
Progress in Understanding Metabolic Syndrome and Knowledge of Its Complex Pathophysiology

 , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
2. Historical Background
3. Definition and Diagnostic Criteria
4. Prevalence of Metabolic Syndrome
5. Pathophysiology
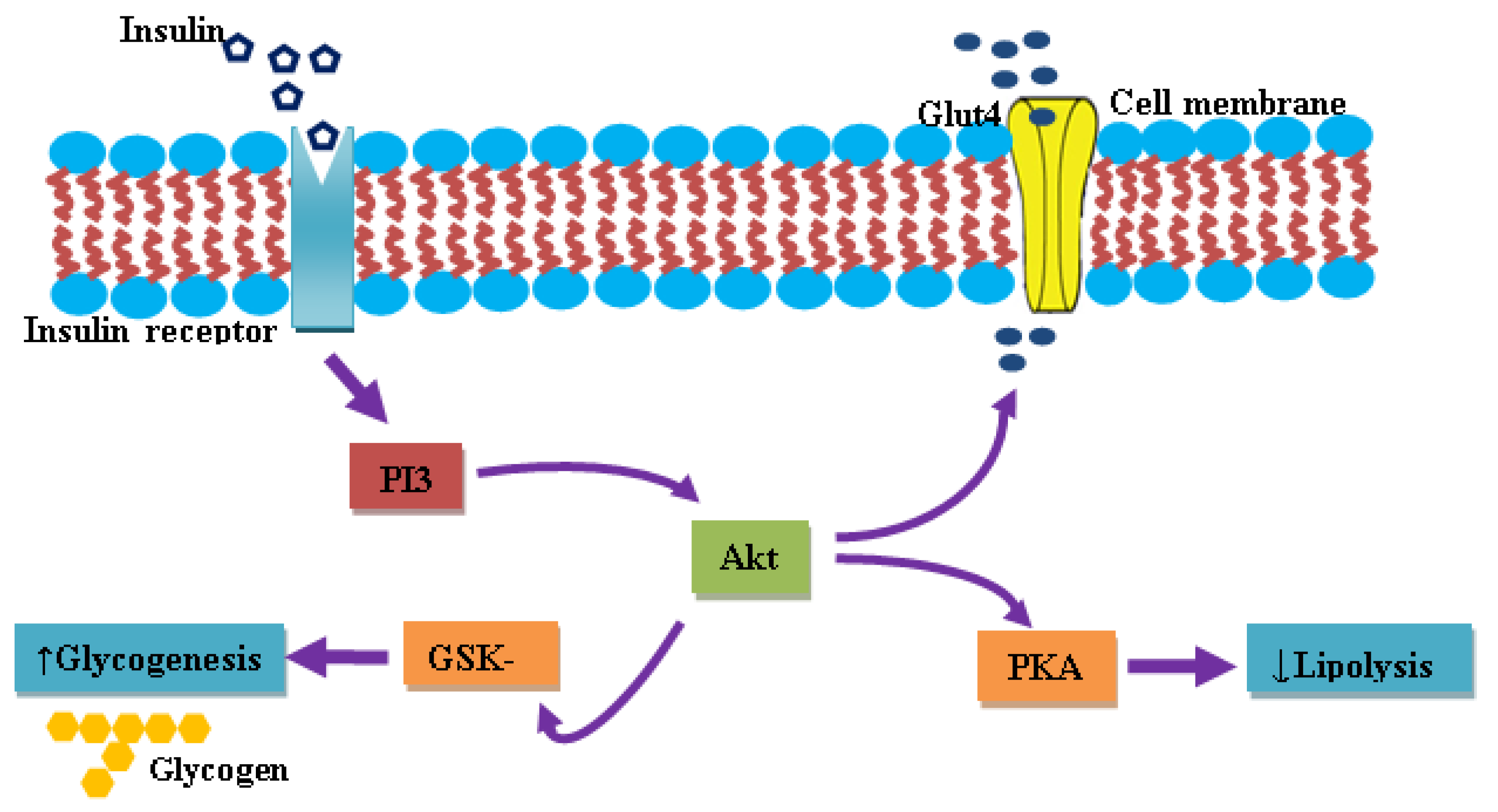
5.1. Insulin and IR
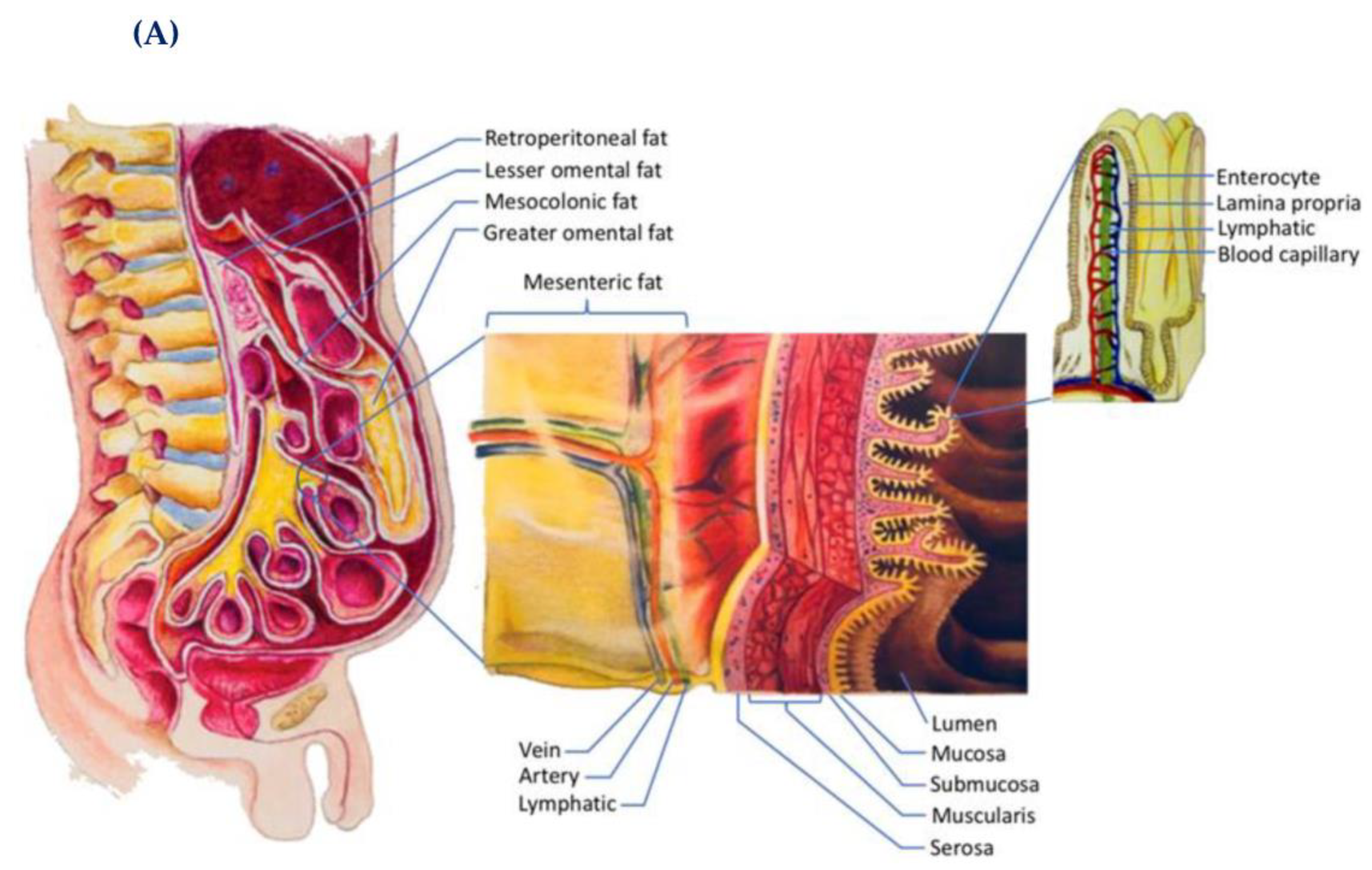
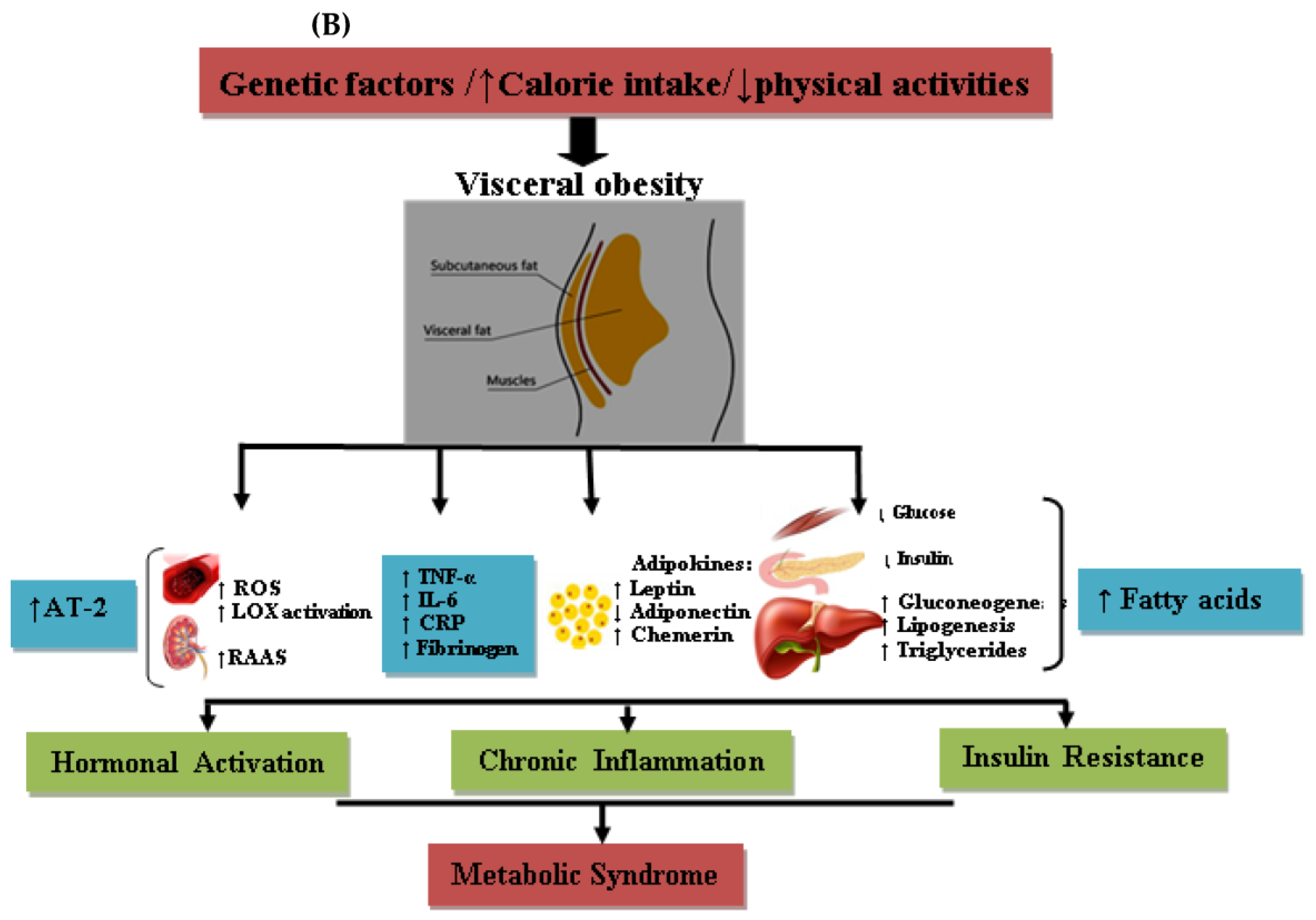
5.2. Adipose Tissue and Its Endocrine Activities
5.3. Leptin
5.4. Adiponectin
5.5. Resistin
5.6. Visfatin
5.7. Chemerin
5.8. Inflammatory Status and Oxidative Stress
6. Metabolomics and Proteomics: Emerging Powerful Tools for Precision Medicine to Treat MetS
6.1. Metabolomics
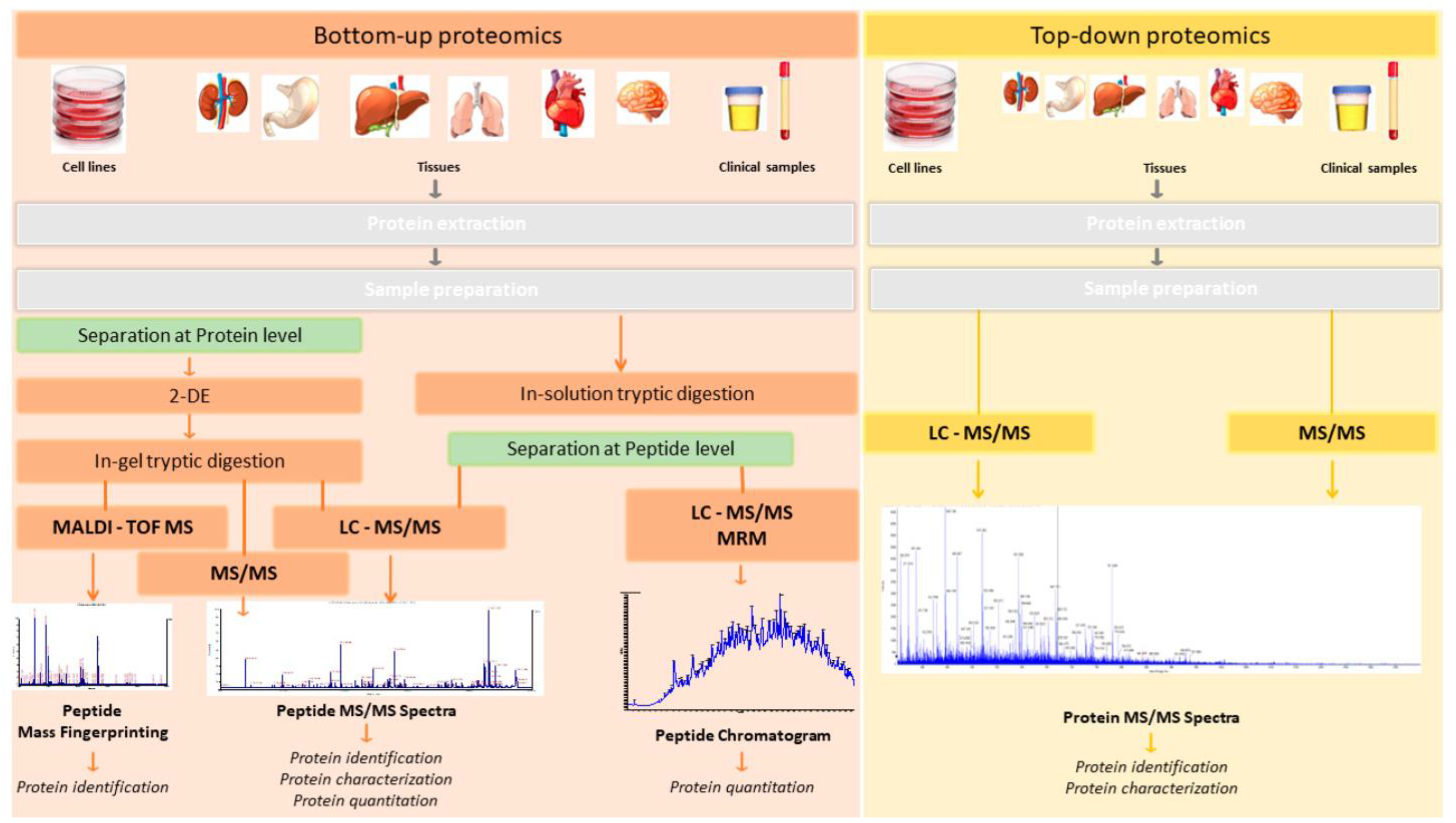
6.2. Proteomics
7. Metabolic Syndrome and Its Association with CVDs
7.1. Impaired Glucose Regulation and CVDs
7.2. Obesity, Central Fat Distribution, and CVDs
7.3. Hypertriglyceridemia and CVDs
7.4. Hypertension and CVDs
7.5. Microalbuminuria and CVDs
7.6. Association between IR and CVDs
7.7. Fibrinolysis in Acute and Chronic CVDs
7.8. Uric Acid and CVDs
8. T2DM and MetS
9. Obesity and MetS
10. MetS and COVID-19 Association
10.1. Diabetes and COVID-19
10.2. Obesity and COVID-19
10.3. Hypertension and COVID-19
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.; Zimmet, P.; Shaw, J.; IDF Epidemiology Task Force Consensus Group. The metabolic syndrome—A new worldwide definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Olijhoek, J.K.; van der Graaf, Y.; Banga, J.D.; Algra, A.; Rabelink, T.J.; Visseren, F.L.; SMART Study Group. The metabolic syndrome is associated with advanced vascular damage in patients with coronary heart disease, stroke, peripheral arterial disease or abdominal aortic aneurysm. Eur. Heart J. 2004, 25, 342–348. [Google Scholar] [CrossRef]
- Kaur, J. A comprehensive review on metabolic syndrome. Cardiol. Res. Pract. 2014, 2014, 943162. [Google Scholar] [CrossRef] [PubMed]
- Apridonidze, T.; Essah, P.A.; Iuorno, M.J.; Nestler, J.E. Prevalence and characteristics of the metabolic syndrome in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2005, 90, 1929–1935. [Google Scholar] [CrossRef]
- Boura-Halfon, S.; Zick, Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E581–E591. [Google Scholar] [CrossRef] [PubMed]
- Sarafidis, P.A.; Nilsson, P.M. The metabolic syndrome: A glance at its history. J. Hypertens. 2006, 24, 621–626. [Google Scholar] [CrossRef]
- Reaven, G.M. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef]
- Oda, E. Metabolic syndrome: Its history, mechanisms, and limitations. Acta Diabetol. 2012, 49, 89–95. [Google Scholar] [CrossRef]
- Vague, J. Sexual differentiation; Factor determining forms of obesity. Presse Med. 1947, 55, 339. [Google Scholar]
- Gupta, A.; Gupta, V. Metabolic syndrome: What are the risks for humans? Biosci. Trends 2010, 4, 204–212. [Google Scholar] [PubMed]
- Avogaro, P.; Crepaldi, G.; Enzi, G.; Tiengo, A. Metabolic aspects of essential obesity. Epatologia 1965, 11, 226–238. [Google Scholar] [PubMed]
- Athyros, V.G.; Tziomalos, K.; Karagiannis, A.; Mikhailidis, D.P. Dyslipidaemia of obesity, metabolic syndrome and type 2 diabetes mellitus: The case for residual risk reduction after statin treatment. Open. Cardiovasc. Med. J. 2011, 5, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, N.M. The deadly quartet and the insulin resistance syndrome: An historical overview. Hypertens. Res. 1996, 19 (Suppl. S1), S9–S11. [Google Scholar] [CrossRef]
- Haffner, S.M.; Valdez, R.A.; Hazuda, H.P.; Mitchell, B.D.; Morales, P.A.; Stern, M.P. Prospective analysis of the insulin-resistance syndrome (syndrome X). Diabetes 1992, 41, 715–722. [Google Scholar] [CrossRef]
- Sypniewska, G. Pro-Inflammatory and Prothrombotic Factors and Metabolic Syndrome. EJIFCC 2007, 18, 39–46. [Google Scholar]
- Alessi, M.C.; Juhan-Vague, I. Contribution of PAI-1 in cardiovascular pathology. Arch. Mal. Coeur Vaiss. 2004, 97, 673–678. [Google Scholar]
- Hollman, G.; Kristenson, M. The prevalence of the metabolic syndrome and its risk factors in a middle-aged Swedish population—Mainly a function of overweight? Eur. J. Cardiovasc. Nurs. 2008, 7, 21–26. [Google Scholar] [CrossRef]
- Alberti, K.G.; Zimmet, P.Z. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet. Med. 1998, 15, 539–553. [Google Scholar] [CrossRef]
- Balkau, B.; Charles, M.A. Comment on the provisional report from the WHO consultation. European Group for the Study of Insulin Resistance (EGIR). Diabet. Med. 1999, 16, 442–443. [Google Scholar] [CrossRef]
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001, 285, 2486–2497. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Spence, C.; Yang, J.W.; Ma, G.X. The IDF Definition Is Better Suited for Screening Metabolic Syndrome and Estimating Risks of Diabetes in Asian American Adults: Evidence from NHANES 2011–2016. J. Clin. Med. 2020, 9, 3871. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.J.; Critchley, J.A.; Chan, J.C.; Cockram, C.S.; Lee, Z.S.; Thomas, G.N.; Tomlinson, B. Factor analysis of the metabolic syndrome: Obesity vs insulin resistance as the central abnormality. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, K.; Iso, H. The criteria for metabolic syndrome and the national health screening and education system in Japan. Epidemiol. Health 2017, 39, e2017003. [Google Scholar] [CrossRef] [PubMed]
- Desroches, S.; Lamarche, B. The evolving definitions and increasing prevalence of the metabolic syndrome. Appl. Physiol. Nutr. Metab. 2007, 32, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Mohanty, P.; Dhindsa, S.; Syed, T.; Ghanim, H.; Aljada, A.; Dandona, P. Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes 2003, 52, 2882–2887. [Google Scholar] [CrossRef]
- Ervin, R.B. Prevalence of metabolic syndrome among adults 20 years of age and over, by sex, age, race and ethnicity, and body mass index: United States, 2003–2006. Natl. Health Stat. Report. 2009, 13, 1–7. [Google Scholar]
- Alberti, K.G.; Zimmet, P.; Shaw, J. Metabolic syndrome—A new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet. Med. 2006, 23, 469–480. [Google Scholar] [CrossRef]
- Nestel, P.; Lyu, R.; Low, L.P.; Sheu, W.H.; Nitiyanant, W.; Saito, I.; Tan, C.E. Metabolic syndrome: Recent prevalence in East and Southeast Asian populations. Asia Pac. J. Clin. Nutr. 2007, 16, 362–367. [Google Scholar]
- Cameron, A.J.; Shaw, J.E.; Zimmet, P.Z. The metabolic syndrome: Prevalence in worldwide populations. Endocrinol. Metab. Clin. N. Am. 2004, 33, 351–375. [Google Scholar] [CrossRef]
- Pan, W.H.; Yeh, W.T.; Weng, L.C. Epidemiology of metabolic syndrome in Asia. Asia Pac. J. Clin. Nutr. 2008, 17 (Suppl. S1), 37–42. [Google Scholar] [PubMed]
- Aryannejad, A.; Eghtesad, S.; Rahimi, Z.; Mohammadi, Z.; Malihi, R.; Danehchin, L.; Paridar, Y.; Abolnezhadian, F.; Cheraghian, B.; Mard, A.; et al. Metabolic syndrome and lifestyle-associated factors in the ethnically diverse population of Khuzestan, Iran: A cross-sectional study. J. Diabetes Metab. Disord. 2021, 20, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Ko, G.T.; Cockram, C.S.; Chow, C.C.; Yeung, V.; Chan, W.B.; So, W.Y.; Chan, N.N.; Chan, J.C. High prevalence of metabolic syndrome in Hong Kong Chinese—Comparison of three diagnostic criteria. Diabetes Res. Clin. Pract. 2005, 69, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Jha, B.K.; Sherpa, M.L.; Dahal, B.K.; Singh, J.K. Prevalence of Metabolic Syndrome and Its Components in Adults with Central Obesity at Janakpur Zone, Nepal. J. Nepal. Health Res. Counc. 2021, 18, 681–685. [Google Scholar] [CrossRef]
- Gupta, R.; Deedwania, P.C.; Gupta, A.; Rastogi, S.; Panwar, R.B.; Kothari, K. Prevalence of metabolic syndrome in an Indian urban population. Int. J. Cardiol. 2004, 97, 257–261. [Google Scholar] [CrossRef]
- Misra, A.; Khurana, L. Obesity and the metabolic syndrome in developing countries. J. Clin. Endocrinol. Metab. 2008, 93, S9–S30. [Google Scholar] [CrossRef]
- Sawant, A.; Mankeshwar, R.; Shah, S.; Raghavan, R.; Dhongde, G.; Raje, H.; D’Souza, S.; Subramanium, A.; Dhairyawan, P.; Todur, S.; et al. Prevalence of metabolic syndrome in urban India. Cholesterol 2011, 2011, 920983. [Google Scholar] [CrossRef]
- Matsuzawa, Y.; Funahashi, T.; Nakamura, T. The concept of metabolic syndrome: Contribution of visceral fat accumulation and its molecular mechanism. J. Atheroscler. Thromb. 2011, 18, 629–639. [Google Scholar] [CrossRef]
- Seneff, S.; Wainwright, G.; Mascitelli, L. Is the metabolic syndrome caused by a high fructose, and relatively low fat, low cholesterol diet? Arch. Med. Sci. 2011, 7, 8–20. [Google Scholar] [CrossRef]
- Nauli, A.M.; Matin, S. Why Do Men Accumulate Abdominal Visceral Fat? Front. Physiol. 2019, 10, 1486. [Google Scholar] [CrossRef]
- Roberts, C.K.; Hevener, A.L.; Barnard, R.J. Metabolic syndrome and insulin resistance: Underlying causes and modification by exercise training. Compr. Physiol. 2013, 3, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar] [PubMed]
- Shepherd, P.R. Mechanisms regulating phosphoinositide 3-kinase signalling in insulin-sensitive tissues. Acta Physiol. Scand. 2005, 183, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Tooke, J.E.; Hannemann, M.M. Adverse endothelial function and the insulin resistance syndrome. J. Intern. Med. 2000, 247, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Shulman, G.I. Free fatty acids in obesity and type 2 diabetes: Defining their role in the development of insulin resistance and beta-cell dysfunction. Eur. J. Clin. Investig. 2002, 32 (Suppl. S3), 14–23. [Google Scholar] [CrossRef]
- Ahima, R.S.; Flier, J.S. Adipose tissue as an endocrine organ. Trends Endocrinol. Metab. 2000, 11, 327–332. [Google Scholar] [CrossRef]
- Lee, D.K.; George, S.R.; O’Dowd, B.F. Unravelling the roles of the apelin system: Prospective therapeutic applications in heart failure and obesity. Trends Pharmacol. Sci. 2006, 27, 190–194. [Google Scholar] [CrossRef]
- Fukuhara, A.; Matsuda, M.; Nishizawa, M.; Segawa, K.; Tanaka, M.; Kishimoto, K.; Matsuki, Y.; Murakami, M.; Ichisaka, T.; Murakami, H.; et al. Visfatin: A protein secreted by visceral fat that mimics the effects of insulin. Science 2005, 307, 426–430. [Google Scholar] [CrossRef]
- Revilla, M.; Puig-Oliveras, A.; Crespo-Piazuelo, D.; Criado-Mesas, L.; Castello, A.; Fernandez, A.I.; Ballester, M.; Folch, J.M. Expression analysis of candidate genes for fatty acid composition in adipose tissue and identification of regulatory regions. Sci. Rep. 2018, 8, 2045. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.; Cline, D.L.; Glavas, M.M.; Covey, S.D.; Kieffer, T.J. Tissue-Specific Effects of Leptin on Glucose and Lipid Metabolism. Endocr. Rev. 2021, 42, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.M.; McMahon, A.D.; Packard, C.J.; Kelly, A.; Shepherd, J.; Gaw, A.; Sattar, N. Plasma leptin and the risk of cardiovascular disease in the west of Scotland coronary prevention study (WOSCOPS). Circulation 2001, 104, 3052–3056. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Ohishi, M.; Kihara, S.; Funahashi, T.; Nakamura, T.; Nagaretani, H.; Kumada, M.; Ohashi, K.; Okamoto, Y.; Nishizawa, H.; et al. Association of hypoadiponectinemia with impaired vasoreactivity. Hypertension 2003, 42, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, I.; Hammer, R.E.; Ikemoto, S.; Brown, M.S.; Goldstein, J.L. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature 1999, 401, 73–76. [Google Scholar] [CrossRef]
- Pischon, T.; Girman, C.J.; Hotamisligil, G.S.; Rifai, N.; Hu, F.B.; Rimm, E.B. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA 2004, 291, 1730–1737. [Google Scholar] [CrossRef]
- Ebihara, K.; Ogawa, Y.; Masuzaki, H.; Shintani, M.; Miyanaga, F.; Aizawa-Abe, M.; Hayashi, T.; Hosoda, K.; Inoue, G.; Yoshimasa, Y.; et al. Transgenic overexpression of leptin rescues insulin resistance and diabetes in a mouse model of lipoatrophic diabetes. Diabetes 2001, 50, 1440–1448. [Google Scholar] [CrossRef]
- Jung, U.J.; Choi, M.S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef]
- Ukkola, O.; Santaniemi, M. Adiponectin: A link between excess adiposity and associated comorbidities? J. Mol. Med. 2002, 80, 696–702. [Google Scholar] [CrossRef]
- Yoon, M.J.; Lee, G.Y.; Chung, J.J.; Ahn, Y.H.; Hong, S.H.; Kim, J.B. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes 2006, 55, 2562–2570. [Google Scholar] [CrossRef]
- Lindsay, R.S.; Funahashi, T.; Hanson, R.L.; Matsuzawa, Y.; Tanaka, S.; Tataranni, P.A.; Knowler, W.C.; Krakoff, J. Adiponectin and development of type 2 diabetes in the Pima Indian population. Lancet 2002, 360, 57–58. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Singh, A.K.; Gupta, V.; Kumar, S.; Srivastava, N.; Jafar, T.; Pant, A.B. Association of circulating resistin with metabolic risk factors in Indian females having metabolic syndrome. Toxicol. Int. 2011, 18, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Way, J.M.; Gorgun, C.Z.; Tong, Q.; Uysal, K.T.; Brown, K.K.; Harrington, W.W.; Oliver, W.R., Jr.; Willson, T.M.; Kliewer, S.A.; Hotamisligil, G.S. Adipose tissue resistin expression is severely suppressed in obesity and stimulated by peroxisome proliferator-activated receptor gamma agonists. J. Biol. Chem. 2001, 276, 25651–25653. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.R.; Lazar, M.A. Human resistin: Found in translation from mouse to man. Trends Endocrinol. Metab. 2011, 22, 259–265. [Google Scholar] [CrossRef]
- Tripathi, D.; Kant, S.; Pandey, S.; Ehtesham, N.Z. Resistin in metabolism, inflammation, and disease. FEBS J. 2020, 287, 3141–3149. [Google Scholar] [CrossRef]
- Abdalla, M.M.I. Role of visfatin in obesity-induced insulin resistance. World J. Clin. Cases 2022, 10, 10840–10851. [Google Scholar] [CrossRef]
- Helfer, G.; Wu, Q.F. Chemerin: A multifaceted adipokine involved in metabolic disorders. J. Endocrinol. 2018, 238, R79–R94. [Google Scholar] [CrossRef]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J. Immunol. 2007, 178, 1748–1758. [Google Scholar] [CrossRef]
- Abdelkader, H.; Alani, A.W.; Alany, R.G. Recent advances in non-ionic surfactant vesicles (niosomes): Self-assembly, fabrication, characterization, drug delivery applications and limitations. Drug. Deliv. 2014, 21, 87–100. [Google Scholar] [CrossRef]
- Okla, M.; Kim, J.; Koehler, K.; Chung, S. Dietary Factors Promoting Brown and Beige Fat Development and Thermogenesis. Adv. Nutr. 2017, 8, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxid. Med. Cell. Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Murray, D.L.; Choy, L.N.; Spiegelman, B.M. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 4854–4858. [Google Scholar] [CrossRef] [PubMed]
- Pant, S.; Deshmukh, A.; Gurumurthy, G.S.; Pothineni, N.V.; Watts, T.E.; Romeo, F.; Mehta, J.L. Inflammation and atherosclerosis—revisited. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Laclaustra, M.; Corella, D.; Ordovas, J.M. Metabolic syndrome pathophysiology: The role of adipose tissue. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 125–139. [Google Scholar] [CrossRef]
- Castell, J.V.; Gomez-Lechon, M.J.; David, M.; Andus, T.; Geiger, T.; Trullenque, R.; Fabra, R.; Heinrich, P.C. Interleukin-6 is the major regulator of acute phase protein synthesis in adult human hepatocytes. FEBS Lett. 1989, 242, 237–239. [Google Scholar] [CrossRef]
- Clish, C.B. Metabolomics: An emerging but powerful tool for precision medicine. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000588. [Google Scholar] [CrossRef]
- Kuehnbaum, N.L.; Britz-McKibbin, P. New advances in separation science for metabolomics: Resolving chemical diversity in a post-genomic era. Chem. Rev. 2013, 113, 2437–2468. [Google Scholar] [CrossRef]
- van der Greef, J.; van Wietmarschen, H.; van Ommen, B.; Verheij, E. Looking back into the future: 30 years of metabolomics at TNO. Mass. Spectrom. Rev. 2013, 32, 399–415. [Google Scholar] [CrossRef]
- Members, M.S.I.B.; Sansone, S.A.; Fan, T.; Goodacre, R.; Griffin, J.L.; Hardy, N.W.; Kaddurah-Daouk, R.; Kristal, B.S.; Lindon, J.; Mendes, P.; et al. The metabolomics standards initiative. Nat. Biotechnol. 2007, 25, 846–848. [Google Scholar] [CrossRef]
- Chantada-Vazquez, M.D.P.; Bravo, S.B.; Barbosa-Gouveia, S.; Alvarez, J.V.; Couce, M.L. Proteomics in Inherited Metabolic Disorders. Int. J. Mol. Sci. 2022, 23, 4744. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.W.; Huang, C.C. Recent Progress in Metabolic Syndrome Research and Therapeutics. Int. J. Mol. Sci. 2021, 22, 6862. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Paudel, K.R.; Kim, D.W. Preventive Effect of traditional korean formulations on intimal thickening of rat carotid artery injured by balloon catheter. Korean J. Plant Resour. 2013, 26, 678–685. [Google Scholar] [CrossRef]
- Jun, M.Y.; Karki, R.; Paudel, K.R.; Sharma, B.R.; Adhikari, D.; Kim, D.-W. Alkaloid rich fraction from Nelumbo nucifera targets VSMC proliferation and migration to suppress restenosis in balloon-injured rat carotid artery. Atherosclerosis 2016, 248, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-H.; Paudel, K.R.; Jeong, J.; Wi, A.-J.; Park, W.-S.; Kim, D.-W.; Oak, M.-H. Antiatherogenic effect of Camellia japonica fruit extract in high fat diet-fed rats. Evid.-Based Complement. Altern. Med. 2016, 2016, 9679867. [Google Scholar]
- Paudel, K.R.; Lee, U.W.; Kim, D.W. Chungtaejeon, a Korean fermented tea, prevents the risk of atherosclerosis in rats fed a high-fat atherogenic diet. J. Integr. Med. 2016, 14, 134–142. [Google Scholar] [CrossRef]
- Panth, N.; Paudel, K.R.; Gong, D.-S.; Oak, M.-H. Vascular protection by ethanol extract of Morus alba root bark: Endothelium-dependent relaxation of rat aorta and decrease of smooth muscle cell migration and proliferation. Evid.-Based Complement. Altern. Med. 2018, 2018, 7905763. [Google Scholar] [CrossRef]
- Ceriello, A. Impaired glucose tolerance and cardiovascular disease: The possible role of post-prandial hyperglycemia. Am. Heart J. 2004, 147, 803–807. [Google Scholar] [CrossRef]
- Hsueh, W.A.; Orloski, L.; Wyne, K. Prediabetes: The importance of early identification and intervention. Postgrad. Med. 2010, 122, 129–143. [Google Scholar] [CrossRef]
- Qiao, Q.; Jousilahti, P.; Eriksson, J.; Tuomilehto, J. Predictive properties of impaired glucose tolerance for cardiovascular risk are not explained by the development of overt diabetes during follow-up. Diabetes Care 2003, 26, 2910–2914. [Google Scholar] [CrossRef]
- Jin, C.; Chen, S.; Vaidya, A.; Wu, Y.; Wu, Z.; Hu, F.B.; Kris-Etherton, P.; Wu, S.; Gao, X. Longitudinal Change in Fasting Blood Glucose and Myocardial Infarction Risk in a Population Without Diabetes. Diabetes Care 2017, 40, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Rijkelijkhuizen, J.M.; Nijpels, G.; Heine, R.J.; Bouter, L.M.; Stehouwer, C.D.; Dekker, J.M. High risk of cardiovascular mortality in individuals with impaired fasting glucose is explained by conversion to diabetes: The Hoorn study. Diabetes Care 2007, 30, 332–336. [Google Scholar] [CrossRef]
- Parizadeh, D.; Rahimian, N.; Akbarpour, S.; Azizi, F.; Hadaegh, F. Sex-specific clinical outcomes of impaired glucose status: A long follow-up from the Tehran Lipid and Glucose Study. Eur. J. Prev. Cardiol. 2019, 26, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Esteghamati, A.; Gouya, M.M.; Abbasi, M.; Delavari, A.; Alikhani, S.; Alaedini, F.; Safaie, A.; Forouzanfar, M.; Gregg, E.W. Prevalence of diabetes and impaired fasting glucose in the adult population of Iran: National Survey of Risk Factors for Non-Communicable Diseases of Iran. Diabetes Care 2008, 31, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Kabootari, M.; Hasheminia, M.; Azizi, F.; Mirbolouk, M.; Hadaegh, F. Change in glucose intolerance status and risk of incident cardiovascular disease: Tehran Lipid and Glucose Study. Cardiovasc. Diabetol. 2020, 19, 41. [Google Scholar] [CrossRef]
- Harris, T.B.; Ballard-Barbasch, R.; Madans, J.; Makuc, D.M.; Feldman, J.J. Overweight, weight loss, and risk of coronary heart disease in older women. The NHANES I Epidemiologic Follow-up Study. Am. J. Epidemiol. 1993, 137, 1318–1327. [Google Scholar] [CrossRef]
- Kannel, W.B.; LeBauer, E.J.; Dawber, T.R.; McNamara, P.M. Relation of body weight to development of coronary heart disease. The Framingham study. Circulation 1967, 35, 734–744. [Google Scholar] [CrossRef]
- Knowler, W.C.; Pettitt, D.J.; Savage, P.J.; Bennett, P.H. Diabetes incidence in Pima indians: Contributions of obesity and parental diabetes. Am. J. Epidemiol. 1981, 113, 144–156. [Google Scholar] [CrossRef]
- Stamler, R.; Stamler, J.; Riedlinger, W.F.; Algera, G.; Roberts, R.H. Weight and blood pressure. Findings in hypertension screening of 1 million Americans. JAMA 1978, 240, 1607–1610. [Google Scholar] [CrossRef]
- Hubert, H.B.; Feinleib, M.; McNamara, P.M.; Castelli, W.P. Obesity as an independent risk factor for cardiovascular disease: A 26-year follow-up of participants in the Framingham Heart Study. Circulation 1983, 67, 968–977. [Google Scholar] [CrossRef]
- Manson, J.E.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C.; Rosner, B.; Monson, R.R.; Speizer, F.E.; Hennekens, C.H. A prospective study of obesity and risk of coronary heart disease in women. N. Engl. J. Med. 1990, 322, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Larsson, B.; Svardsudd, K.; Welin, L.; Wilhelmsen, L.; Bjorntorp, P.; Tibblin, G. Abdominal adipose tissue distribution, obesity, and risk of cardiovascular disease and death: 13 year follow up of participants in the study of men born in 1913. Br. Med. J. (Clin. Res. Ed) 1984, 288, 1401–1404. [Google Scholar] [CrossRef] [PubMed]
- Ohlson, L.O.; Larsson, B.; Svardsudd, K.; Welin, L.; Eriksson, H.; Wilhelmsen, L.; Bjorntorp, P.; Tibblin, G. The influence of body fat distribution on the incidence of diabetes mellitus. 13.5 years of follow-up of the participants in the study of men born in 1913. Diabetes 1985, 34, 1055–1058. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Benjamin, I.J.; Burke, G.L.; Chait, A.; Eckel, R.H.; Howard, B.V.; Mitch, W.; Smith, S.C., Jr.; Sowers, J.R. Diabetes and cardiovascular disease: A statement for healthcare professionals from the American Heart Association. Circulation 1999, 100, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Pouliot, M.C.; Despres, J.P.; Lemieux, S.; Moorjani, S.; Bouchard, C.; Tremblay, A.; Nadeau, A.; Lupien, P.J. Waist circumference and abdominal sagittal diameter: Best simple anthropometric indexes of abdominal visceral adipose tissue accumulation and related cardiovascular risk in men and women. Am. J. Cardiol. 1994, 73, 460–468. [Google Scholar] [CrossRef]
- Paudel, K.R.; Karki, R.; Kim, D.W. Cepharanthine inhibits in vitro VSMC proliferation and migration and vascular inflammatory responses mediated by RAW264.7. Toxicol. In Vitro 2016, 34, 16–25. [Google Scholar] [CrossRef]
- Paudel, K.R.; Kim, D.W. Microparticles-Mediated Vascular Inflammation and its Amelioration by Antioxidant Activity of Baicalin. Antioxidants 2020, 9, 890. [Google Scholar] [CrossRef]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Bairey Merz, C.N.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2014, 63, 2889–2934. [Google Scholar] [CrossRef]
- Mora, S.; Caulfield, M.P.; Wohlgemuth, J.; Chen, Z.; Superko, H.R.; Rowland, C.M.; Glynn, R.J.; Ridker, P.M.; Krauss, R.M. Atherogenic Lipoprotein Subfractions Determined by Ion Mobility and First Cardiovascular Events After Random Allocation to High-Intensity Statin or Placebo: The Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) Trial. Circulation 2015, 132, 2220–2229. [Google Scholar] [CrossRef]
- Mora, S.; Wenger, N.K.; Demicco, D.A.; Breazna, A.; Boekholdt, S.M.; Arsenault, B.J.; Deedwania, P.; Kastelein, J.J.; Waters, D.D. Determinants of residual risk in secondary prevention patients treated with high- versus low-dose statin therapy: The Treating to New Targets (TNT) study. Circulation 2012, 125, 1979–1987. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Varbo, A. Triglycerides and cardiovascular disease. Lancet 2014, 384, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, S.E. Hypertension and cardiovascular risk: General aspects. Pharmacol. Res. 2018, 129, 95–99. [Google Scholar] [CrossRef]
- Weir, M.R. Microalbuminuria and cardiovascular disease. Clin. J. Am. Soc. Nephrol. 2007, 2, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuniga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef]
- Kietsiriroje, N.; Ariens, R.A.S.; Ajjan, R.A. Fibrinolysis in Acute and Chronic Cardiovascular Disease. Semin. Thromb. Hemost. 2021, 47, 490–505. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsen, L.; Svardsudd, K.; Korsan-Bengtsen, K.; Larsson, B.; Welin, L.; Tibblin, G. Fibrinogen as a risk factor for stroke and myocardial infarction. N. Engl. J. Med. 1984, 311, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Thogersen, A.M.; Jansson, J.H.; Boman, K.; Nilsson, T.K.; Weinehall, L.; Huhtasaari, F.; Hallmans, G. High plasminogen activator inhibitor and tissue plasminogen activator levels in plasma precede a first acute myocardial infarction in both men and women: Evidence for the fibrinolytic system as an independent primary risk factor. Circulation 1998, 98, 2241–2247. [Google Scholar] [CrossRef]
- Kohler, H.P.; Grant, P.J. Plasminogen-activator inhibitor type 1 and coronary artery disease. N. Engl. J. Med. 2000, 342, 1792–1801. [Google Scholar] [CrossRef]
- Muiesan, M.L.; Agabiti-Rosei, C.; Paini, A.; Salvetti, M. Uric Acid and Cardiovascular Disease: An Update. Eur. Cardiol. 2016, 11, 54–59. [Google Scholar] [CrossRef]
- Ndrepepa, G. Uric acid and cardiovascular disease. Clin. Chim. Acta 2018, 484, 150–163. [Google Scholar] [CrossRef]
- Souto, E.B.; Souto, S.B.; Campos, J.R.; Severino, P.; Pashirova, T.N.; Zakharova, L.Y.; Silva, A.M.; Durazzo, A.; Lucarini, M.; Izzo, A.A.; et al. Nanoparticle Delivery Systems in the Treatment of Diabetes Complications. Molecules 2019, 24, 4209. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.Y.; Al-Salami, H.; Dass, C.R. Potential of insulin nanoparticle formulations for oral delivery and diabetes treatment. J. Control. Release 2017, 264, 247–275. [Google Scholar] [CrossRef] [PubMed]
- Neef, T.; Miller, S.D. Tolerogenic Nanoparticles to Treat Islet Autoimmunity. Curr. Diab Rep. 2017, 17, 84. [Google Scholar] [CrossRef] [PubMed]
- Veiseh, O.; Tang, B.C.; Whitehead, K.A.; Anderson, D.G.; Langer, R. Managing diabetes with nanomedicine: Challenges and opportunities. Nat. Rev. Drug. Discov. 2015, 14, 45–57. [Google Scholar] [CrossRef]
- Lin, C.H.; Chen, C.H.; Lin, Z.C.; Fang, J.Y. Recent advances in oral delivery of drugs and bioactive natural products using solid lipid nanoparticles as the carriers. J. Food Drug. Anal. 2017, 25, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Bahman, F.; Greish, K.; Taurin, S. Nanotechnology in Insulin Delivery for Management of Diabetes. Pharm. Nanotechnol. 2019, 7, 113–128. [Google Scholar] [CrossRef]
- Mohsen, A.M. Nanotechnology Advanced Strategies for the Management of Diabetes Mellitus. Curr. Drug. Targets 2019, 20, 995–1007. [Google Scholar] [CrossRef]
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front. Endocrinol. 2013, 4, 37. [Google Scholar] [CrossRef]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K.; International Agency for Research on Cancer Handbook Working Group. Body Fatness and Cancer--Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef]
- Bouchard, C. Defining the genetic architecture of the predisposition to obesity: A challenging but not insurmountable task. Am. J. Clin. Nutr. 2010, 91, 5–6. [Google Scholar] [CrossRef]
- Speakman, J.R. Thrifty genes for obesity and the metabolic syndrome--time to call off the search? Diab Vasc. Dis. Res. 2006, 3, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Tsou, Y.H.; Wang, B.; Ho, W.; Hu, B.; Tang, P.; Sweet, S.; Zhang, X.Q.; Xu, X. Nanotechnology-Mediated Drug Delivery for the Treatment of Obesity and Its Related Comorbidities. Adv. Healthc. Mater. 2019, 8, e1801184. [Google Scholar] [CrossRef] [PubMed]
- Yameen, B.; Choi, W.I.; Vilos, C.; Swami, A.; Shi, J.; Farokhzad, O.C. Insight into nanoparticle cellular uptake and intracellular targeting. J. Control. Release 2014, 190, 485–499. [Google Scholar] [CrossRef]
- Rosen, E.D.; Spiegelman, B.M. What we talk about when we talk about fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef]
- Rytka, J.M.; Wueest, S.; Schoenle, E.J.; Konrad, D. The portal theory supported by venous drainage-selective fat transplantation. Diabetes 2011, 60, 56–63. [Google Scholar] [CrossRef]
- Vishvanath, L.; Gupta, R.K. Contribution of adipogenesis to healthy adipose tissue expansion in obesity. J. Clin. Investig. 2019, 129, 4022–4031. [Google Scholar] [CrossRef]
- Geurts, L.; Neyrinck, A.M.; Delzenne, N.M.; Knauf, C.; Cani, P.D. Gut microbiota controls adipose tissue expansion, gut barrier and glucose metabolism: Novel insights into molecular targets and interventions using prebiotics. Benef. Microbes 2014, 5, 3–17. [Google Scholar] [CrossRef]
- Panth, N.; Paudel, K.R.; Parajuli, K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv. Med. 2016, 2016, 9152732. [Google Scholar] [CrossRef]
- Imran, M.; Jin, X.; Ali, M.; Tapfumaneyi, P.; Lelasseur, P.; Carlo, L.; Jude, A.; Bourg, A.L.; Panchal, B.; Dick, A. The Pandemic and Your Skin—Direct and Indirect Impact of COVID-19. Cosmetics 2023, 10, 34. [Google Scholar] [CrossRef]
- Ashique, S.; Gupta, K.; Gupta, G.; Mishra, N.; Singh, S.K.; Wadhwa, S.; Gulati, M.; Dureja, H.; Zacconi, F.; Oliver, B.G.; et al. Vitamin D-A prominent immunomodulator to prevent COVID-19 infection. Int. J. Rheum. Dis. 2023, 26, 13–30. [Google Scholar] [CrossRef]
- Paudel, K.R.; Patel, V.; Vishwas, S.; Gupta, S.; Sharma, S.; Chan, Y.; Jha, N.K.; Shrestha, J.; Imran, M.; Panth, N.; et al. Nutraceuticals and COVID-19: A mechanistic approach toward attenuating the disease complications. J. Food Biochem. 2022, e14445. [Google Scholar] [CrossRef]
- Sathish, T.; Kapoor, N.; Cao, Y.; Tapp, R.J.; Zimmet, P. Proportion of newly diagnosed diabetes in COVID-19 patients: A systematic review and meta-analysis. Diabetes Obes. Metab. 2021, 23, 870–874. [Google Scholar] [CrossRef]
- Roy, S. Metabolic Syndrome and COVID-19. 2021. Available online: https://www.vumc.org/viiii/immuknow/metabolic-syndrome-and-covid-19 (accessed on 11 March 2023).
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1alpha/Glycolysis-Dependent Axis. Cell. Metab. 2020, 32, 437–446.e435. [Google Scholar] [CrossRef]
- Thomas, T.; Stefanoni, D.; Reisz, J.A.; Nemkov, T.; Bertolone, L.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hansen, K.C.; Hod, E.A.; et al. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight 2020, 5, e140327. [Google Scholar] [CrossRef]
- Xie, J.; Zu, Y.; Alkhatib, A.; Pham, T.T.; Gill, F.; Jang, A.; Radosta, S.; Chaaya, G.; Myers, L.; Zifodya, J.S.; et al. Metabolic Syndrome and COVID-19 Mortality Among Adult Black Patients in New Orleans. Diabetes Care 2020, 44, 188–193. [Google Scholar] [CrossRef]
- Metwally, A.A.; Mehta, P.; Johnson, B.S.; Nagarjuna, A.; Snyder, M.P. COVID-19-Induced New-Onset Diabetes: Trends and Technologies. Diabetes 2021, 70, 2733–2744. [Google Scholar] [CrossRef]
- Hosseinzadeh, R.; Goharrizi, M.; Bahardoust, M.; Alvanegh, A.G.; Ataee, M.R.; Bagheri, M.; Navidiyan, E.S.; Zijoud, S.R.H.; Heiat, M. Should all patients with hypertension be worried about developing severe coronavirus disease 2019 (COVID-19)? Clin. Hypertens. 2021, 27, 3. [Google Scholar] [CrossRef]
- Akpek, M. Does COVID-19 Cause Hypertension? Angiology 2022, 73, 682–687. [Google Scholar] [CrossRef]
- Muntner, P.; Hardy, S.T.; Fine, L.J.; Jaeger, B.C.; Wozniak, G.; Levitan, E.B.; Colantonio, L.D. Trends in Blood Pressure Control Among US Adults With Hypertension, 1999-2000 to 2017-2018. JAMA 2020, 324, 1190–1200. [Google Scholar] [CrossRef]
- Singh, R.; Rathore, S.S.; Khan, H.; Karale, S.; Chawla, Y.; Iqbal, K.; Bhurwal, A.; Tekin, A.; Jain, N.; Mehra, I.; et al. Association of Obesity With COVID-19 Severity and Mortality: An Updated Systemic Review, Meta-Analysis, and Meta-Regression. Front. Endocrinol. 2022, 13, 780872. [Google Scholar] [CrossRef]
- Ohno, M.; Dzurova, D. Body Mass Index and Risk for COVID-19-Related Hospitalization in Adults Aged 50 and Older in Europe. Nutrients 2022, 14, 4001. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; He, J.; Xue, Y.; Yang, X.; Liu, S.; Gong, Z. Role of Hypertension on the Severity of COVID-19: A Review. J. Cardiovasc. Pharmacol. 2021, 78, e648–e655. [Google Scholar] [CrossRef]
- Bress, A.P.; Cohen, J.B.; Anstey, D.E.; Conroy, M.B.; Ferdinand, K.C.; Fontil, V.; Margolis, K.L.; Muntner, P.; Millar, M.M.; Okuyemi, K.S.; et al. Inequities in Hypertension Control in the United States Exposed and Exacerbated by COVID-19 and the Role of Home Blood Pressure and Virtual Health Care During and After the COVID-19 Pandemic. J. Am. Heart Assoc. 2021, 10, e020997. [Google Scholar] [CrossRef]
- Mackman, N.; Antoniak, S.; Wolberg, A.S.; Kasthuri, R.; Key, N.S. Coagulation abnormalities and thrombosis in patients infected with SARS-CoV-2 and other pandemic viruses. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2033–2044. [Google Scholar] [CrossRef]
- Mohamed, N.E.; Benn, E.K.T.; Astha, V.; Okhawere, K.E.; Korn, T.G.; Nkemdirim, W.; Rambhia, A.; Ige, O.A.; Funchess, H.; Mihalopoulos, M.; et al. Association between chronic kidney disease and COVID-19-related mortality in New York. World J. Urol. 2021, 39, 2987–2993. [Google Scholar] [CrossRef]
- Rovetta, A.; Bhagavathula, A.S. The Impact of COVID-19 on Mortality in Italy: Retrospective Analysis of Epidemiological Trends. JMIR Public Health Surveill. 2022, 8, e36022. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Parameters | Criteria | ||||||
|---|---|---|---|---|---|---|---|
| Central Obesity | FBS | ↑ TG | ↓ HDL-C | ↑ BP | Other | Diagnosed as MetS, If | |
| WHO (1998) [19] | Waist/hip ratio Male: >0.9 cm Female: >0.85 or BMI > 30 kg/m2 | ≥110 mg/dL or IR or T2DM or Rx | ≥150 mg/dL | Male: <40 mg/dL Female: <50 mg/dL | Diastolic ≥ 140 and systolic ≥ 90 mmHg | Microalbuminuria | Absolutely required IR plus ≥ 2 criteria |
| EGIR (1999) [20] | WC Male: ≥94 cm Female: ≥80 cm | ≥108.11 mg/dL | ≥150 mg/dL | <39 mg/dL | Diastolic ≥ 140 and/or systolic ≥ 90 mmHg or Rx | Absolutely required IR plus ≥ 2 criteria | |
| IDF (2005) [22] | WC defined in terms of ethnicity specific values | ≥100 mg/dL or Rx | ≥150 mg/dL or Rx | Male: <40 mg/dL Female: <50 mg/dL | Diastolic ≥ 130 and/or systolic ≥ 85 mmHg or Rx | Absolutely required central obesity plus ≥ 2 criteria | |
| AHA/NHLBI (2005) [24] | WC Male: ≥102 cm Female: ≥88 cm | ≥100 mg/dL or Rx | ≥150 mg/dL or Rx | Male: <40 mg/dL Female: <50 mg/dL | Diastolic ≥ 130 and/or systolic ≥ 85 mmHg or Rx | ≥3 criteria | |
| AHA/NHLBI and IDF:2009 [25] | WC defined in terms of population- and country-based specific definition | ≥100 mg/dL or Rx | ≥150 mg/dL or Rx | Male: <40 mg/dL Female: <50 mg/dL | Diastolic ≥ 130 and/or systolic ≥ 85 mmHg or Rx | ≥3 criteria | |
| Country/Ethnic group | Waist Circumference | |
|---|---|---|
| Male | Female | |
| Europids | ≥94 cm | ≥80 cm |
| South Asians Based on Chinese, Malay, and Asian–Indian population | ≥90 cm | ≥80 |
| Chinese | ≥90 cm | 80 cm |
| Japanese | ≥90 cm | ≥80 cm |
| Ethnic South and Central Americans | Use South Asian recommendation until more specific data are available | |
| Sub-Saharan Africans | Use European data until more specific data are available. | |
| Eastern Mediterranean and Middle East (Arab) population | Use European data until more specific data are available. | |
| Population | Organization | Recommended Waist Circumference | |
|---|---|---|---|
| Male | Female | ||
| Caucasian | WHO | ≥94 cm (increased risk) ≥102 cm (still higher risk) | ≥80 cm (increased risk) ≥88 cm (still higher risk) |
| United States, Canada, and European | AHA/NHLBI (“ATP III”), Health Canada, and European Cardiovascular Societies | ≥102 cm | ≥88 cm |
| Japanese | Japanese Obesity Society | ≥85 cm | ≥90 cm |
| China | Cooperative Task Force | ≥80 cm | |
| Middle East, Mediterranean, Europid, Asian, and Sub-Saharan African | IDF | ≥94 cm | ≥80 cm |
| Ethnic Central and South American | ≥90 cm | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jha, B.K.; Sherpa, M.L.; Imran, M.; Mohammed, Y.; Jha, L.A.; Paudel, K.R.; Jha, S.K. Progress in Understanding Metabolic Syndrome and Knowledge of Its Complex Pathophysiology. Diabetology 2023, 4, 134-159. https://doi.org/10.3390/diabetology4020015
Jha BK, Sherpa ML, Imran M, Mohammed Y, Jha LA, Paudel KR, Jha SK. Progress in Understanding Metabolic Syndrome and Knowledge of Its Complex Pathophysiology. Diabetology. 2023; 4(2):134-159. https://doi.org/10.3390/diabetology4020015
Chicago/Turabian StyleJha, Birendra Kumar, Mingma Lhamu Sherpa, Mohammad Imran, Yousuf Mohammed, Laxmi Akhileshwar Jha, Keshav Raj Paudel, and Saurav Kumar Jha. 2023. "Progress in Understanding Metabolic Syndrome and Knowledge of Its Complex Pathophysiology" Diabetology 4, no. 2: 134-159. https://doi.org/10.3390/diabetology4020015
APA StyleJha, B. K., Sherpa, M. L., Imran, M., Mohammed, Y., Jha, L. A., Paudel, K. R., & Jha, S. K. (2023). Progress in Understanding Metabolic Syndrome and Knowledge of Its Complex Pathophysiology. Diabetology, 4(2), 134-159. https://doi.org/10.3390/diabetology4020015