
Effects of Acute Muscle Contraction on the Key Molecules in Insulin and Akt Signaling in Skeletal Muscle in Health and in Insulin Resistant States


Abstract
:1. Introduction
2. Methods
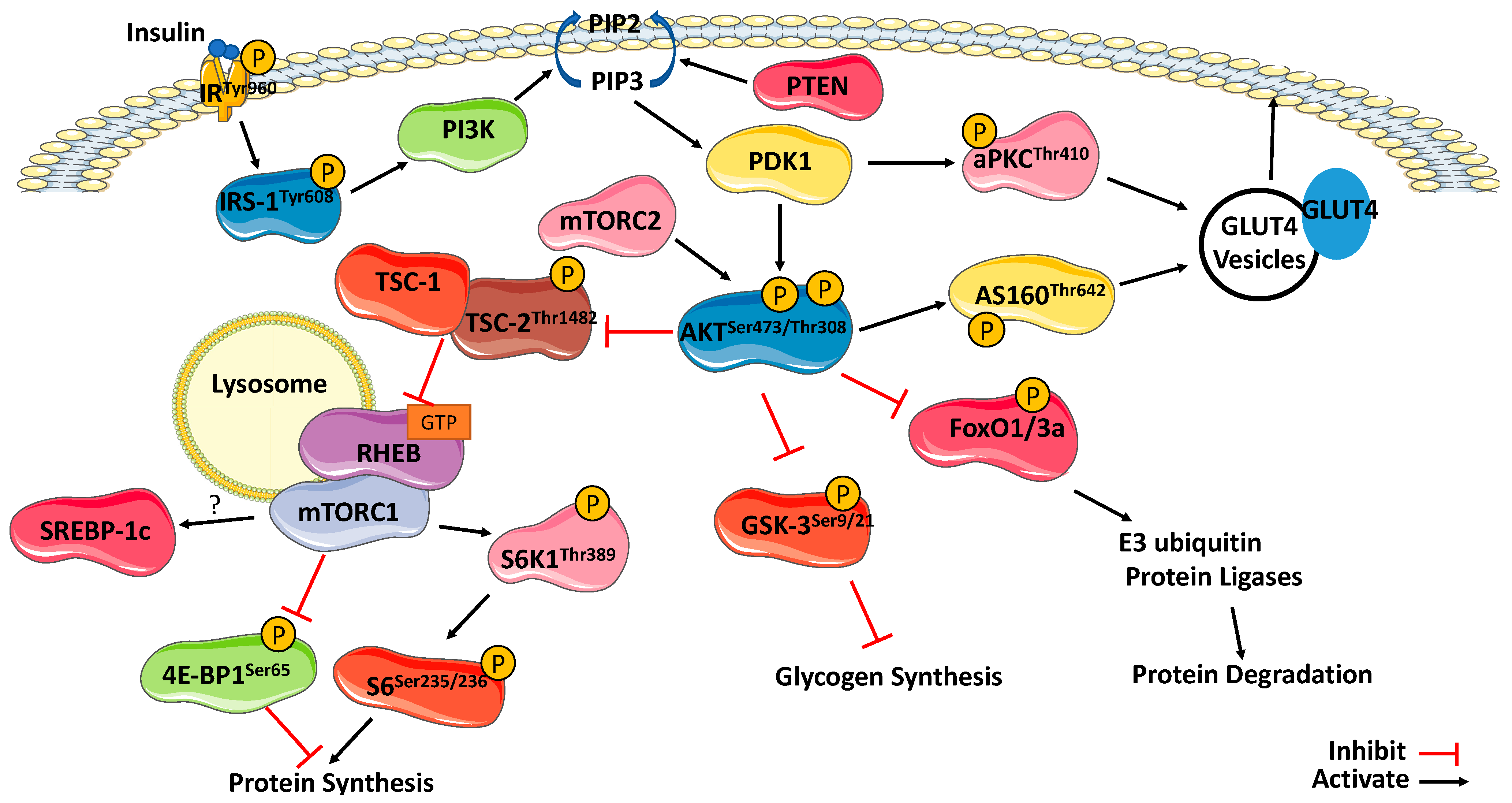
3. Insulin Signaling within Skeletal Muscle
3.1. Insulin Receptor and Insulin Receptor Substrates
3.2. IRS-1/PI3K/Akt Pathway
3.3. Substrates of Akt
3.4. mTORC1
3.5. Substrates of mTORC1
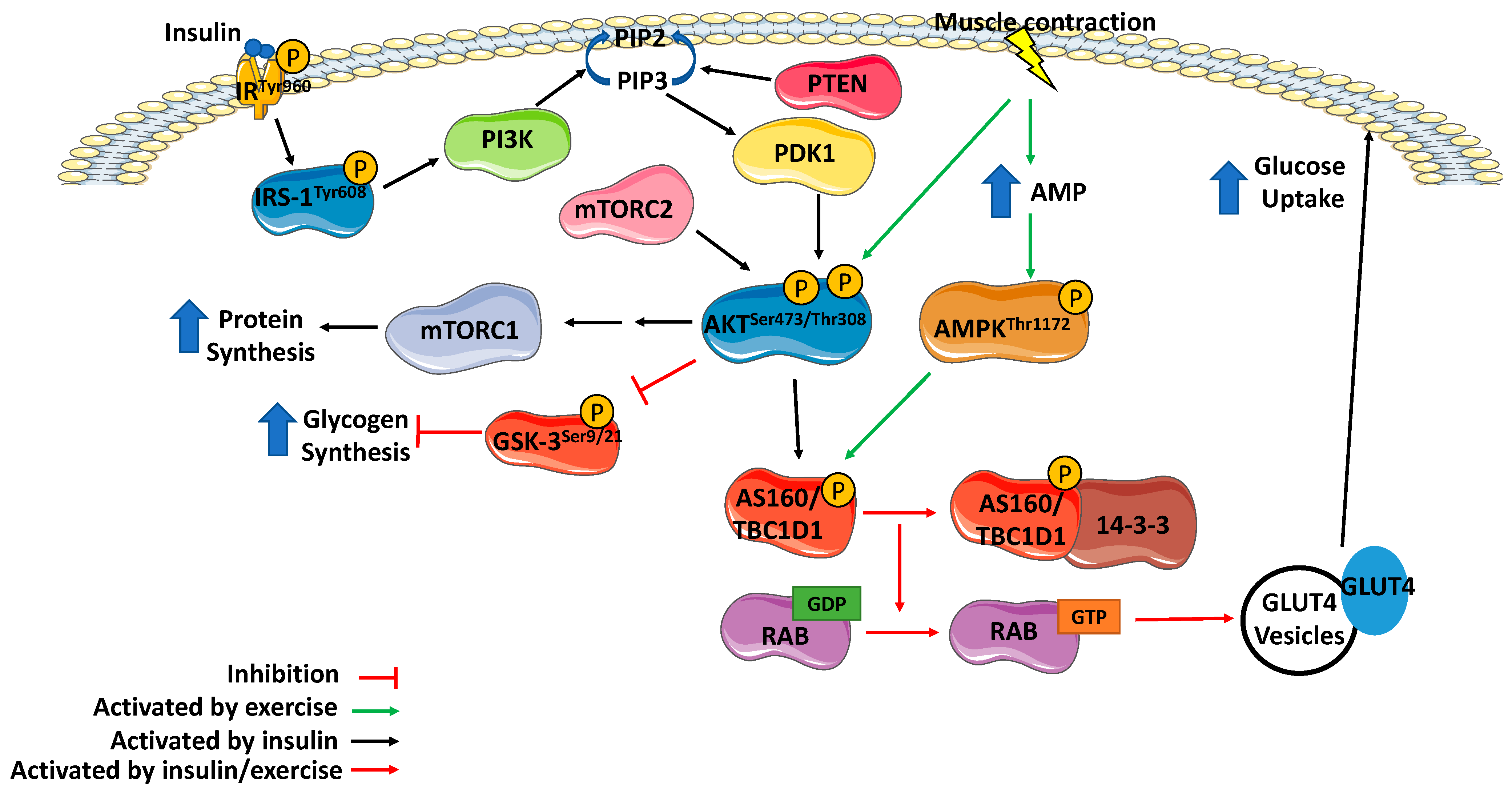
4. Effect of Exercise on Insulin Signaling
4.1. Effect of Exercise on Insulin Receptor and Insulin Receptor Substrate
4.2. Effect of Exercise on PI3K
4.3. Effect of Exercise on AKT
4.4. Effect of Exercise on Akt substrates
4.5. Additional Substates of Akt
4.6. Effect of Exercise on mTORC1 and Its Substrates
4.7. Exercise Prescription
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Yamakawa, H.; Kusumoto, D.; Hashimoto, H.; Yuasa, S. Stem cell aging in skeletal muscle regeneration and disease. Int. J. Mol. Sci. 2020, 21, 1830. [Google Scholar] [CrossRef] [Green Version]
- Baron, A.D.; Brechtel, G.; Wallace, P.; Edelman, S.V. Rates and Tissue Sites of Noninsulin- and Insulin-Mediated Glucose Uptake in Humans. Am. J. Physiol.-Endocrinol. Metab. 1988, 255, 769–774. [Google Scholar] [CrossRef] [PubMed]
- De Fronzo, R.A.; Ferrannini, E.; Sato, Y.; Felig, P.; Wahren, J. Synergistic interaction between exercise and insulin on peripheral glucose uptake. J. Clin. Investig. 1981, 68, 1468–1474. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- Kleinert, M.; Sylow, L.; Richter, E.A. Regulation of glycogen synthase in muscle and its role in Type 2 diabetes. Diabetes Manag. 2013, 3, 81–90. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.A.; Defronzo, R.A. Pathogenesis of insulin resistance in skeletal muscle. J. Biomed. Biotechnol. 2010, 2010, 476279. [Google Scholar] [CrossRef] [Green Version]
- Frøsig, C.; Richter, E.A. Improved insulin sensitivity after exercise: Focus on insulin signaling. Obesity 2009, 17 (Suppl. S3), S15–S20. [Google Scholar] [CrossRef] [PubMed]
- Sylow, L.; Kleinert, M.; Richter, E.A.; Jensen, T.E. Exercise-stimulated glucose uptake-regulation and implications for glycaemic control. Nat. Rev. Endocrinol. 2017, 13, 133–148. [Google Scholar] [CrossRef]
- Sellami, M.; Gasmi, M.; Denham, J.; Hayes, L.D.; Stratton, D.; Padulo, J.; Bragazzi, N. Effects of acute and chronic exercise on immunological parameters in the elderly aged: Can physical activity counteract the effects of aging? Front. Immunol. 2018, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Dela, F.; Larsen, J.J.; Mikines, K.J.; Ploug, T.; Petersen, L.N. Insulin-stimulated muscle glucose clearance in patients with NIDDM. Effects of One-Legged Physical Training. Diabetes 1995, 44, 1010–1020. [Google Scholar] [CrossRef]
- Houmard, J.A.; Egan, P.C.; Neufer, P.D.; Friedman, J.E.; Wheeler, W.S.; Israel, R.G.; Dohm, G.L. Elevated skeletal muscle glucose transporter levels in exercise-trained middle-aged men. Am. J. Physiol.-Endocrinol. Metab. 1991, 261, E437–E443. [Google Scholar] [CrossRef] [PubMed]
- Hughes, V.A.; Fiatarone, M.A.; Fielding, R.A.; Kahn, B.B.; Ferrara, C.M.; Shepherd, P.; Fisher, E.C.; Wolfe, R.R.; Elahi, D.; Evans, W.J. Exercise increases muscle GLUT-4 levels and insulin action in subjects with impaired glucose tolerance. Am. J. Physiol.-Endocrinol. Metab. 1993, 264, 855–862. [Google Scholar] [CrossRef]
- Yu, M.; Blomstrand, E.; Chibalin, A.V.; Wallberg-Henriksson, H.; Zierath, J.R.; Krook, A. Exercise-associated differences in an array of proteins involved in signal transduction and glucose transport. J. Appl. Physiol. 2001, 90, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Chibalin, A.V.; Yu, M.; Ryder, J.W.; Song, X.M.; Galuska, D.; Krook, A.; Wallberg-Henriksson, H.; Zierath, J.R. Exercise-induced changes in expression and activity of proteins involved in insulin signal transduction in skeletal muscle: Differential effects on insulin-receptor substrates 1 and 2. Proc. Natl. Acad. Sci. USA 2000, 97, 38–43. [Google Scholar] [CrossRef] [Green Version]
- Musi, N.; Fujii, N.; Hirshman, M.F.; Ekberg, I.; Fröberg, S.; Ljungqvist, O.; Thorell, A.; Goodyear, L.J. AMP-Activated Protein Kinase (AMPK) Is Activated in Muscle of Subjects with Type 2 Diabetes During Exercise. Diabetes 2001, 50, 921–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, I.K.; Katz, A.; Wahren, J. Splanchnic and muscle metabolism during exercise in NIDDM patients. Am. J. Physiol.-Endocrinol. Metab. 1995, 269, 583–590. [Google Scholar] [CrossRef]
- Kahn, C.R.; White, M.F. The insulin receptor and the molecular mechanism of insulin action. J. Clin. Investig. 1988, 82, 1151–1156. [Google Scholar] [CrossRef] [Green Version]
- Soliman, G.A. The integral role of mTOR in lipid metabolism. Cell Cycle 2011, 10, 861–862. [Google Scholar] [CrossRef] [Green Version]
- Hoxjah, G.; Hughes-Hallett, J.; Timson, R.; Ilagan, E.; Yuan, M.; Asara, J.; Ben-Sahra, I.; Manning, B. The mTORC1 signaling network senses changes in cellular purine nucleotide levels. Cell Rep. 2017, 21, 1331–1346. [Google Scholar]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Pederson, T.M.; Kramer, D.L.; Rondinone, C.M. Serine/threonine phosphorylation of IRS-1 triggers its degradation: Possible regulation by tyrosine phosphorylation. Diabetes 2001, 50, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Thirone, A.C.P.; Huang, X.; Klip, A. Differential contribution of insulin receptor substrates 1 versus 2 to insulin signaling and glucose uptake in L6 myotubes. J. Biol. Chem. 2005, 280, 19426–19435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckstein, S.S.; Weigert, C.; Lehmann, R. Divergent Roles of IRS (Insulin Receptor Substrate) 1 and 2 in Liver and Skeletal Muscle. Curr. Med. Chem. 2017, 24, 1827–1852. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; Lavan, B.E.; Wang, Q.; Jenkins, N.A.; Gilbert, D.J.; Copeland, N.G.; Keller, S.R.; Lienhard, G.E. Cloning, tissue expression, and chromosomal location of the mouse insulin receptor substrate 4 gene. Endocrinology 1999, 140, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Dhe-Paganon, S.; Melendez, P.A.; Lee, J.; Shoelson, S.E. Two new substrates in insulin signaling, IRS5/DOK4 and IRS6/DOK5. J. Biol. Chem. 2003, 278, 25323–25330. [Google Scholar] [CrossRef] [Green Version]
- Metz, H.E.; Houghton, A.M.G. Insulin receptor substrate regulation of phosphoinositide 3-kinase. Clin. Cancer Res. 2011, 17, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Vadas, O.; Burke, J.E.; Zhang, X.; Berndt, A.; Williams, R.L. Structural biology structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci. Signal. 2011, 4, re2. [Google Scholar] [CrossRef]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.J.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultze, S.M.; Jensen, J.; Hemmings, B.A.; Tschopp, O.; Niessen, M. Promiscuous affairs of PKB/AKT isoforms in metabolism. Arch. Physiol. Biochem. 2011, 117, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B.; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef]
- Dugani, C.B.; Klip, A. Glucose transporter 4: Cycling, compartments and controversies. EMBO Rep. 2005, 6, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Farese, R.V. Function and dysfunction of aPKC isoforms for glucose transport in insulin-sensitive and insulin-resistant states. Am. J. Physiol.-Endocrinol. Metab. 2002, 283, E1–E11. [Google Scholar] [CrossRef]
- Standaert, M.L.; Bandyopadhyay, G.; Kanoh, Y.; Sajan, M.P.; Farese, R.V. Insulin and PIP3 activate PKC-ζ by mechanisms that are both dependent and independent of phosphorylation of activation loop (T410) and autophosphorylation (T560) sites. Biochemistry 2001, 40, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Standaert, M.L.; Bandyopadhyay, G.; Perez, L.; Price, D.; Galloway, L.; Poklepovic, A.; Sajan, M.P.; Cenni, V.; Sirri, A.; Moscat, J.; et al. Insulin activates protein kinases C-ζ and C-λ by an autophosphorylation-dependent mechanism and stimulates their translocation to GLUT4 vesicles and other membrane fractions in rat adipocytes. J. Biol. Chem. 1999, 274, 25308–25316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanoh, Y.; Sajan, M.P.; Bandyopadhyay, G.; Miura, A.; Standaert, M.L.; Farese, R.V. Defective activation of atypical protein kinase C ζ and λ by insulin and phosphatidylinositol-3,4,5-(PO4)3 in skeletal muscle of rats following high-fat feeding and streptozotocin-induced diabetes. Endocrinology 2003, 144, 947–954. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, N.; Gavin, M.G.; Quinn, W.J.; Luongo, T.S.; Gelfer, R.G.; Baur, J.A.; Titchenell, P.M. The role of skeletal muscle Akt in the regulation of muscle mass and glucose homeostasis. Mol. Metab. 2019, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.; Abdullah, N.; Thowfeik, F.S.; Altorki, N.K.; McGraw, T.E. Distinct Akt phosphorylation states are required for insulin regulated Glut4 and Glut1-mediated glucose uptake. eLife 2017, 6, e26896. [Google Scholar] [CrossRef]
- Kohn, A.D.; Summers, S.A.; Birnbaum, M.J.; Roth, R.A. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J. Biol. Chem. 1996, 271, 31372–31378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.M.; Clark, S.F.; Tucker, D.F.; Birnbaum, M.J.; James, D.E.; Macaulay, S.L. A Role for Protein Kinase Bβ/Akt2 in Insulin-Stimulated GLUT4 Translocation in Adipocytes. Mol. Cell Biol. 1999, 19, 7771–7781. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Somwar, R.; Bilan, P.J.; Liu, Z.; Jin, J.; Woodgett, J.R.; Klip, A. Protein Kinase B/Akt Participates in GLUT4 Translocation by Insulin in L6 Myoblasts. Mol. Cell Biol. 1999, 19, 4008–4018. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, E.; McGraw, T.E. Insulin Signaling Diverges into Akt-dependent and -independent Signals to Regulate the Recruitment/Docking and the Fusion of GLUT4 Vesicles to the Plasma Membrane. Mol. Biol. Cell 2006, 17, 4484–4493. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Holman, G.D. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am. J. Physiol.–Endocrinol. Metab. 2008, 295, E29–E37. [Google Scholar] [CrossRef]
- An, D.; Toyoda, T.; Taylor, E.B.; Yu, H.; Fujii, N.; Hirshman, M.F.; Goodyear, L.J. TBC1D1 regulates insulin- and contraction-induced glucose transport in mouse skeletal muscle. Diabetes 2010, 59, 1358–1365. [Google Scholar] [CrossRef] [Green Version]
- Taylor, E.B.; An, D.; Kramer, H.F.; Yu, H.; Fujii, N.L.; Roeckl, K.S.C.; Bowles, N.; Hirshman, M.F.; Xie, J.; Feener, E.P.; et al. Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. J. Biol. Chem. 2008, 283, 9787–9796. [Google Scholar] [CrossRef] [Green Version]
- Sano, H.; Kane, S.; Sano, E.; Mîinea, C.P.; Asara, J.M.; Lane, W.S.; Garner, C.W.; Lienhard, G.E. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J. Biol. Chem. 2003, 278, 14599–14602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.Y.; Ducommun, S.; Quan, C.; Xie, B.; Li, M.; Wasserman, D.H.; Sakamoto, K.; Mackintosh, C.; Chen, S. AS160 deficiency causes whole-body insulin resistance via composite effects in multiple tissues. Biochem. J. 2013, 2, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Physiol. Behav. 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McManus, E.J.; Sakamoto, K.; Armit, L.J.; Ronaldson, L.; Shpiro, N.; Marquez, R.; Alessi, D.R. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005, 24, 1571–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apró, W.; Wang, L.; Pontén, M.; Blomstrand, E.; Sahlin, K. Resistance exercise induced mTORC1 signaling is not impaired by subsequent endurance exercise in human skeletal muscle. Am. J. Physiol.–Endocrinol. Metab. 2013, 305, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Atherton, P.J.; Babraj, J.A.; Smith, K.; Singh, J.; Rennie, M.J.; Wackerhage, H. Selective activation of AMPK-PGC-1α or PKB-TSC2-mTOR signaling can explain specific adaptive responses to endurance or resistance training-like electrical muscle stimulation. FASEB J. 2005, 19, 1–23. [Google Scholar] [CrossRef]
- Camera, D.M.; Edge, J.; Short, M.J.; Hawley, J.A.; Coffey, V.G. Early time course of akt phosphorylation after endurance and resistance exercise. Med. Sci. Sports Exerc. 2010, 42, 1843–1852. [Google Scholar] [CrossRef]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell. 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, T.P.; Suman, S.; Alatassi, H.; Ankem, M.K.; Damodaran, C. Inhibition of AKT promotes FOXO3a-dependent apoptosis in prostate cancer. Cell Death Dis. 2016, 7, e2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzivion, G.; Dobson, M.; Ramakrishnan, G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta-Mol. Cell. Res. 2011, 1813, 1938–1945. [Google Scholar] [CrossRef] [Green Version]
- Southgate, R.J.; Bruce, C.R.; Carey, A.L.; Steinberg, G.R.; Walder, K.; Monks, R.; Watt, M.J.; Hawley, J.A.; Birnbaum, M.J.; Febbraio, M.A. PGC-1α gene expression is down-regulated by Akt-mediated phosphorylation and nuclear exclusion of FoxO1 in insulin- stimulated skeletal muscle Robert. FASEB J. 2005, 24, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Kamei, Y.; Miura, S.; Suzuki, M.; Kai, Y.; Mizukami, J.; Taniguchi, T.; Mochida, K.; Hata, T.; Matsuda, J.; Aburatani, H.; et al. Skeletal muscle FOXO1 (FKHR) transgenic mice have less skeletal muscle mass, down-regulated type I (slow twitch/red muscle) fiber genes, and impaired glycemic control. J. Biol. Chem. 2004, 279, 41114–41123. [Google Scholar] [CrossRef] [Green Version]
- Ruegsegger, G.N.; Creo, A.L.; Cortes, T.M.; Dasari, S.; Nair, K.S. Altered mitochondrial function in insulin-deficient and insulin-resistant states. J. Clin. Investig. 2018, 128, 3671–3681. [Google Scholar] [CrossRef] [Green Version]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S.; Sandhu, M.A. PGC-1 α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krycer, J.R.; Sharpe, L.J.; Luu, W.; Brown, A.J. The Akt-SREBP nexus: Cell signaling meets lipid metabolism. Trends Endocrinol. Metab. 2010, 21, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Guillet-Deniau, I.; Mieulet, V.; Le Lay, S.; Achouri, Y.; Carré, D.; Girard, J.; Foufelle, F.; Ferré, P. Sterol regulatory element binding protein-1c expression and action in rat muscles: Insulin-like effects on the control of glycolytic and lipogenic enzymes and UCP3 gene expression. Diabetes 2002, 51, 1722–1728. [Google Scholar] [CrossRef] [Green Version]
- Dif, N.; Euthine, V.; Gonnet, E.; Laville, M.; Vidal, H.; Lefai, E. Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem. J. 2006, 400, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Boonsong, T.; Norton, L.; Chokkalingam, K.; Jewell, K.; Macdonald, I.; Bennett, A.; Tsintzas, K. Effect of exercise and insulin on SREBP-1c expression in human skeletal muscle: Potential roles for the ERK1/2 and Akt signalling pathways. Biochem. Soc. Trans. 2007, 35, 1310–1311. [Google Scholar] [CrossRef]
- Thomson, D.M.; Winder, W.W. AMP-activated protein kinase control of fat metabolism in skeletal muscle. Acta Physiol. 2009, 196, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Jhanwar-Uniyal, M.; Gillick, J.L.; Neil, J.; Tobias, M.; Thwing, Z.E.; Murali, R. Distinct signaling mechanisms of mTORC1 and mTORC2 in glioblastoma multiforme: A tale of two complexes. Adv. Biol. Regul. 2015, 57, 64–74. [Google Scholar] [CrossRef]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR Inhibitor Whose Frequent Overexpression in Multiple Myeloma Cells Promotes their Survival Timothy. Cell 2009, 137, 873–886. Available online: http://books.google.com/books?hl=en&lr=&id=xDJoXDOweRwC&oi=fnd&pg=PA85&dq=NF-kB+Signaling+in+Skeletal+Muscle+Health+and+Disease&ots=5evHiNdA6b&sig=rF9rldt8sby9Z4v2PtqR6hqV-l0 (accessed on 10 November 2020). [CrossRef] [PubMed] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in Mice of the mTORC Components raptor, rictor, or mLST8 Reveals that mTORC2 Is Required for Signaling to Akt-FOXO and PKCα, but Not S6K1. Dev. Cell. 2006, 11, 859–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avruch, J.; Hara, K.; Lin, Y.; Liu, M.; Long, X.; Ortiz-Vega, S.; Yonezawa, K. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene 2006, 25, 6361–6372. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 Is an Insulin-Regulated Inhibitor of the mTORC1 Protein Kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knuiman, P.; Hopman, M.T.E.; Verbruggen, C.; Mensink, M. Protein and the adaptive response with endurance training: Wishful thinking or a competitive edge? Front. Physiol. 2018, 9, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damas, F.; Phillips, S.; Vechin, F.C.; Ugrinowitsch, C. A Review of Resistance Training-Induced Changes in Skeletal Muscle Protein Synthesis and Their Contribution to Hypertrophy. Sports Med. 2015, 45, 801–807. [Google Scholar] [CrossRef]
- Yoon, M.S. The role of mammalian target of rapamycin (mTOR) in insulin signaling. Nutrients 2017, 9, 1176. [Google Scholar] [CrossRef] [PubMed]
- Holz, M.K.; Blenis, J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J. Biol. Chem. 2005, 280, 26089–26093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and bTRCP-Mediated Degradation of PDCD4 Promotes Protein Translation and Cell Growth. Science 2006, 314, 467–472. [Google Scholar] [CrossRef]
- Biever, A.; Valjent, E.; Puighermanal, E. Ribosomal protein S6 phosphorylation in the nervous system: From regulation to function. Front. Mol. Neurosci. 2015, 8, 75. [Google Scholar] [CrossRef] [Green Version]
- Ruvinsky, I.; Meyuhas, O. Ribosomal protein S6 phosphorylation: From protein synthesis to cell size. Trends Biochem. Sci. 2006, 31, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Jiang, B.; Zhang, Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle 2016, 15, 781–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashida, K.; Kim, S.H.; Higuchi, M.; Holloszy, J.O.; Han, D.H. Normal adaptations to exercise despite protection against oxidative stress. Am. J. Physiol.-Endocrinol. Metab. 2011, 301, 779–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef] [Green Version]
- Howlett, K.F.; Andrikopoulos, S.; Proietto, J.; Hargreaves, M. Exercise-induced muscle glucose uptake in mice with graded, muscle-specific GLUT-4 deletion. Physiol. Rep. 2013, 1, e00065. [Google Scholar] [CrossRef]
- Hussey, S.E.; Mcgee, S.L.; Garnham, A.; Mcconell, G.K.; Hargreaves, M. Exercise increases skeletal muscle GLUT4 gene expression in patients with type 2 diabetes. Diabetes Obes. Metab. 2012, 14, 768–771. [Google Scholar] [CrossRef]
- Kramer, H.F.; Witczak, C.A.; Taylor, E.B.; Fujii, N.; Hirshman, M.F.; Goodyear, L.J. AS160 regulates insulin- and contraction-stimulated glucose uptake in mouse skeletal muscle. J. Biol. Chem. 2006, 56, 2854–2862. [Google Scholar]
- Flockhart, M.; Nilsson, L.C.; Tais, S.; Ekblom, B.; Apró, W.; Larsen, F.J. Excessive exercise training causes mitochondrial functional impairment and decreases glucose tolerance in healthy volunteers. Cell Metab. 2021, 33, 957–970.e6. Available online: https://www.sciencedirect.com/science/article/pii/S1550413121001029 (accessed on 21 September 2021). [CrossRef]
- Kurauti, M.A.; Freitas-Dias, R.; Ferreira, S.M.; Vettorazzi, J.F.; Nardelli, T.R.; Araujo, H.N.; Santos, G.J.; Carneiro, E.M.; Boschero, A.C.; Rezende, L.F.; et al. Acute exercise improves insulin clearance and increases the expression of Insulin-Degrading enzyme in the liver and skeletal muscle of swiss mice. PLoS ONE 2016, 11, e0160239. [Google Scholar]
- Cusi, K.; Maezono, K.; Osman, A.; Pendergrass, M.; Patti, M.E.; Pratipanawatr, T.; Defronzo, R.A.; Kahn, C.R.; Mandarino, L.J. Insulin resistance differentially affects the PI 3-kinase– and MAP kinase–mediated signaling in human muscle. J. Clin. Investig. 2000, 105, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Howlett, K.F.; Sakamoto, K.; Yu, H.; Goodyear, L.J.; Hargreaves, M. Insulin-stimulated insulin receptor substrate-2-associated phosphatidylinositol 3-kinase activity is enhanced in human skeletal muscle after exercise. Metabolism 2006, 55, 1046–1052. [Google Scholar] [CrossRef]
- Goodyear, L.J.; Giorgino, F.; Balon, T.W.; Condorelli, G.; Smith, R.J. Effects of contractile activity on tyrosine phosphoproteins and PI 3- kinase activity in rat skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 1995, 268, E987–E995. [Google Scholar] [CrossRef]
- Wojtaszewski, J.F.P.; Hansen, B.F.; Ursø, B.; Richter, E.A. Wortmannin inhibits both insulin- and contraction-stimulated glucose uptake and transport in rat skeletal muscle. J. Appl. Physiol. 1996, 81, 1501–1509. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, J.P.; Soos, M.A.; Aslesen, R.; O’Rahilly, S.; Jensen, J. Contraction inhibits insulin-stimulated insulin receptor substrate-1/2-associated phosphoinositide 3-kinase activity, but not protein kinase B activation or glucose uptake, in rat muscle. Biochem. J. 2000, 349, 775–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soos, M.A.; Jensen, J.; Brown, R.A.; O’Rahilly, S.; Shepherd, P.R.; Whitehead, J.P. Research report: Class II phosphoinositide 3-kinase is activated by insulin but not by contraction in skeletal muscle. Arch Biochem. Biophys. 2001, 396, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Frøsig, C.; Sajan, M.P.; Maarbjerg, S.J.; Brandt, N.; Roepstorff, C.; Wojtaszewski, J.F.P.; Kiens, B.; Farese, R.V.; Richter, E.A. Exercise improves phosphatidylinositol-3,4,5-trisphosphate responsiveness of atypical protein kinase C and interacts with insulin signalling to peptide elongation in human skeletal muscle. J. Physiol. 2007, 582, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Lund, S.; Pryor, P.R.; Østergaard, S.; Schmitz, O.; Pedersen, O.; Holman, G.D. Evidence against protein kinase B as a mediator of contraction-induced glucose transport and GLUT4 translocation in rat skeletal muscle. FEBS Lett. 1998, 425, 472–474. [Google Scholar] [CrossRef] [Green Version]
- Brozinick, J.T.; Birnbaum, M.J. Insulin, but not contraction, activates Akt/PKB in isolated rat skeletal muscle. J. Biol. Chem. 1998, 273, 14679–14682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.; Hirshman, M.F.; Aschenbach, W.G.; Goodyear, L.J. Contraction regulation of Akt in rat skeletal muscle. J. Biol. Chem. 2002, 277, 11910–11917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widegren, U.; Jiang, X.J.; Krook, A.; Chibalin, A.V.; Björnholm, M.; Tally, M.; Roth, R.A.; Henriksson, J.; Wallberg-Henriksson, H.; Zierath, J.R. Divergent effects of exercise on metabolic and mitogenic signaling pathways in human skeletal muscle. FASEB J. 1998, 12, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.; Coffey, V.G.; Zhong, Z.; Chibalin, A.V.; Hawley, J.A.; Zierath, J.R. Exercise-induced phosphorylation of the novel Akt substrates AS160 and filamin A in human skeletal muscle. Diabetes 2006, 55, 1776–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.; Aschenbach, W.G.; Hirshman, M.F.; Goodyear, L.J. Akt signaling in skeletal muscle: Regulation by exercise and passive stretch. Am. J. Physiol.-Endocrinol. Metab. 2003, 285, 1081–1088. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Arnolds, D.E.W.; Ekberg, I.; Thorell, A.; Goodyear, L.J. Exercise regulates Akt and glycogen synthase kinase-3 activities in human skeletal muscle. Biochem. Biophys. Res. Commun. 2004, 319, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Castorena, C.M.; Arias, E.B.; Sharma, N.; Cartee, G.D. Postexercise improvement in insulin-stimulated glucose uptake occurs concomitant with greater AS160 phosphorylation in muscle from normal and insulin-resistant rats. Diabetes 2014, 63, 2297–2308. [Google Scholar] [CrossRef] [Green Version]
- Wojtaszewski, J.F.P.; Higaki, Y.; Hirshman, M.F.; Michael, M.D.; Dufresne, S.D.; Kahn, C.R.; Goodyear, L.J. Exercise modulates post receptor insulin signaling and glucose transport in muscle-specific insulin receptor knockout mice. J. Clin. Investig. 1999, 104, 1257–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlett, K.F.; Mathews, A.; Garnham, A.; Sakamoto, K. The effect of exercise and insulin on AS160 phosphorylation and 14-3-3 binding capacity in human skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 2008, 294, E401–E407. [Google Scholar] [CrossRef]
- Bruss, M.D.; Arias, E.B.; Lienhard, G.E.; Cartee, G.D. Increased phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle in response to insulin or contractile activity. Diabetes 2005, 54, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Arias, E.B.; Treebak, J.T.; Cartee, G.D. Exercise Effects on y3-AMPK Activity, Akt Substrate of 160 kDa Phosphorylation, and Glucose Uptake in Muscle of Normal and Insulin-Resistant Female Rats. Diabetes 2022, 132, 140–153. [Google Scholar]
- Dreyer, H.C.; Drummond, M.J.; Glynn, E.L.; Fujita, S.; Chinkes, D.L.; Volpi, E.; Rasmussen, B.B. Resistance exercise increases human skeletal muscle AS160/TBC1D4 phosphorylation in association with enhanced leg glucose uptake during postexercise recovery. J. Appl. Physiol. 2008, 105, 1967–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlett, K.F.; Sakamoto, K.; Garnham, A.; Cameron-Smith, D.; Hargreaves, M. Resistance exercise and insulin regulate AS160 and interaction with 14-3-3 in human skeletal muscle. Diabetes 2007, 56, 1608–1614. [Google Scholar] [CrossRef] [Green Version]
- Treebak, J.T.; Birk, J.B.; Rose, A.J.; Kiens, B.; Richter, E.A.; Wojtaszewski, J.F.P. AS160 phosphorylation is associated with activation of α 2β2γ1- but not α 2β2γ3-AMPK trimeric complex in skeletal muscle during exercise in humans. Am. J. Physiol.-Endocrinol. Metab. 2007, 292, 715–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funai, K.; Cartee, G.D. Inhibition of contraction-stimulated amp-activated protein kinase inhibits contraction-stimulated increases in pas-tbc1d1 and glucose transport without altering pas-as160 in rat skeletal muscle. Diabetes 2009, 58, 1096–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funai, K.; Schweitzer, G.G.; Sharma, N.; Kanzaki, M.; Cartee, G.D. Increased AS160 phosphorylation, but not TBC1D1 phosphorylation, with increased postexercise insulin sensitivity in rat skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 2009, 297, 242–251. [Google Scholar] [CrossRef] [Green Version]
- Edgett, B.A.; Fortner, M.L.; Bonen, A.; Gurd, B.J. mTOR Pathway Is Up-Regulated by Both Acute Endurance Exercise and Chronic Muscle Contraction in Rat Skeletal Muscle; Queen’s University: Kingston, ON, Canada, 2013; Volume 38, pp. 862–869. [Google Scholar]
- Mascher, H.; Ekblom, B.; Rooyackers, O.; Blomstrand, E. Enhanced rates of muscle protein synthesis and elevated mTOR signalling following endurance exercise in human subjects. Acta Physiol. 2011, 202, 175–184. [Google Scholar] [CrossRef]
- Mascher, H.; Andersson, H.; Nilsson, P.A.; Ekblom, B.; Blomstrand, E. Changes in signalling pathways regulating protein synthesis in human muscle in the recovery period after endurance exercise. Acta Physiol. 2007, 191, 67–75. [Google Scholar] [CrossRef]
- Knudsen, J.R.; Li, Z.; Persson, K.W.; Li, J.; Henriquez-Olguin, C.; Jensen, T.E. Contraction-regulated mTORC1 and protein synthesis: Influence of AMPK and glycogen. J. Physiol. 2020, 598, 2637–2649. [Google Scholar] [CrossRef]
- You, J.S.; Mcnally, R.M.; Jacobs, B.L.; Privett, R.E.; Gundermann, D.M.; Lin, K.H.; Steinert, N.D.; Goodman, C.A.; Hornberger, T.A. The role of raptor in the mechanical load-induced regulation of mTOR signaling, protein synthesis, and skeletal muscle hypertrophy. FASEB J. 2019, 33, 4021–4034. [Google Scholar] [CrossRef] [PubMed]
- Philp, A.; Hamilton, D.L.; Baar, K. Signals mediating skeletal muscle remodeling by resistance exercise: PI3-kinase independent activation of mTORC1. J. Appl. Physiol. 2011, 110, 561–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duplanty, A.A.; Budnar, R.G.; Luk, H.Y.; Levitt, D.E.; Hill, D.W.; McFarlin, B.K.; Huggett, D.B.; Vigren, J.L. Effect of Acute Alcohol Ingestion on Resistance Exercise-Induced mTORC1 Signaling in Human Muscle. J. Strength Cond. Res. 2016, 31, 54–61. [Google Scholar] [CrossRef]
- Kim, Y.B.; Inoue, T.; Nakajima, R.; Nakae, K.; Tamura, T.; Tokuyama, K.; Suzuki, M. Effects of endurance training of gene expression on insulin signal transduction pathway. Biochem. Biophys. Res. Commun. 1995, 210, 766–773. [Google Scholar] [CrossRef]
- Glynn, E.L.; Lujan, H.L.; Kramer, V.J.; Drummond, M.J.; DiCarlo, S.E.; Rasmussen, B.B. A chronic increase in physical activity inhibits fedstate mTOR/S6K1 signaling and reduces IRS-1 serine phosphorylation in rat skeletal muscle. Appl. Physiol. Nutr. Metab. 2008, 33, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Surapongchai, J.; Rattanavichit, Y.; Buniam, J.; Saengsirisuwan, V. Exercise protects against defective insulin signaling and insulin resistance of glucose transport in skeletal muscle of angiotensin II-infused rat. Front. Physiol. 2018, 9, 358. [Google Scholar] [CrossRef] [Green Version]
- Kido, K.; Sase, K.; Yokokawa, T.; Fujita, S. Enhanced skeletal muscle insulin sensitivity after acute resistance-type exercise is upregulated by rapamycin-sensitive mTOR complex 1 inhibition. Sci. Rep. 2020, 10, 8509. [Google Scholar] [CrossRef]
- Pauli, J.R.; Ropelle, E.R.; Cintra, D.E.; De Souza, C.T.; da Silva, A.S.R.; Moraes, J.C.; Prada, P.O.; de Almedia Leme, J.A.C.; Luciano, E.; Velloso, L.A.; et al. Acute exercise reverses aged-induced impairments in insulin signaling in rodent skeletal muscle. Mech. Ageing Dev. 2010, 131, 323–329. [Google Scholar] [CrossRef]
- Parker, L.; Stepto, N.K.; Shaw, C.S.; Serpiello, F.R.; Anderson, M.; Hare, D.L.; Levinger, I. Acute high-intensity interval exercise-induced redox signaling is associated with enhanced insulin sensitivity in obese middle-aged men. Front. Physiol. 2016, 7, 411. [Google Scholar] [CrossRef] [Green Version]
- Aoi, W.; Naito, Y.; Tokuda, H.; Tanimura, Y.; Oya-Ito, T.; Yoshikawa, T. Exercise-induced muscle damage impairs insulin signaling pathway associated with IRS-1 oxidative modification. Physiol. Res. 2012, 61, 81–88. [Google Scholar] [CrossRef]
- Wadley, G.D.; Tunstall, R.J.; Sanigorski, A.; Collier, G.R.; Hargreaves, M.; Cameron-Smith, D. Differential effects of exercise on insulin-signaling gene expression in human skeletal muscle. J. Appl. Physiol. 2001, 90, 436–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirwan, J.P.; Del Aguila, L.F.; Hernandez, J.M.; Williamson, D.L.; O’Gorman, D.J.; Lewis, R.; Krishnan, R.K. Regular exercise enhances insulin activation of IRS-1-associated PI3- kinase in human skeletal muscle. J. Appl. Physiol. 2000, 88, 797–803. [Google Scholar] [CrossRef]
- Frøsig, C.; Rose, A.J.; Treebak, J.T.; Kiens, B.; Richter, E.A.; Wojtaszewski, J.F.P. Effects of endurance exercise training on insulin signaling in human skeletal muscle: Interactions at the level of phosphatidylinositol 3-kinase, Akt, and AS160. Diabetes 2007, 56, 2093–2102. [Google Scholar] [CrossRef]
- Lee, A.D.; Hansen, P.A.; Holloszy, J.O. Wortmannin inhibits insulin-stimulated but not contraction-stimulated glucose transport activity in skeletal muscle. FEBS Lett. 1995, 361, 51–54. [Google Scholar] [CrossRef] [Green Version]
- Lund, S.; Holman, G.D.; Schmitz, O.; Pedersen, O. Contraction stimulates translocation of glucose transporter GLUT4 in skeletal muscle through a mechanism distinct from that of insulin. Proc. Natl. Acad. Sci. USA 1995, 92, 5817–5821. [Google Scholar] [CrossRef] [Green Version]
- Margolis, L.M.; Berryman, C.E.; Murphy, N.E.; Carrigan, C.T.; Young, A.J.; Carbone, J.W.; Pasiakos, S.M. PI3K-AKT-FOXO1 pathway targeted by skeletal muscle microRNA to suppress proteolytic gene expression in response to carbohydrate intake during aerobic exercise. Physiol. Rep. 2018, 6, e13931. [Google Scholar] [CrossRef] [Green Version]
- Rice, K.M.; Katta, A.; Manne, N.D.P.K.; Arvapalli, R.; Ginjupalli, G.K.; Wu, M.; Asano, S.; Blough, E.R. Lean and Obese Zucker Rat Extensor Digitorum Longus Muscle high-frequency electrical stimulation (HFES) Data: Regulation of p70S6kinase Associated Proteins. Data Br. 2018, 16, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.J.; Michell, B.J.; Kemp, B.E.; Hargreaves, M. Effect of exercise on protein kinase C activity and localization in human skeletal muscle. J. Physiol. 2004, 561, 861–870. [Google Scholar] [CrossRef]
- Beeson, M.; Sajan, M.P.; Dizon, M.; Grebenev, D.; Gomez-daspet, J.; Miura, A.; Kanoh, Y.; Powe, J.; Bandyopadhyay, G.; Standaert, M.L.; et al. Activation of Protein Kinase C-ζ by insulin and phosphatidylinositol 3,4,5-(PO4)3 Is Defective in Muscle in Type 2 Diabetes and Impaired Glucose Tolerance: Amelioration by Rosiglitazone and Exercise. Rev. Lit Arts Am. 2003, 52, 1926–1934. [Google Scholar] [CrossRef] [Green Version]
- Richter, E.A.; Vistisen, B.; Maarbjerg, S.J.; Sajan, M.; Farese, R.B.; Kiens, B. Differential effect of bicycling exercise intensity on activity and phosphorylation of atypical protein kinase C and extracellular signal-regulated protein kinase in skeletal muscle. J. Physiol. 2004, 560, 909–918. [Google Scholar] [CrossRef]
- Nielsen, J.N.; Frøsig, C.; Sajan, M.P.; Miura, A.; Standaert, M.L.; Graham, D.A.; Wojtaszewski, J.F.P.; Farese, R.V.; Richter, E.A. Increased atypical PKC activity in endurance-trained human skeletal muscle. Biochem. Biophys. Res. Commun. 2003, 312, 1147–1153. [Google Scholar] [CrossRef]
- Lai, K.-M.V.; Gonzalez, M.; Poueymirou, W.T.; Kline, W.O.; Na, E.; Zlotchenko, E.; Stitt, T.N.; Economides, A.N.; Yancopolous, G.D.; Glass, D.J. Conditional Activation of Akt in Adult Skeletal Muscle Induces Rapid Hypertrophy. Mol. Cell Biol. 2004, 24, 9295–9304. [Google Scholar] [CrossRef] [Green Version]
- Cleasby, M.E.; Reinten, T.A.; Cooney, G.J.; James, D.E.; Kraegen, E.W. Functional studies of Akt isoform specificity in skeletal muscle in vivo; maintained insulin sensitivity despite reduced insulin receptor substrate-1 expression. Mol. Endocrinol. 2007, 21, 215–228. [Google Scholar] [CrossRef] [Green Version]
- Holten, M.K.; Zacho, M.; Gaster, M.; Juel, C.; Wojtaszewski, J.F.P.; Dela, F. Strength Training Increases Insulin-Mediated Glucose Uptake, GLUT4 Content, and Insulin Signaling in Skeletal Muscle in Patients with Type 2 Diabetes. Diabetes 2004, 53, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.W.; Chang, S.J. Moderate exercise suppresses NF-κB signaling and activates the SIRT1-AMPK-PGC1α axis to attenuate muscle loss in diabetic db/db mice. Front. Physiol. 2018, 9, 636. [Google Scholar] [CrossRef]
- Wang, H.; Bei, Y.; Lu, Y.; Sun, W.; Liu, Q.; Wang, Y.; Cao, Y.; Chen, P.; Xiao, J.; Kong, X. Exercise prevents cardiac injury and improves mitochondrial biogenesis in advanced diabetic cardiomyopathy with PGC-1α and Akt activation. Cell Physiol. Biochem. 2015, 35, 2159–2168. [Google Scholar] [CrossRef]
- Horii, N.; Hasegawa, N.; Fujie, S.; Uchida, M.; Iemitsu, M. Resistance exercise-induced increase in muscle 5α-dihydrotestosterone contributes to the activation of muscle Akt/mTOR/p70S6K- and Akt/AS160/GLUT4-signaling pathways in type 2 diabetic rats. FASEB J. 2020, 34, 11047–11057. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.T.; Song, Z.; Zhang, W.C.; Jiao, B.; Yu, Z.B. Impaired translocation of GLUT4 results in insulin resistance of atrophic soleus muscle. Biomed. Res. Int. 2015, 2015, 291987. [Google Scholar] [CrossRef] [Green Version]
- Leguisamo, N.M.; Lehnen, A.M.; Machado, U.F.; Okamoto, M.M.; Markoski, M.M.; Pinto, G.H.; Schann, B. GLUT4 content decreases along with insulin resistance and high levels of inflammatory markers in rats with metabolic syndrome. Cardiovasc. Diabetol. 2012, 11, 1. [Google Scholar] [CrossRef] [Green Version]
- Mueckler, M. Insulin resistance and the disruption of glut4 trafficking in skeletal muscle. J. Clin. Investig. 2001, 107, 1211–1213. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, B.J.; Griesel, B.A.; King, C.D.; Josey, M.A.; Olson, A.L. Moderate glut4 overexpression improves insulin sensitivity and fasting triglyceridemia in high-fat diet-fed transgenic mice. Diabetes 2013, 62, 2249–2258. [Google Scholar] [CrossRef] [Green Version]
- Zisman, A.; Peroni, O.D.; Abel, D.E.; Michael, M.D.; Mauvais-Jarvis, F.; Lowell, B.B.; Wojtaszewski, J.F.P.; Hirshman, M.F.; Virkamaki, A.; Goodyear, L.J.; et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat. Med. 2000, 6, 924–928. [Google Scholar] [CrossRef]
- Coderre, L.; Kandror, K.V.; Vallega, G.; Pilch, P.F. Identification and characterization of an exercise-sensitive pool of glucose transporters in skeletal muscle. J. Biol. Chem. 1995, 270, 27584–27588. [Google Scholar] [CrossRef] [Green Version]
- Lemieux, K.; Han, X.X.; Dombrowski, L.; Bonen, A.; Marette, A. The transferrin receptor defines two distinct contraction-responsive GLUT4 vesicle populations in skeletal muscle. Diabetes 2000, 49, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Ploug, T.; Van Deurs, B.; Ai, H.; Cushman, S.W.; Ralston, E. Analysis of GLUT4 distribution in whole skeletal muscle fibers: Identification of distinct storage compartments that are recruited by insulin and muscle contractions. J. Cell Biol. 1998, 142, 1429–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consitt, L.A.; Van Meter, J.; Newton, C.A.; Collier, D.N.; Dar, M.S.; Wojtaszewski, J.F.P.; Treebak, J.T.; Tanner, C.J.; Houmard, J.A. Impairments in site-specific AS160 phosphorylation and effects of exercise training. Diabetes 2013, 62, 3437–3447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, A.; Arias, E.B.; Wang, H.; Kwak, S.E.; Pan, X.; Duan, D.; Cartee, G.D. Exercise-Induced Improvement in Insulin-Stimulated Glucose Uptake by Rat Skeletal Muscle Is Absent in Male AS160-Knockout Rats, Partially Restored by Muscle Expression of Phosphomutated AS160, and Fully Restored by Muscle Expression of Wild-Type AS160. Diabetes 2022, 71, 219–232. [Google Scholar] [CrossRef]
- Broberg, S.; Sahlin, K. Adenine nucleotide degradation in human skeletal muscle during prolonged exercise. J. Appl. Physiol. 1989, 67, 116–122. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Cartee, G.D.; Funai, K. Exercise and insulin: Convergence or divergence at AS160 and TBC1D1? Exerc. Sport Sci. Rev. 2009, 37, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Lu, A.M.; Wang, Y.; Hong, A.; Chen, Y.; Hu, J.; Li, X.; Qin, Z. Chronic resistance training activates autophagy and reduces apoptosis of muscle cells by modulating IGF-1 and its receptors, Akt/mTOR and Akt/FOXO3a signaling in aged rats. Exp. Gerontol. 2013, 48, 427–436. [Google Scholar] [CrossRef]
- Egan, B.; Zierath, J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [Green Version]
- Vissing, K.; Mcgee, S.L.; Farup, J.; Kjølhede, T.; Vendelbo, M.H.; Jessen, N. Differentiated mTOR but not AMPK signaling after strength vs endurance exercise in training-accustomed individuals. Scand. J. Med. Sci. Sports 2013, 23, 355–366. [Google Scholar] [CrossRef]
- Arias, E.B.; Kim, J.; Funai, K.; Cartee, G.D. Prior exercise increases phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 2007, 292, E1191–E1200. [Google Scholar] [CrossRef]
- Frøsig, C.; Pehmøller, C.; Birk, J.B.; Richter, E.A.; Wojtaszewski, J.F.P. Exercise-induced TBC1D1 Ser237 phosphorylation and 14-3-3 protein binding capacity in human skeletal muscle. J. Physiol. 2010, 588, 4539–4548. [Google Scholar] [CrossRef]
- Pehmøller, C.; Treebak, J.T.; Birk, J.B.; Chen, S.; MacKintosh, C.; Hardie, D.G.; Richter, E.A.; Wojtaszewski, J.F.P. Genetic disruption of AMPK signaling abolishes both contraction- and insulin-stimulated TBC1D1 phosphorylation and 14-3-3 binding in mouse skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 2009, 297, E665–E675. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, H.M.; Maarbjerg, S.J.; Crane, J.D.; Jeppesen, J.; Jørgensen, S.B.; Schertzer, J.D.; Shyroka, O.; Kiens, B.; Van Denderen, B.J.; Tarnopolsky, M.A.; et al. AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. USA 2011, 108, 16092–16097. [Google Scholar] [CrossRef] [Green Version]
- Vichaiwong, K.; Purohit, S.; An, D.; Toyoda, T.; Jessen, N.; Hirshman, M.F.; Goodyear, L.J. Contraction regulates site-specific phosphorylation of TBC1D1 in skeletal muscle. Biochem. J. 2010, 431, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, C.K.; Barnard, R.J.; Jasman, A.; Balon, T.W. Acute exercise increases nitric oxide synthase activity in skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 1999, 277, E390–E394. [Google Scholar] [CrossRef]
- Merry, T.L.; Steinberg, G.R.; Lynch, G.S.; McConell, G.K. Skeletal muscle glucose uptake during contraction is regulated by nitric oxide and ROS independently of AMPK. Am. J. Physiol.-Endocrinol. Metab. 2010, 298, E577–E585. [Google Scholar] [CrossRef] [PubMed]
- Bradley, S.J.; Kingwell, B.A.; McConell, G.K. Nitric oxide synthase inhibition reduces leg glucose uptake but not blood flow during dynamic exercise in humans. Diabetes 1999, 48, 1815–1821. [Google Scholar] [CrossRef] [PubMed]
- Kingwell, B.A.; Formosa, M.; Muhlmann, M.; Bradley, S.J.; McConell, G.K. Nitric oxide synthase inhibition reduces glucose uptake during exercise in individuals with type 2 diabetes more than in control subjects. Diabetes 2002, 51, 2572–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, R.M.; Wadley, G.D.; Clark, M.G.; Rattigan, S.; McConell, G.K. Local nitric oxide synthase inhibition reduces skeletal muscle glucose uptake but not capillary blood flow during in situ muscle contraction in rats. Diabetes 2007, 56, 2885–2892. [Google Scholar] [CrossRef] [Green Version]
- Heinonen, I.; Saltin, B.; Kemppainen, J.; Nuutila, P.; Knuuti, J.; Kalliokoski, K.; Hellsten, Y. Effect of nitric oxide synthase inhibition on the exchange of glucose and fatty acids in human skeletal muscle. Nutr. Metab. 2013, 10, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higaki, Y.; Hirshman, M.F.; Fujii, N.; Goodyear, L.J. Nitric oxide increases glucose uptake through a mechanism that is distinct from the insulin and contraction pathways in rat skeletal muscle. Diabetes 2001, 50, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Kalliokoski, K.K.; Langberg, H.; Ryberg, A.K.; Scheede-Bergdahl, C.; Doessing, S.; Kjær, A.; Kjær, M.; Boushel, M. Nitric oxide and prostaglandins influence local skeletal muscle blood flow during exercise in humans: Coupling between local substrate uptake and blood flow. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2006, 291, R803–R809. [Google Scholar] [CrossRef] [Green Version]
- Patwell, D.M.; McArdle, A.; Morgan, J.E.; Patridge, T.A.; Jackson, M.J. Release of reactive oxygen and nitrogen species from contracting skeletal muscle cells. Free Radic. Biol. Med. 2004, 37, 1064–1072. [Google Scholar] [CrossRef]
- Sandström, M.E.; Zhang, S.J.; Bruton, J.; Silva, J.P.; Reid, M.B.; Westerblad, H.; Katz, A. Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. J. Physiol. 2006, 575, 251–262. [Google Scholar] [CrossRef]
- Reid, M.B.; Haack, K.E.; Franchek, K.M.; Valberg, P.A.; Kobzik, L.; West, M.S. Reactive oxygen in skeletal muscle. I. Intracellular oxidant kinetics and fatigue in vitro. J. Appl. Physiol. 1992, 73, 1797–1804. [Google Scholar] [CrossRef]
- Svensson, M.B.; Ekblom, B.; Cotgreave, I.A.; Norman, B.; Sjöberg, B.; Ekblom, Ö.; Sjödin, B.; Sjödin, A. Adaptive stress response of glutathione and uric acid metabolism in man following controlled exercise and diet. Acta Physiol. Scand. 2002, 176, 43–56. [Google Scholar] [CrossRef]
- Sen, C.K.; Rankinen, T.; Vaisanen, S.; Rauramaa, R. Oxidative stress after human exercise: Effect of N-acetylcysteine supplementation. J. Appl. Physiol. 1994, 76, 2570–2577. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Borrás, C.; Pallardo, F.V.; Sastre, J.; Ji, L.L.; Viña, J. Decreasing xanthine oxidase-mediated oxidative stress prevents useful cellular adaptations to exercise in rats. J. Physiol. 2005, 567, 113–120. [Google Scholar] [CrossRef]
- Henríquez-Olguin, C.; Knudsen, J.R.; Raun, S.H.; Li, Z.; Dalbram, E.; Treebak, J.T.; Sylow, L.; Holmdahl, R.; Richter, E.A.; Jaimovich, E.; et al. Cytosolic ROS production by NADPH oxidase 2 regulates muscle glucose uptake during exercise. Nat. Commun. 2019, 10, 4623. [Google Scholar] [CrossRef] [Green Version]
- Friedrichsen, M.; Birk, J.B.; Richter, E.A.; Ribel-Madsen, R.; Pehmøller, C.; Hansen, B.F.; Beck-Nielsen, H.; Hirshman, M.F.; Goodyear, M.J.; Vaag, A.; et al. Akt2 influences glycogen synthase activity in human skeletal muscle through regulation of NH2-terminal (sites 2 + 2a) phosphorylation. Am. J. Physiol.-Endocrinol. Metab. 2013, 304, 631–639. [Google Scholar] [CrossRef] [Green Version]
- Shulman, G.I.; Rothman, D.; Jue, T.; Stein, P.; DeFronzo, R.; Shulman, R. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N. Engl. J. Med. 1990, 322, 223–228. Available online: http://content.nejm.org/cgi/content/abstract/329/14/977%5Cnhttp://www.nejm.org/doi/abs/10.1056/NEJM199309303291401 (accessed on 10 May 2021). [CrossRef] [PubMed]
- Jensen, J.; Rustad, P.I.; Kolnes, A.J.; Lai, Y.C. The role of skeletal muscle glycogen breakdown for regulation of insulin sensitivity by exercise. Front. Physiol. 2011, 2, 112. [Google Scholar] [CrossRef] [Green Version]
- Manabe, Y.; Gollisch, K.S.C.; Holton, L.; Kim, Y.B.; Brandauer, J.; Fujii, N.L.; Hirshman, M.F.; Goodyear, L.J. Exercise training-induced adaptations associated with increases in skeletal muscle glycogen content. FEBS J. 2013, 280, 916–926. [Google Scholar] [CrossRef] [Green Version]
- Markuns, J.F.; Wojtaszewski, J.F.P.; Goodyear, L.J. Insulin and exercise decrease glycogen synthase kinase-3 activity by different mechanisms in rat skeletal muscle. J. Biol. Chem. 1999, 274, 24896–24900. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.C.; Lin, F.C.; Jensen, J. Glycogen content regulates insulin- but not contraction-mediated glycogen synthase activation in the rat slow-twitch soleus muscles. Acta Physiol. 2009, 197, 139–150. [Google Scholar] [CrossRef]
- Lai, Y.C.; Zarrinpashneh, E.; Jensen, J. Additive effect of contraction and insulin on glucose uptake and glycogen synthase in muscle with different glycogen contents. J. Appl. Physiol. 2010, 108, 1106–1115. [Google Scholar] [CrossRef] [Green Version]
- Jensen, J.; Tantiwong, P.; Stuenæs, J.T.; Molina-Carrion, M.; de Fronzo, R.A.; Sakamoto, K.; Musi, N. Effect of acute exercise on glycogen synthase in muscle from obese and diabetic subjects. Am. J. Physiol.-Endocrinol. Metab. 2012, 303, E82–E89. [Google Scholar] [CrossRef] [Green Version]
- Brandt, N.; Dethlefsen, M.M.; Bangsbo, J.; Pilegaard, H. PGC-1α and exercise intensity dependent adaptations in mouse skeletal muscle. PLoS ONE 2017, 12, e0185993. [Google Scholar]
- Wang, J.; Wang, F.; Zhang, P.; Liu, H.; He, J.; Zhang, C.; Fan, M.; Chan, X. PGC-1α over-expression suppresses the skeletal muscle atrophy and myofiber-type composition during hindlimb unloading. Biosci. Biotechnol. Biochem. 2017, 81, 500–513. [Google Scholar] [CrossRef] [Green Version]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruas, J.L.; White, J.P.; Rao, R.R.; Kleiner, S.; Brannan, K.T.; Harrison, B.C.; Greene, N.P.; Wu, J.; Estall, J.L.; Irving, B.A.; et al. A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 2012, 151, 1319–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriwijitkamol, A.; Ivy, J.L.; Christ-Roberts, C.; DeFronzo, R.A.; Mandarino, L.J.; Musi, N. LKB1-AMPK signaling in muscle from obese insulin-resistant Zucker rats and effects of training. Am. J. Physiol.-Endocrinol. Metab. 2006, 290, E925–E932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akimoto, T.; Pohnert, S.C.; Li, P.; Zhang, M.; Gumbs, C.; Rosenberg, P.B.; Williams, R.S.; Yan, Z. Exercise stimulates Pgc-1α transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 2005, 280, 19587–19593. [Google Scholar] [CrossRef] [Green Version]
- Taylor, E.B.; Lamb, J.D.; Hurst, R.W.; Chesser, D.G.; Ellingson, W.J.; Greenwood, L.J.; Porter, B.B.; Herway, S.T.; Winder, W. Endurance training increases skeletal muscle LKB1 and PGC-1α protein abundance: Effects of time and intensity. Am. J. Physiol.-Endocrinol. Metab. 2005, 289, 960–968. [Google Scholar] [CrossRef] [Green Version]
- Leick, L.; Wojtaszewski, J.F.P.; Johansen, S.T.; Kiilerich, K.; Comes, G.; Hellsten, Y.; Hidalgo, J.; Pilegaard, H. PGC-1α is not mandatory for exercise- and training-induced adaptive gene responses in mouse skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 2008, 294, 463–474. [Google Scholar] [CrossRef] [Green Version]
- Baar, K.; Wende, A.R.; Jones, T.E.; Marison, M.; Nolte, L.A.; Chen, M.; Kelly, D.P.; Holloszy, J.O. Adaptations of skeletal muscle to exercise: Rapid increase in the transcriptional coactivator PGC-1. FASEB J. 2002, 16, 1879–1886. [Google Scholar] [CrossRef]
- Terada, S.; Goto, M.; Kato, M.; Kawanaka, K.; Shimokawa, T.; Tabata, I. Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochem. Biophys. Res. Commun. 2002, 296, 350–354. [Google Scholar] [CrossRef]
- Pilegaard, H.; Saltin, B.; Neufer, D.P. Exercise induces transient transcriptional activation of the PGC-1α gene in human skeletal muscle. J. Physiol. 2003, 546, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.; Carson, B.P.; Garcia-Roves, P.M.; Chibalin, A.V.; Sarsfield, F.M.; Barron, N.; McCaffrey, N.; Moyna, N.M.; Zierath, J.R.; O’Gorman, D.J. Exercise intensity-dependent regulation of peroxisome proliferator-activated receptor γ coactivator-1α mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J. Physiol. 2010, 588, 1779–1790. [Google Scholar] [CrossRef]
- Barrès, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gormon, D.J.; Zierath, J.R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Nordsborg, N.B.; Lundby, C.; Leick, L.; Pilegaard, H. Relative workload determines exercise-induced increases in PGC-1α mRNA. Med. Sci. Sports Exerc. 2010, 42, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Lira, V.A.; Benton, C.R.; Yan, Z.; Bonen, A. PGC-1α regulation by exercise training and its influences on muscle function and insulin sensitivity. Am. J. Physiol.-Endocrinol. Metab. 2010, 299, E145–E161. [Google Scholar] [CrossRef] [Green Version]
- Suwa, M.; Nakano, H.; Kumagai, S. Effects of chronic AICAR treatment on fiber composition, enzyme activity, UCP3, and PGC-1 in rat muscles. J. Appl. Physiol. 2003, 95, 960–968. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Gerhart-hines, Z.; Feige, J.N.; Lagouge, M.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD + metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Gurd, B.J. Deacetylation of PGC-1a by SIRT1: Importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl. Physiol. Nutr. Metab. 2011, 36, 589–597. [Google Scholar] [CrossRef]
- Cantó, C.; Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Jiang, L.Q.; Deshmukh, A.S.; Mataki, C.; Coste, A.; Lagouge, M.; Zierath, J.R.; Auwerx, J. Interdependence of AMPK and SIRT1 for Metabolic Adaptation to Fasting and Exercise in Skeletal Muscle. Cell Metab. 2010, 11, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Price, N.L.; Gomes, A.P.; Ling, A.J.Y.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavazis, A.N.; Smuder, A.J.; Powers, S.K. Effects of short-term endurance exercise training on acute doxorubicin-induced FoxO transcription in cardiac and skeletal muscle. J. Appl. Physiol. 2014, 117, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Azad, M.; Khaledi, N.; Hedayati, M. Effect of acute and chronic eccentric exercise on FOXO1 mRNA expression as fiber type transition factor in rat skeletal muscles. Gene 2016, 584, 180–184. [Google Scholar] [CrossRef]
- Louis, E.; Raue, U.; Yang, Y.; Jemiolo, B.; Trappe, S. Time course of proteolytic, cytokine, and myostatin gene expression after acute exercise in human skeletal muscle. J. Appl. Physiol. 2007, 103, 1744–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanetti, R.J.; Lamon, S.; Rahbek, S.K.; Farup, J.; Zacharewicz, E.; Wallace, M.A.; Vendelbo, M.H.; Russell, A.P.; Vissing, K. Influence of divergent exercise contraction mode and whey protein supplementation on atrogin-1, MuRF1, and FOXO1/3A in human skeletal muscle. J. Appl. Physiol. 2014, 116, 1491–1502. [Google Scholar] [CrossRef] [Green Version]
- Lysenko, E.A.; Popov, D.V.; Vepkhvadze, T.F.; Lednev, E.M.; Vinogradova, O.L. Effect of combined aerobic and strength exercises on the regulation of mitochondrial biogenesis and protein synthesis and degradation in human skeletal muscle. Hum. Physiol. 2016, 42, 634–644. [Google Scholar] [CrossRef]
- Nadeau, K.J.; Ehlers, L.B.; Aguirre, L.E.; Moore, R.L.; Jew, K.N.; Ortmeyer, H.K.; Hansen, B.C.; Reusch, J.E.B.; Draznin, B. Exercise training and calorie restriction increase SREBP-1 expression and intramuscular triglyceride in skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 2006, 291, E90–E98. [Google Scholar] [CrossRef]
- Ikeda, S.; Miyazaki, H.; Nakatani, T.; Kai, Y.; Kamei, Y.; Miura, S.; Tsuboyama-Kasaoka, N.; Ezaki, O. Up-regulation of SREBP-1c and lipogenic genes in skeletal muscles after exercise training. Biochem. Biophys. Res. Commun. 2002, 296, 395–400. [Google Scholar] [CrossRef]
- Rabøl, R.; Petersen, K.F.; Dufour, S.; Flannery, C.; Shulman, G.I. Reversal of muscle insulin resistance with exercise reduces postprandial hepatic de novo lipogenesis in insulin resistant individuals. Proc. Natl. Acad. Sci. USA 2011, 108, 13705–13709. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Zhou, L.; Chen, S.; Shangguan, R.; Qu, Y.; Sun, J. Acute and chronic effects of high-intensity interval training (HIIT) on postexercise intramuscular lipid metabolism in rats. Physiol. Res. 2021, 70, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Risson, V.; Mazelin, L.; Roceri, M.; Sanchez, H.; Moncollin, V.; Corneloup, C.; Richard-Bulteau, H.; Vignaud, A.; Baas, D.; Defour, A.; et al. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J. Cell Biol. 2009, 187, 859–874. [Google Scholar] [CrossRef] [Green Version]
- Bentzinger, C.F.; Romanino, K.; Cloëtta, D.; Lin, S.; Mascarenhas, J.B.; Oliveri, F.; Xia, J.; Casanova, E.; Costa, C.F.; Brink, M.; et al. Skeletal Muscle-Specific Ablation of raptor, but not of rictor, Causes Metabolic Changes and Results in Muscle Dystrophy. Cell Metab. 2008, 8, 411–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, M.S. mTOR as a key regulator in maintaining skeletal muscle mass. Front. Physiol. 2017, 8, 788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogasawara, R.; Fujita, S.; Hornberger, T.A.; Kitaoka, Y.; Makanae, Y.; Nakazato, K.; Naokata, I. The role of mTOR signalling in the regulation of skeletal muscle mass in a rodent model of resistance exercise. Sci. Rep. 2016, 6, 31142. [Google Scholar] [CrossRef]
- Stuart, C.A.; Howell, M.E.A.; Baker, J.D.; Dykes, R.J.; Duffourc, M.M.; Ramsey, M.W.; Stone, M.H. Cycle training increased glut4 and activation of mammalian target of rapamycin in fast twitch muscle fibers. Med. Sci. Sports Exerc. 2010, 42, 96–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philp, A.; Schenk, S.; Perez-Schindler, J.; Hamilton, D.L.; Breen, L.; Laverone, E.; Jeromson, S.; Phillips, S.M.; Baar, K. Rapamycin does not prevent increases in myofibrillar or mitochondrial protein synthesis following endurance exercise. J. Physiol. 2015, 593, 4275–4284. [Google Scholar] [CrossRef] [Green Version]
- Morrison, P.J.; Hara, D.; Ding, Z.; Ivy, J.L. Adding protein to a carbohydrate supplement provided after endurance exercise enhances 4E-BP1 and RPS6 signaling in skeletal muscle. J. Appl. Physiol. 2008, 104, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Mothe-Satney, I.; Gautiert, N.; Hinault, C.; Lawrence, J.C.; Van Obberghen, E. In rat hepatocytes glucagon increases mammalian target of rapamycin phosphorylation on serine 2448 but antagonizes the phosphorylation of its downstream targets induced by insulin and amino acids. J. Biol. Chem. 2004, 279, 42628–42637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Donato, D.M.; West, D.W.D.; Churchward-Venne, T.A.; Breen, L.; Baker, S.K.; Phillips, S.M. Influence of aerobic exercise intensity on myofibrillar and mitochondrial protein synthesis in young men during early and late postexercise recovery. Am. J. Physiol.-Endocrinol. Metab. 2014, 306, 1025–1032. [Google Scholar] [CrossRef] [Green Version]
- Coffey, V.G.; Moore, D.R.; Burd, N.A.; Rerecich, T.; Stellingwerff, T.; Garnham, A.P.; Phillips, S.M.; Hawley, J.A. Nutrient provision increases signalling and protein synthesis in human skeletal muscle after repeated sprints. Eur. J. Appl. Physiol. 2011, 111, 1473–1483. [Google Scholar] [CrossRef] [PubMed]
- Gual, P.; Le Marchand-Brustel, Y.; Tanti, J.F. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie 2005, 87, 99–109. [Google Scholar] [CrossRef]
- Tang, H.; Shrager, J.B.; Goldman, D. Rapamycin protects aging muscle. Aging 2019, 11, 5868–5870. [Google Scholar] [CrossRef]
- Molitoris, J.K.; McColl, K.S.; Swerdlow, S.; Matsuyama, M.; Lam, M.; Finkel, T.H.; Matsuyama, S.; Distelhorst, C.W. Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J. Biol. Chem. 2011, 286, 30181–30189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofer, A.; Lei, K.; Johannessen, C.M.; Ellisen, L.W. Regulation of mTOR and Cell Growth in Response to Energy Stress by REDD1. Mol. Cell Biol. 2005, 25, 5834–5845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimball, S.R.; Jefferson, L.S. Induction of REDD1 Gene Expression in the Liver in Response to Endoplasmic Reticulum Stress is Mediated through a PERK, eIF2α Phosphorylation, ATF4-dependent Cascade. Bone 2012, 427, 485–489. [Google Scholar] [CrossRef] [Green Version]
- D’Hulst, G.; Jamart, C.; Van Thienen, R.; Hespel, P.; Francaux, M.; Deldicque, L. Effect of acute environmental hypoxia on protein metabolism in human skeletal muscle. Acta Physiol. 2013, 208, 251–264. [Google Scholar] [CrossRef]
- McGhee, N.K.; Jefferson, L.S.; Kimball, S.R. Elevated corticosterone associated with food deprivation upregulates expression in rat skeletal muscle of the mTORC1 repressor, REDD1. J. Nutr. 2009, 139, 828–834. [Google Scholar] [CrossRef] [Green Version]
- Wolff, N.C.; Vega-Rubin-de-Celis, S.; Xie, X.-J.; Castrillon, D.H.; Kabbani, W.; Brugarolas, J. Cell-Type-Dependent Regulation of mTORC1 by REDD1 and the Tumor Suppressors TSC1/TSC2 and LKB1 in Response to Hypoxia. Mol. Cell Biol. 2011, 31, 1870–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, M.J.; Fujita, S.; Takashi, A.; Dreyer, H.C.; Volpi, E.; Rasmussen, B.B. Human Muscle Gene Expression Following Resistance Exercise and Blood Flow Restriction. Physiol. Behav. 2008, 40, 691–698. Available online: http://internal-pdf//Nishijima (accessed on 10 May 2021). [CrossRef] [Green Version]
- Gordon, B.S.; Steiner, J.L.; Lang, C.H.; Jefferson, L.S.; Kimball, S.R. Reduced REDD1 expression contributes to activation of mTORC1 following electrically induced muscle contraction. Am. J. Physiol.-Endocrinol. Metab. 2014, 307, E703–E711. [Google Scholar] [CrossRef] [Green Version]
- Williamson, D.L.; Kubica, N.; Kimball, S.R.; Jefferson, L.S. Exercise-induced alterations in extracellular signal-regulated kinase 1/2 and mammalian target of rapamycin (mTOR) signalling to regulatory mechanisms of mRNA translation in mouse muscle. J. Physiol. 2006, 573, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreyer, H.C.; Fujita, S.; Cadenas, J.G.; Chinkes, D.L.; Volpi, E.; Rasmussen, B.B. Resistance exercise increases AMPK activity and reduces 4E-BP1 phosphorylation and protein synthesis in human skeletal muscle. J. Physiol. 2006, 576, 613–624. [Google Scholar] [CrossRef]
- Moore, G.E. The role of exercise prescription in chronic disease. Br. J. Sports Med. 2004, 38, 6–7. [Google Scholar]
- Lakey, W.C.; Barnard, K.; Batch, B.C.; Chiswell, K.; Tasneem, A.; Green, J.B. Are current clinical trials in diabetes addressing important issues in diabetes care? Diabetologia 2013, 56, 1226–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes, R.; Sousa, N.; Almeida, A.; Subtil, P.; Guedes-Marques, F.; Reis, V.M.; Themudo-Barata, J.L. Exercise prescription for patients with type 2 diabetes—a synthesis of international recommendations: Narrative review. Br. J. Sports Med. 2016, 50, 1379–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, T.D.; Clark, L.A.; Clark, B.C. Resistance Exercise to Prevent and Manage Sarcopenia and Dynapenia. Annu. Rev. Gerontol. Geriatr. 2016, 36, 205–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santilli, V.; Bernetti, A.; Mangone, M.; Paoloni, M. Clinical definition of sarcopenia. Clin. Cases Miner. Bone Metab. 2014, 11, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Sahay, R. Diabetes Fatigue Syndrome. Diabetes Ther. 2016, 9, 1421–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Protein | Effect of Exercise | Reference |
|---|---|---|
| Insulin receptor phosphorylation | No change | [89,90,91] |
| IRS-1 tyrosine phosphorylation | No change | [90,92] |
| PI3K activity | No change | [92,93,94,95] |
| aPKC activity | No change | [96] |
| Akt S473 phosphorylation | No change | [97,98,99,100,101,102] |
| Akt S473 phosphorylation | Increased | [99,103,104,105] |
| AS160 phosphorylation | Increased | [101,106,107,108] |
| AS160 phosphorylation | No change | [101,109,110,111] |
| TBC1D1 phosphorylation | Increased | [46,112,113] |
| mTOR activation | Increased | [51,52,53,114,115,116,117,118,119,120] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mann, G.; Riddell, M.C.; Adegoke, O.A.J. Effects of Acute Muscle Contraction on the Key Molecules in Insulin and Akt Signaling in Skeletal Muscle in Health and in Insulin Resistant States. Diabetology 2022, 3, 423-446. https://doi.org/10.3390/diabetology3030032
Mann G, Riddell MC, Adegoke OAJ. Effects of Acute Muscle Contraction on the Key Molecules in Insulin and Akt Signaling in Skeletal Muscle in Health and in Insulin Resistant States. Diabetology. 2022; 3(3):423-446. https://doi.org/10.3390/diabetology3030032
Chicago/Turabian StyleMann, Gagandeep, Michael C. Riddell, and Olasunkanmi A. J. Adegoke. 2022. "Effects of Acute Muscle Contraction on the Key Molecules in Insulin and Akt Signaling in Skeletal Muscle in Health and in Insulin Resistant States" Diabetology 3, no. 3: 423-446. https://doi.org/10.3390/diabetology3030032
APA StyleMann, G., Riddell, M. C., & Adegoke, O. A. J. (2022). Effects of Acute Muscle Contraction on the Key Molecules in Insulin and Akt Signaling in Skeletal Muscle in Health and in Insulin Resistant States. Diabetology, 3(3), 423-446. https://doi.org/10.3390/diabetology3030032