alternative to Direct Iodination
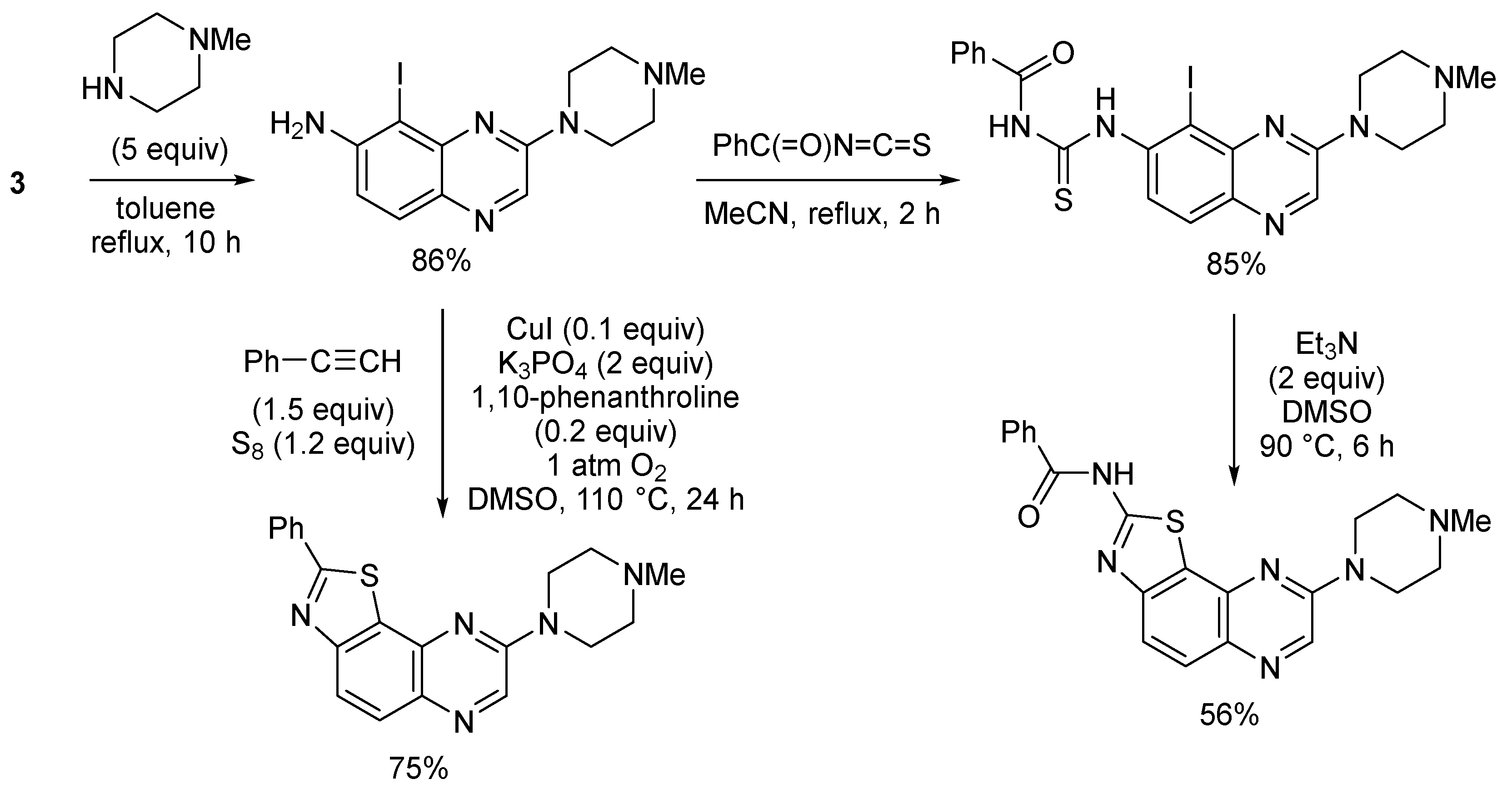
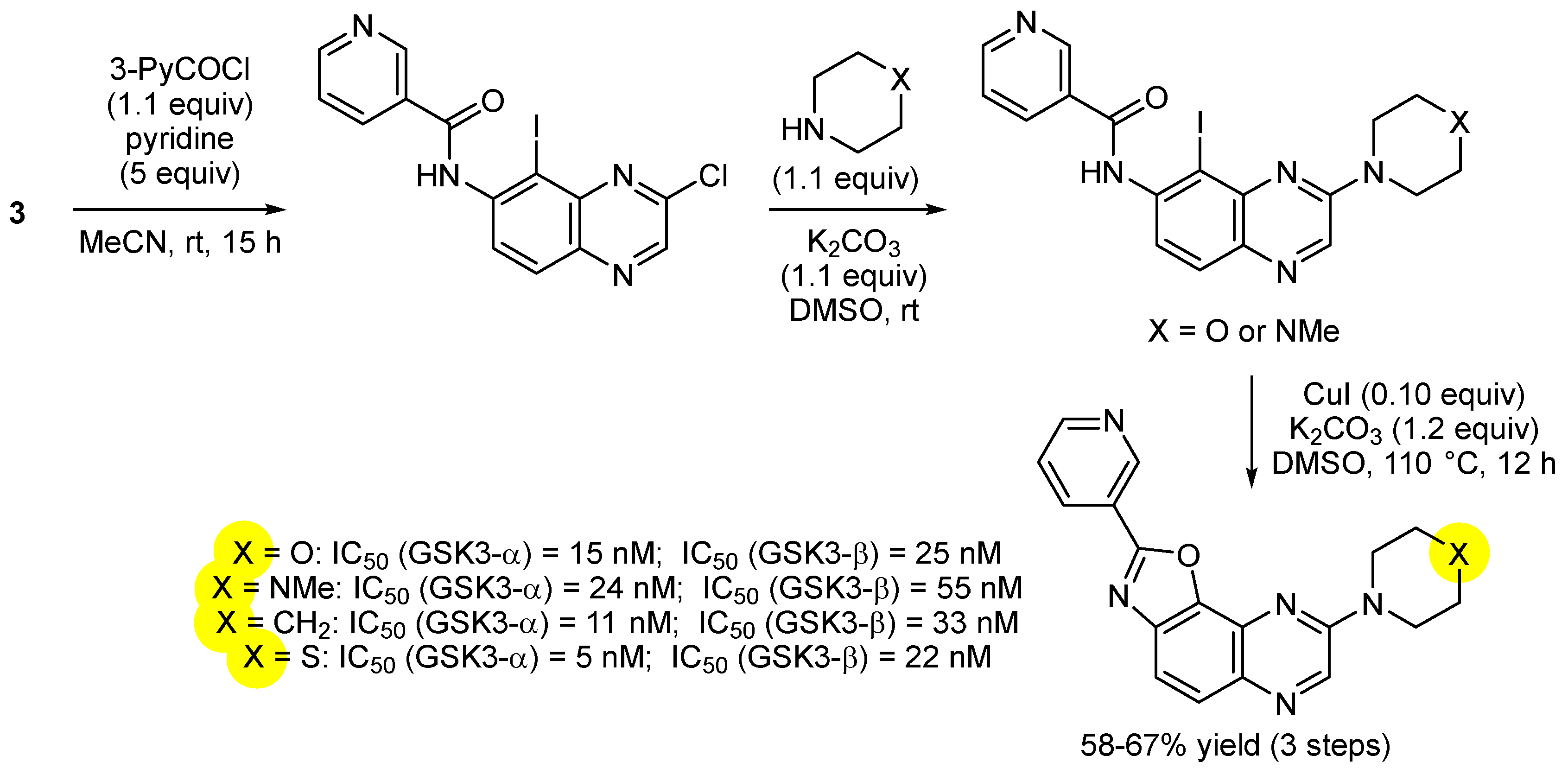
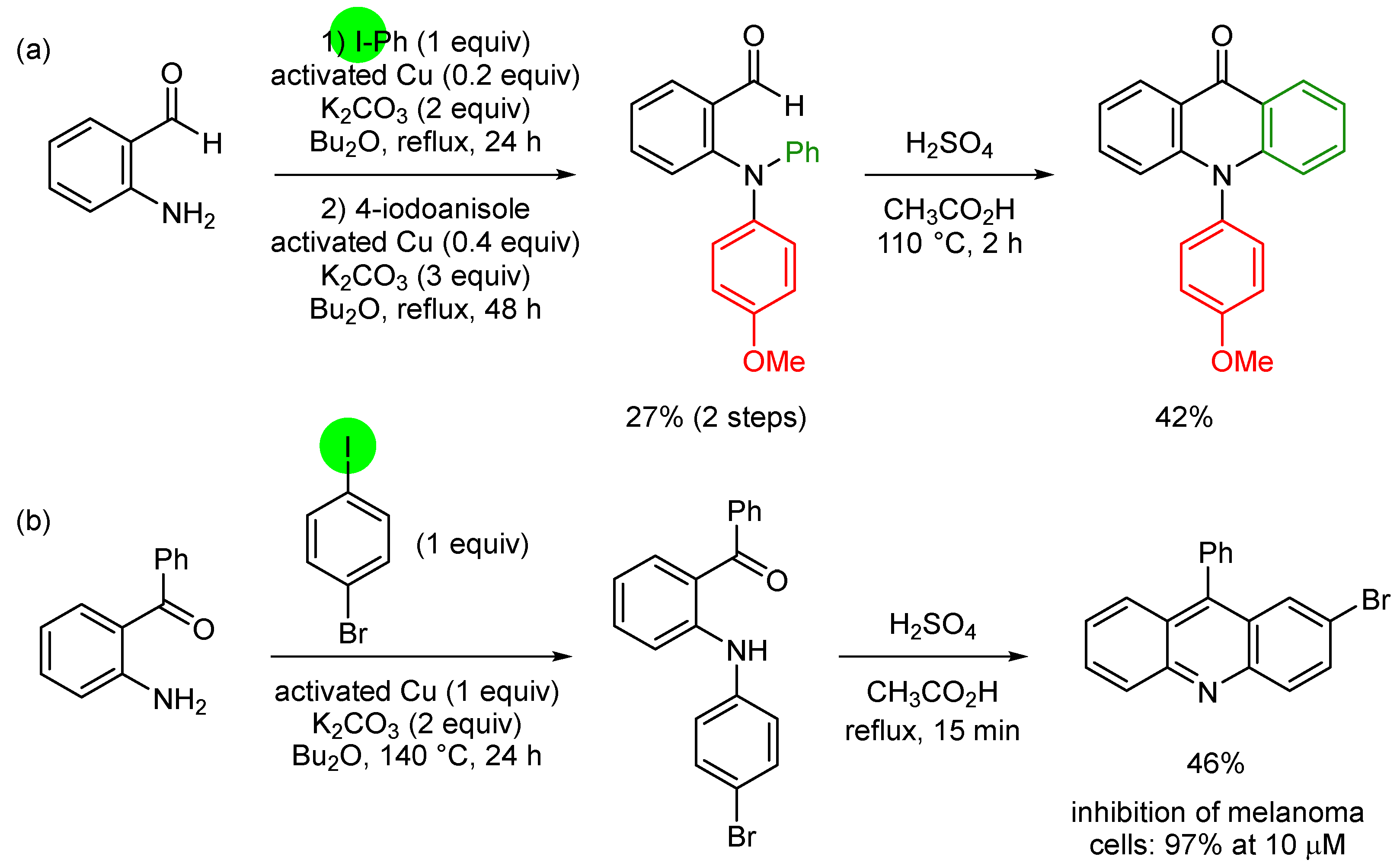
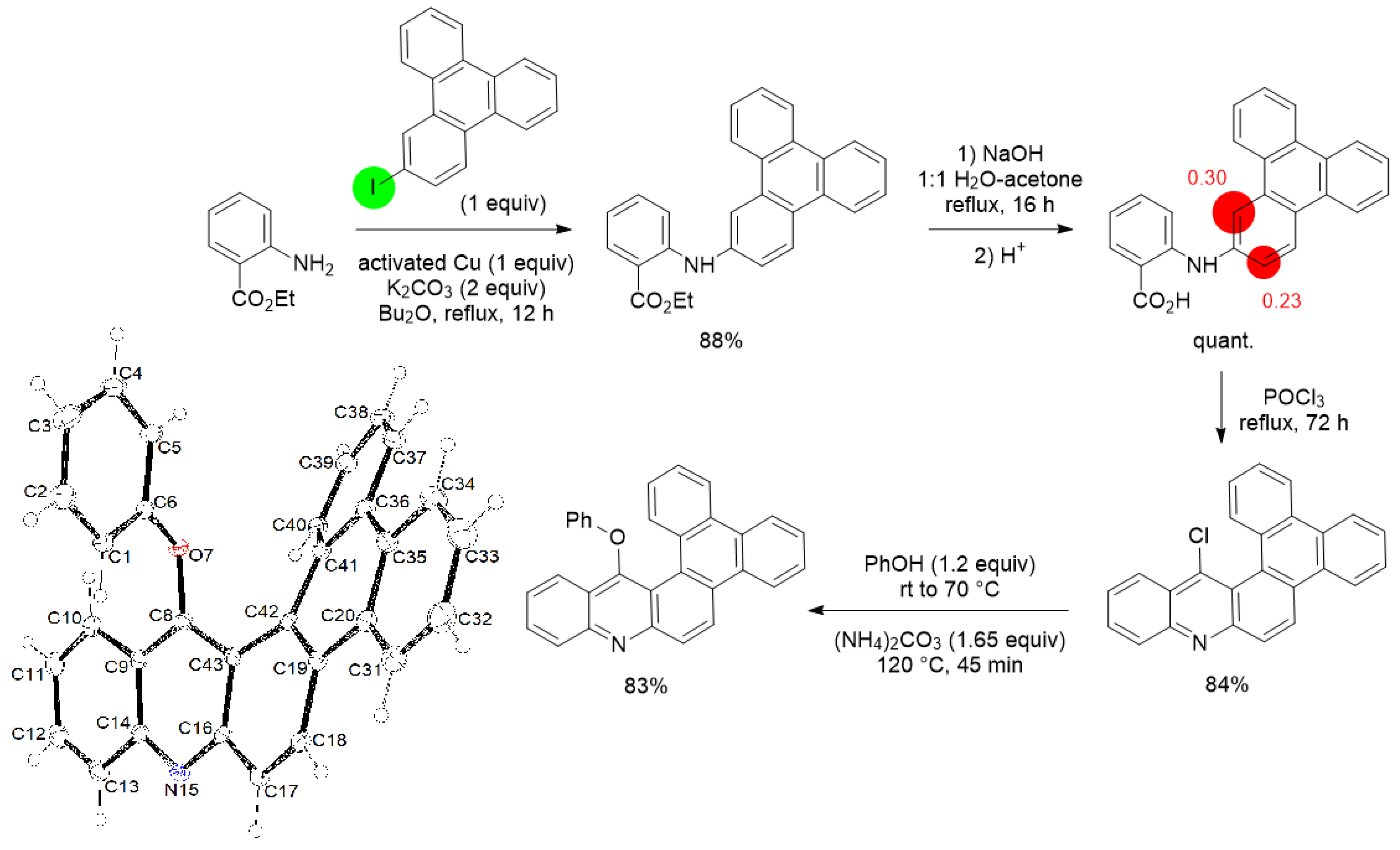
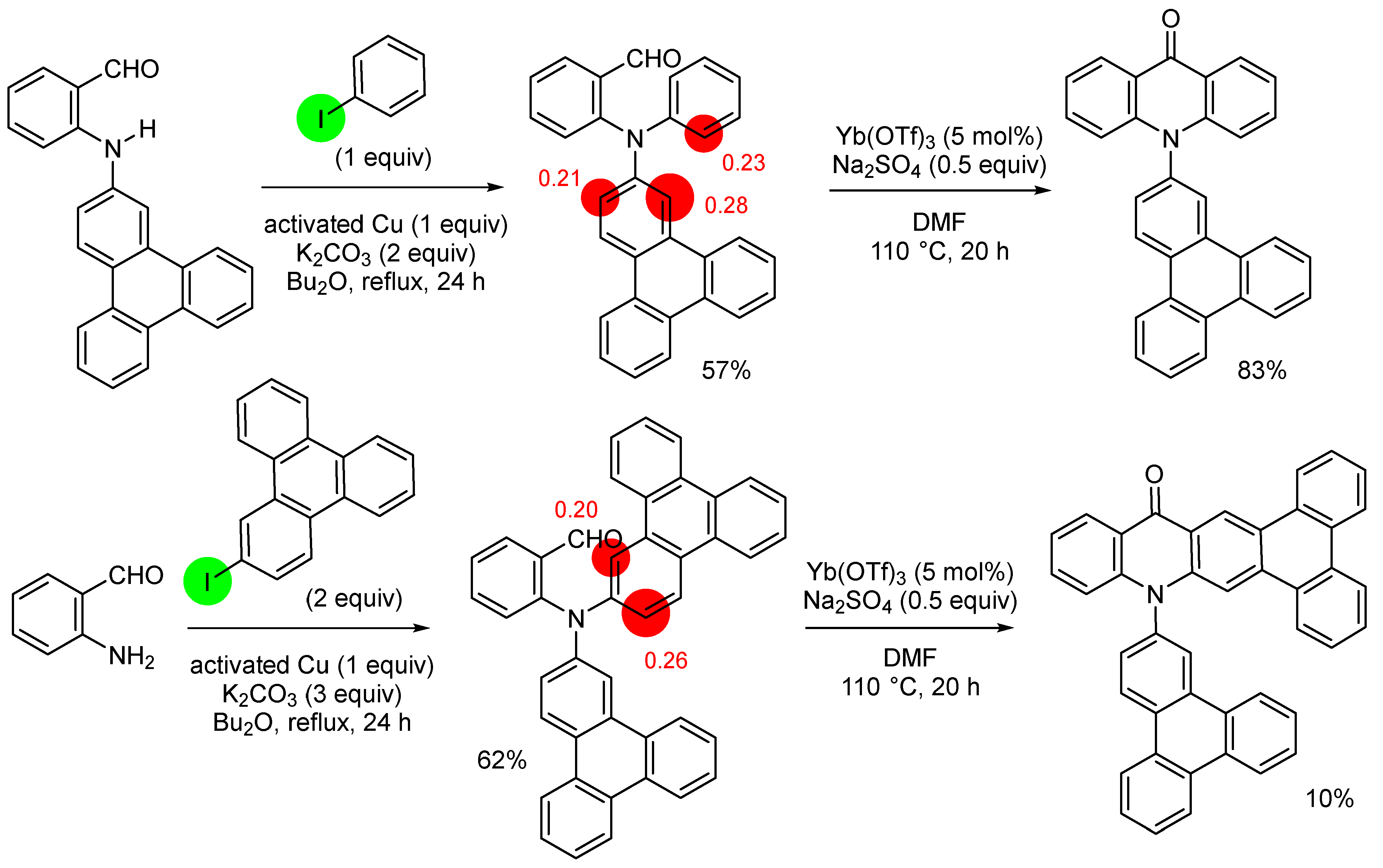
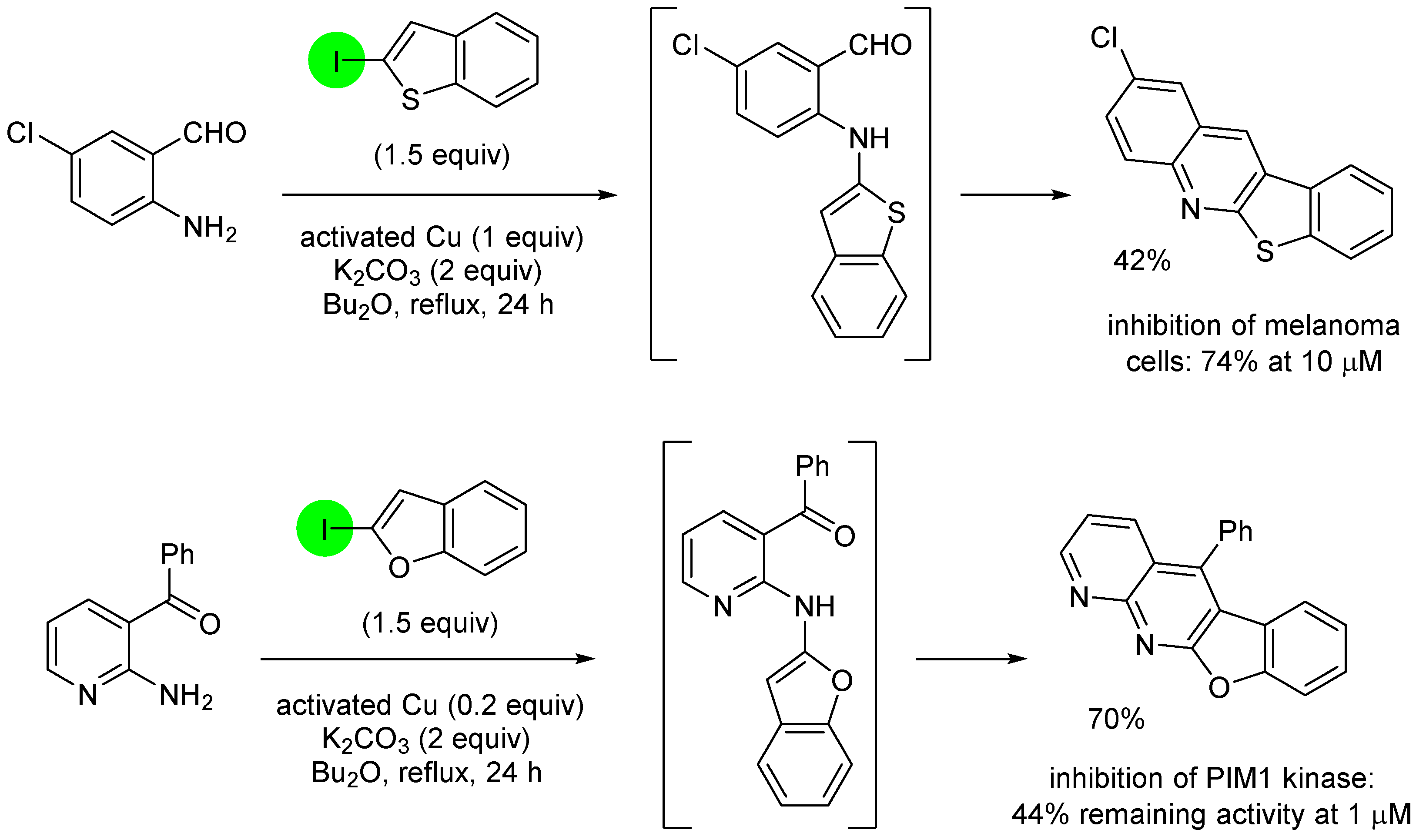
3. Aromatic Iodides in Copper-Mediated N-Arylation of Anilines
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Begunov, R.S.; Sokolov, A.A.; Belova, V.O.; Solov’ev, M.E. Quantum chemical study of regioselectivity of reactions of substituted pyrido[1,2-a]benzimidazoles with electrophiles. Russ. Chem. Bull. 2016, 65, 644–647. [Google Scholar] [CrossRef]
- Fukui, K.; Yonezawa, T.; Nagata, C.; Shingu, H. Molecular-orbital theory of orientation in aromatic, heteroaromatic, and other conjugated molecules. J. Chem. Phys. 1954, 22, 1433–1442. [Google Scholar] [CrossRef]
- Carissan, Y.; Hagebaum-Reignier, D.; Goudard, N.; Humbel, S. Hückel-Lewis projection method: A "weights watcher" for mesomeric structures. J. Phys. Chem. A 2008, 112, 13256–13262. [Google Scholar] [CrossRef]
- Goudard, N.; Carissan, Y.; Hagebaum-Reignier, D.; Humbel, S. HuLiS 3.3.4: Simple HÜckel and LewIS Embedded in the Hückel Theory. Available online: https://ism2.univ-amu.fr/hulis (accessed on 1 January 2008).
- Ameur Messaoud, M.Y.; Bentabed-Ababsa, G.; Fajloun, Z.; Hamze, M.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Roisnel, T.; Dorcet, V.; Mongin, F. Deprotometalation-iodolysis and direct iodination of 1-arylated 7-azaindoles: Reactivity studies and molecule properties. Molecules 2021, 26, 6314. [Google Scholar] [CrossRef]
- Lassagne, F.; Chevallier, F.; Mongin, F. Saccharin as an organocatalyst for quinoxalines and pyrido[2,3-b]pyrazines syntheses. Synth. Commun. 2014, 44, 141–149. [Google Scholar] [CrossRef]
- Saini, K.M.; Saunthwal, R.K.; Kumar, S.; Verma, A.K. On water: Metal-free synthesis of highly functionalized benzothiazolylidene from ortho-haloanilines. J. Org. Chem. 2019, 84, 2689–2698. [Google Scholar] [CrossRef]
- Huang, Y.; Yan, D.; Wang, X.; Zhou, P.; Wu, W.; Jiang, H. Controllable assembly of the benzothiazole framework using a C≡C bond as a one-carbon synthon. Chem. Commun. 2018, 54, 1742–1745. [Google Scholar] [CrossRef]
- Lassagne, F.; Sims, J.M.; Erb, W.; Mongin, O.; Richy, N.; El Osmani, N.; Fajloun, Z.; Picot, L.; Thiery, V.; Robert, T.; et al. Thiazolo[5,4-f]quinoxalines, oxazolo[5,4-f]quinoxalines and pyrazino[b,e]isatins: Synthesis from 6-aminoquinoxalines and properties. Eur. J. Org. Chem. 2021, 2021, 2756–2763. [Google Scholar] [CrossRef]
- Demmer, C.S.; Bunch, L. Benzoxazoles and oxazolopyridines in medicinal chemistry studies. Eur. J. Med. Chem. 2015, 97, 778–785. [Google Scholar] [CrossRef]
- Viirre, R.D.; Evindar, G.; Batey, R.A. Copper-catalyzed domino annulation approaches to the synthesis of benzoxazoles under microwave-accelerated and conventional thermal conditions. J. Org. Chem. 2008, 73, 3452–3459. [Google Scholar] [CrossRef]
- Lassagne, F.; Duguépéroux, C.; Roca, C.; Perez, C.; Martinez, A.; Baratte, B.; Robert, T.; Ruchaud, S.; Bach, S.; Erb, W.; et al. From simple quinoxalines to potent oxazolo[5,4-f]quinoxaline inhibitors of glycogen-synthase kinase 3 (GSK3). Org. Biomol. Chem. 2020, 18, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A Convenient synthesis of acetylenes: Catalytic substitutions of acetylenic hydrogen with bromo alkenes, iodo arenes, and bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar] [CrossRef]
- Aburano, D.; Yoshida, T.; Miyakoshi, N.; Mukai, C. Synthesis of hexahydropyrrolo[2,3-b]indole alkaloids based on the aza-pauson-khand-type reaction of alkynecarbodiimides. J. Org. Chem. 2007, 72, 6878–6884. [Google Scholar] [CrossRef] [PubMed]
- Gschwend, H.W.; Rodriguez, H.R. Heteroatom-facilitated lithiations. Org. React. 1979, 26, 1–360. [Google Scholar]
- Gant, T.G.; Meyers, A.I. The chemistry of 2-oxazolines (1985-present). Tetrahedron 1994, 50, 2297–2360. [Google Scholar] [CrossRef]
- Schlosser, M. Organometallics in Synthesis, 2nd ed.; Schlosser, M., Ed.; Wiley: New York, NY, USA, 2002; Chapter I. [Google Scholar]
- Hartung, C.G.; Snieckus, V. The directed ortho metalation reaction—A point of departure for new synthetic aromatic chemistry. In Modern Arene Chemistry; Astruc, D., Ed.; Wiley: New York, NY, USA, 2002; Chapter 10; pp. 330–367. [Google Scholar]
- Clayden, J. Directed metalation of aromatic compounds. Chem. Organolithium Compd. 2004, 1, 495–646. [Google Scholar] [CrossRef]
- Tilly, D.; Magolan, J.; Mortier, J. Directed remote aromatic metalations: Mechanisms and driving forces. Chem. Eur. J. 2012, 18, 3804–3820. [Google Scholar] [CrossRef]
- Mulvey, R.E.; Mongin, F.; Uchiyama, M.; Kondo, Y. Deprotonative metalation using ate compounds: Synergy, synthesis, and structure building. Angew. Chem. Int. Ed. 2007, 46, 3802–3824. [Google Scholar] [CrossRef]
- Mulvey, R.E. Avant-garde metalating agents: Structural basis of alkali-metal-mediated metalation. Acc. Chem. Res. 2009, 42, 743–755. [Google Scholar] [CrossRef]
- Haag, B.; Mosrin, M.; Ila, H.; Malakhov, V.; Knochel, P. Regio- and chemoselective metalation of arenes and heteroarenes using hindered metal amide bases. Angew. Chem. Int. Ed. 2011, 50, 9794–9824. [Google Scholar] [CrossRef]
- Mongin, F.; Harrison-Marchand, A. Mixed AggregAte (MAA): A single concept for all dipolar organometallic aggregates. 2. Syntheses and reactivities of homo/heteroMAAs. Chem. Rev. 2013, 113, 7563–7727. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, R.E. An alternative picture of alkali-metal-mediated metallation: Cleave and capture chemistry. Dalton Trans. 2013, 42, 6676–6693. [Google Scholar] [CrossRef] [PubMed]
- Harford, P.J.; Peel, A.J.; Chevallier, F.; Takita, R.; Mongin, F.; Uchiyama, M.; Wheatley, A.E.H. New avenues in the directed deprotometallation of aromatics: Recent advances in directed cupration. Dalton Trans. 2014, 43, 14181–14203. [Google Scholar] [CrossRef]
- Lysén, M.; Hansen, H.M.; Begtrup, M.; Kristensen, J.L. Synthesis of substituted 2-cyanoarylboronic esters. J. Org. Chem. 2006, 71, 2518–2520. [Google Scholar] [CrossRef]
- Mokhtari Brikci-Nigassa, N.; Bentabed-Ababsa, G.; Erb, W.; Mongin, F. In situ ‘trans-metal trapping’: An efficient way to extend the scope of aromatic deprotometalation. Synthesis 2018, 50, 3615–3633. [Google Scholar]
- Lassagne, F.; Langlais, T.; Caytan, E.; Limanton, E.; Paquin, L.; Boullard, M.; Courtel, C.; Curbet, I.; Gedeon, C.; Lebreton, J.; et al. From quinoxaline, pyrido[2,3-b]pyrazine and pyrido[3,4-b]pyrazine to pyrazino-fused carbazoles and carbolines. Molecules 2018, 23, 2961. [Google Scholar] [CrossRef]
- Van Baelen, G.; Meyers, C.; Lemière, G.L.F.; Hostyn, S.; Dommisse, R.; Maes, L.; Augustyns, K.; Haemers, A.; Pieters, L.; Maes, B.U.W. Synthesis of 6-methyl-6H-indolo[3,2-c]isoquinoline and 6-methyl-6H-indolo[2,3-c]isoquinoline: Two new unnatural isoquinoline isomers of the cryptolepine series. Tetrahedron 2008, 64, 11802–11809. [Google Scholar] [CrossRef]
- Van Baelen, G.; Hostyn, S.; Dhooghe, L.; Tapolcsányi, P.; Mátyus, P.; Lemière, G.; Dommisse, R.; Kaiser, M.; Brun, R.; Cos, P.; et al. Structure-activity relationship of antiparasitic and cytotoxic indoloquinoline alkaloids, and their tricyclic and bicyclic analogues. Bioorg. Med. Chem. 2009, 17, 7209–7217. [Google Scholar] [CrossRef]
- Queguiner, G.; Marsais, F.; Snieckus, V.; Epsztajn, J. Directed metalation of pi-deficient azaaromatics: Strategies of functionalization of pyridines, quinolines, and diazines. Adv. Heterocycl. Chem. 1991, 52, 187–304. [Google Scholar]
- Turck, A.; Plé, N.; Mongin, F.; Queguiner, G. Advances in the directed metallation of azines and diazines (pyridines, pyrimidines, pyrazines, pyridazines, quinolines, benzodiazines and carbolines). Part 2: Metallation of pyrimidines, pyrazines, pyridazines and benzodiazines. Tetrahedron 2001, 57, 4489–4505. [Google Scholar] [CrossRef]
- Chevallier, F.; Mongin, F. Functionalization of diazines and benzo derivatives through deprotonated intermediates. Chem. Soc. Rev. 2008, 37, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Bisballe, N.; Hedidi, M.; Demmer, C.S.; Chevallier, F.; Roisnel, T.; Dorcet, V.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Bentabed-Ababsa, G.; et al. Functionalization of oxazolo[4,5-b]pyrazines by deprotometallation. Eur. J. Org. Chem. 2018, 2018, 3904–3913. [Google Scholar] [CrossRef]
- Hedidi, M.; Erb, W.; Lassagne, F.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Roisnel, T.; Bentabed-Ababsa, G.; Mongin, F. Functionalization of pyridyl ketones using deprotolithiation-in situ zincation. RSC Adv. 2016, 6, 63185–63189. [Google Scholar] [CrossRef]
- Hedidi, M.; Maillard, J.; Erb, W.; Lassagne, F.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Roisnel, T.; Dorcet, V.; Hamzé, M.; et al. Fused systems based on 2-aminopyrimidines: Synthesis combining deprotolithiation-in situ zincation with N-arylation reactions and biological properties. Eur. J. Org. Chem. 2017, 2017, 5903–5915. [Google Scholar] [CrossRef]
- Antilla, J.C.; Baskin, J.M.; Barder, T.E.; Buchwald, S.L. Copper-diamine-catalyzed n-arylation of pyrroles, pyrazoles, indazoles, imidazoles, and triazoles. J. Org. Chem. 2004, 69, 5578–5587. [Google Scholar] [CrossRef]
- Gao, L.; Song, Y.; Zhang, X.; Guo, S.; Fan, X. Copper-catalyzed tandem reactions of 2-bromobenzaldehydes/ketones with aminopyrazoles toward the synthesis of pyrazolo[1,5-a]quinazolines. Tetrahedron Lett. 2014, 55, 4997–5002. [Google Scholar] [CrossRef]
- Teo, Y.-C.; Yong, F.-F.; Sim, S. Ligand-free cu2o-catalyzed cross coupling of nitrogen heterocycles with iodopyridines. Tetrahedron 2013, 69, 7279–7284. [Google Scholar] [CrossRef]
- Akimoto, G.; Otsuka, M.; Takita, R.; Uchiyama, M.; Hedidi, M.; Bentabed-Ababsa, G.; Lassagne, F.; Erb, W.; Mongin, F. Deprotonative metalation of methoxy-substituted arenes using lithium 2,2,6,6-tetramethylpiperidide: Experimental and computational study. J. Org. Chem. 2018, 83, 13498–13506. [Google Scholar] [CrossRef]
- Plé, N.; Turck, A.; Couture, K.; Quéguiner, G. Diazines 13: Metalation without ortho-directing group—Functionalization of diazines via direct metalation. J. Org. Chem. 1995, 60, 3781–3786. [Google Scholar] [CrossRef]
- Seggio, A.; Chevallier, F.; Vaultier, M.; Mongin, F. Lithium-mediated zincation of pyrazine, pyridazine, pyrimidine, and quinoxaline. J. Org. Chem. 2007, 72, 6602–6605. [Google Scholar] [CrossRef]
- Anderson, R.J.; Morris, J.C. Total synthesis of variolin B. Tetrahedron Lett. 2001, 42, 8697–8699. [Google Scholar] [CrossRef]
- Anderson, R.J.; Hill, J.B.; Morris, J.C. Concise total syntheses of variolin B and deoxyvariolin B. J. Org. Chem. 2005, 70, 6204–6212. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.J.; Morris, J.C. Studies toward the total synthesis of the variolins: Rapid entry to the core structure. Tetrahedron Lett. 2001, 42, 311–313. [Google Scholar] [CrossRef]
- Marquise, N.; Nguyen, T.T.; Chevallier, F.; Picot, L.; Thiery, V.; Lozach, O.; Bach, S.; Ruchaud, S.; Mongin, F. Azine and diazine functionalization using 2,2,6,6-tetramethylpiperidino-based lithium-metal combinations: Application to the synthesis of 5,9-disubstituted pyrido[3′,2′:4,5]pyrrolo[1,2-c]pyrimidines. Synlett 2015, 26, 2811–2816. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Kotha, S.; Lahiri, K.; Kashinath, D. Recent applications of the Suzuki-Miyaura cross-coupling reaction in organic synthesis. Tetrahedron 2002, 58, 9633–9695. [Google Scholar] [CrossRef]
- Sreeshailam, A.; Dayaker, G.; Ramana, D.V.; Chevallier, F.; Roisnel, T.; Komagawa, S.; Takita, R.; Uchiyama, M.; Radha Krishna, P.; Mongin, F. Synthesis of both enantiomers of ferrocene[1,2-c]1H-quinolin-2-one by diastereoselective deproto-zincation of sugar-derived ferrocene esters. RSC Adv. 2012, 2, 7030–7032. [Google Scholar] [CrossRef][Green Version]
- Alberico, D.; Scott, M.E.; Lautens, M. Aryl-aryl bond formation by transition-metal-catalyzed direct arylation. Chem. Rev. 2007, 107, 174–238. [Google Scholar] [CrossRef]
- Chen, X.; Engle, K.M.; Wang, D.-H.; Yu, J.-Q. Palladium(II)-catalyzed C-H activation/C-C cross-coupling reactions: Versatility and practicality. Angew. Chem. Int. Ed. 2009, 48, 5094–5115. [Google Scholar] [CrossRef]
- Ackermann, L.; Vicente, R.; Kapdi, A.R. Transition metal-catalyzed direct arylation of (hetero)arenes by C-H bond cleavage. Angew. Chem. Int. Ed. 2009, 48, 9792–9826. [Google Scholar] [CrossRef]
- Kadiyala, R.R.; Tilly, D.; Nagaradja, E.; Roisnel, T.; Matulis, V.E.; Ivashkevich, O.A.; Halauko, Y.S.; Chevallier, F.; Gros, P.C.; Mongin, F. Computed CH acidity of biaryl compounds and their deprotonative metalation by using a mixed lithium/zinc-TMP base. Chem. Eur. J. 2013, 19, 7944–7960. [Google Scholar] [CrossRef]
- Hedidi, M.; Bentabed-Ababsa, G.; Derdour, A.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Chevallier, F.; Roisnel, T.; Dorcet, V.; Mongin, F. Deprotometalation of substituted pyridines and regioselectivity-computed CH acidity relationships. Tetrahedron 2016, 72, 2196–2205. [Google Scholar] [CrossRef]
- Elslager, E.F.; Haley, N.F.; McLean, J.R.; Potoczak, D.; Veloso, H.; Wheelock, R.H. Inhibitors of platelet aggregation. 3. {[(dialkylamino)alkyl]thio} heterocyclic compounds. J. Med. Chem. 1972, 15, 61–65. [Google Scholar] [CrossRef]
- Yang, B.; Mao, Z.; Zhu, X.; Wan, Y. Functionalised chitosan as a green, recyclable, supported catalyst for the copper-catalysed ullmann C-N coupling reaction in water. Catal. Commun. 2015, 60, 92–95. [Google Scholar] [CrossRef]
- Lu, Z.; Twieg, R.J. A mild and practical copper-catalyzed amination of halothiophenes. Tetrahedron 2005, 61, 903–918. [Google Scholar] [CrossRef]
- Hellwinkel, D.; Ittemann, P. 12-Organyldibenz[b,g]azocine-5,7-diones. Chem. Ber. 1986, 119, 3165–3197. [Google Scholar] [CrossRef]
- Bouarfa, S.; Bentabed-Ababsa, G.; Erb, W.; Picot, L.; Thiery, V.; Roisnel, T.; Dorcet, V.; Mongin, F. Iodothiophenes and related compounds as coupling partners in copper-mediated n-arylation of anilines. Synthesis 2021, 53, 1271–1284. [Google Scholar]
- Bouarfa, S.; Grassl, S.; Ivanova, M.; Langlais, T.; Bentabed-Ababsa, G.; Lassagne, F.; Erb, W.; Roisnel, T.; Dorcet, V.; Knochel, P.; et al. Copper- and cobalt-catalyzed syntheses of thiophene-based tertiary amines. Eur. J. Org. Chem. 2019, 2019, 3244–3258. [Google Scholar] [CrossRef]
- Zeghada, S.; Bentabed-Ababsa, G.; Mongin, O.; Erb, W.; Picot, L.; Thiery, V.; Roisnel, T.; Dorcet, V.; Mongin, F. 2-Aminobenzaldehyde, a common precursor to acridines and acridones endowed with bioactivities. Tetrahedron 2020, 76, 131435. [Google Scholar] [CrossRef]
- Bouarfa, S.; Jehan, P.; Erb, W.; Mongin, O.; Poree, F.-H.; Roisnel, T.; Bentabed-Ababsa, G.; Le Yondre, N.; Mongin, F. Aza-aromatic polycycles based on triphenylene and acridine or acridone: Synthesis and properties. New J. Chem. 2021, 45, 14414–14424. [Google Scholar] [CrossRef]
- Li, X.-A.; Wang, H.-L.; Yang, S.-D. Sc(OTf)3-catalyzed dehydrogenative cyclization for synthesis of N-methylacridones. Org. Lett. 2013, 15, 1794–1797. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari Brikci-Nigassa, N.; Bentabed-Ababsa, G.; Erb, W.; Chevallier, F.; Picot, L.; Vitek, L.; Fleury, A.; Thiéry, V.; Souab, M.; Robert, T.; et al. 2-Aminophenones, a common precursor to N-aryl isatins and acridines endowed with bioactivities. Tetrahedron 2018, 74, 1785–1801. [Google Scholar] [CrossRef]
- Mokhtari Brikci-Nigassa, N.; Nauton, L.; Moreau, P.; Mongin, O.; Duval, R.E.; Picot, L.; Thiery, V.; Souab, M.; Baratte, B.; Ruchaud, S.; et al. Functionalization of 9-thioxanthone at the 1-position: From arylamino derivatives to [1]benzo(thio)pyrano[4,3,2-de]benzothieno[2,3-b]quinolines of biological interest. Bioorg. Chem. 2020, 94, 103347. [Google Scholar] [CrossRef] [PubMed]


























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mongin, F.; Erb, W.; Lassagne, F. Aromatic Iodides: Synthesis and Conversion to Heterocycles. Chem. Proc. 2022, 12, 20. https://doi.org/10.3390/ecsoc-26-13641
Mongin F, Erb W, Lassagne F. Aromatic Iodides: Synthesis and Conversion to Heterocycles. Chemistry Proceedings. 2022; 12(1):20. https://doi.org/10.3390/ecsoc-26-13641
Chicago/Turabian StyleMongin, Florence, William Erb, and Frédéric Lassagne. 2022. "Aromatic Iodides: Synthesis and Conversion to Heterocycles" Chemistry Proceedings 12, no. 1: 20. https://doi.org/10.3390/ecsoc-26-13641
APA StyleMongin, F., Erb, W., & Lassagne, F. (2022). Aromatic Iodides: Synthesis and Conversion to Heterocycles. Chemistry Proceedings, 12(1), 20. https://doi.org/10.3390/ecsoc-26-13641