

Improved and Novel Methods for Investigating Organophosphate Esters in Particulate Matter

Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Optimization
3.2. Validation
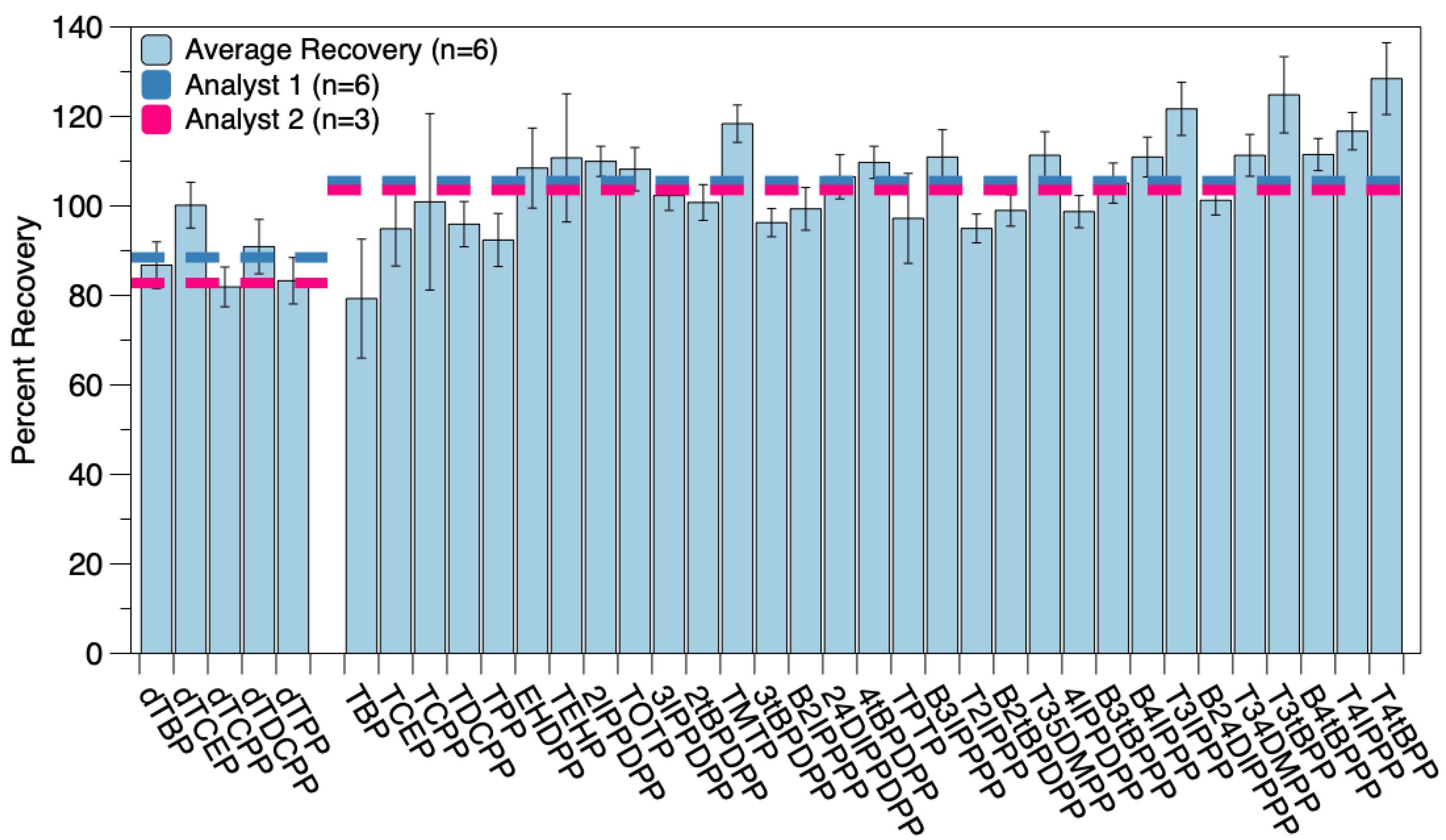
3.2.1. Reproducibility Studies, SRM, and MDLs
3.2.2. Environmental Samples
3.3. Additional Considerations (PAHs)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, K.-H.; Kabir, E.; Kabir, S. A review on the human health impact of airborne particulate matter. Environ. Int. 2015, 74, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Lao, J.-Y.; Lin, H.; Qin, X.; Ruan, Y.; Leung, K.M.Y.; Zeng, E.Y.; Lam, P.K.S. Insights into the Atmospheric Persistence, Transformation, and Health Implications of Organophosphate Esters in Urban Ambient Air. Environ. Sci. Technol. Lib. 2022, 56, 12003–12013. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Cai, Z. Chapter 9—Airborne particulate matter and its organic components: Complex triggers of human disease. In Air Pollution, Climate, and Health; Gao, M., Wang, Z., Carmichael, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 193–206. [Google Scholar]
- Yang, J.; Zhao, Y.; Li, M.; Du, M.; Li, X.; Li, Y. A Review of a Class of Emerging Contaminants: The Classification, Distribution, Intensity of Consumption, Synthesis Routes, Environmental Effects and Expectation of Pollution Abatement to Organophosphate Flame Retardants (OPFRs). Int. J. Mol. Sci. 2019, 20, 2874. [Google Scholar] [CrossRef] [PubMed]
- Olivero-Verbel, R.; Moreno, T.; Fernández-Arribas, J.; Reche, C.; Minguillón, M.C.; Martins, V.; Querol, X.; Johnson-Restrepo, B.; Eljarrat, E. Organophosphate esters in airborne particles from subway stations. Sci. Total Environ. 2021, 769, 145105. [Google Scholar] [CrossRef] [PubMed]
- Marklund, A.; Andersson, B.; Haglund, P. Screening of organophosphorus compounds and their distribution in various indoor environments. Chemosphere 2003, 53, 1137–1146. [Google Scholar] [CrossRef]
- Reemtsma, T.; Quintana, J.B.; Rodil, R.; Garcı’a-López, M.; Rodrı´guez, I. Organophosphorus flame retardants and plasticizers in water and air I. Occurrence and fate. TrAC 2008, 27, 727–737. [Google Scholar] [CrossRef]
- van der Veen, I.; de Boer, J. Phosphorus flame retardants: Properties, production, environmental occurrence, toxicity and analysis. Chemosphere 2012, 88, 1119–1153. [Google Scholar] [CrossRef]
- Hoffman, K.; Butt, C.M.; Chen, A.; Limkakeng, A.T., Jr.; Stapleton, H.M. High Exposure to Organophosphate Flame Retardants in Infants: Associations with Baby Products. Environ. Sci. Technol. 2015, 49, 14554–14559. [Google Scholar] [CrossRef]
- Wei, G.-L.; Li, D.-Q.; Zhuo, M.-N.; Liao, Y.-S.; Xie, Z.-Y.; Guo, T.-L.; Li, J.-J.; Zhang, S.-Y.; Liang, Z.-Q. Organophosphorus flame retardants and plasticizers: Sources, occurrence, toxicity and human exposure. Environ. Pollut. 2015, 196, 29–46. [Google Scholar] [CrossRef]
- Li, J.; Zhao, L.; Letcher, R.J.; Zhang, Y.; Jian, K.; Zhang, J.; Su, G. A review on organophosphate Ester (OPE) flame retardants and plasticizers in foodstuffs: Levels, distribution, human dietary exposure, and future directions. Environ. Int. 2019, 127, 35–51. [Google Scholar] [CrossRef]
- Sheldon, L.S.; Hites, R.A. Organic compounds in the Delaware River. Environ. Sci. Technol. 1978, 12, 1188–1194. [Google Scholar] [CrossRef]
- Cristale, J.; Dantas, R.F.; De Luca, A.; Sans, C.; Esplugas, S.; Lacorte, S. Role of oxygen and DOM in sunlight induced photodegradation of organophosphorous flame retardants in river water. J. Hazard. Mater. 2017, 323, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Kim, E.; Strathmann, T.J. Mineral- and Base-Catalyzed Hydrolysis of Organophosphate Flame Retardants: Potential Major Fate-Controlling Sink in Soil and Aquatic Environments. Environ. Sci. Technol. 2018, 52, 1997–2006. [Google Scholar] [CrossRef] [PubMed]
- Salamova, A.; Hermanson, M.H.; Hites, R.A. Organophosphate and Halogenated Flame Retardants in Atmospheric Particles from a European Arctic Site. Environ. Sci. Technol. 2014, 48, 6133–6140. [Google Scholar] [CrossRef]
- Sühring, R.; Scheringer, M.; Rodgers, T.F.M.; Jantunen, L.M.; Diamond, M.L. Evaluation of the OECD POV and LRTP screening tool for estimating the long-range transport of organophosphate esters. Environ. Sci. Process. Impacts 2020, 22, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Chen, D.; Peng, C.; Liu, X.; Wu, Y.; Li, X.; Du, R.; Wang, B.; Guo, Y.; Zeng, E.Y. Novel and Traditional Organophosphate Esters in House Dust from South China: Association with Hand Wipes and Exposure Estimation. Environ. Sci. Technol. 2018, 52, 11017–11026. [Google Scholar] [CrossRef]
- Liu, X.; Chen, D.; Yu, Y.; Zeng, X.; Li, L.; Xie, Q.; Yang, M.; Wu, Q.; Dong, G. Novel Organophosphate Esters in Airborne Particulate Matters: Occurrences, Precursors, and Selected Transformation Products. Environ. Sci. Technol. 2020, 54, 13771–13777. [Google Scholar] [CrossRef]
- Zeng, Y.; Chen, S.J.; Liang, Y.H.; Zhu, C.Y.; Liu, Z.; Guan, Y.F.; Ma, H.M.; Mai, B.X. Traditional and novel organophosphate esters (OPEs) in PM(2.5) of a megacity, southern China: Spatioseasonal variations, sources, and influencing factors. Environ. Pollut. 2021, 284, 117208. [Google Scholar] [CrossRef]
- Schantz, M.M. Pressurized liquid extraction in environmental analysis. Anal. Bioanal. Chem. 2006, 386, 1043–1047. [Google Scholar] [CrossRef]
- Primbs, T.; Genualdi, S.; Simonich, S.M. Solvent selection for pressurized liquid extraction of polymeric sorbents used in air sampling. Environ. Toxicol. Chem. 2008, 27, 1267–1272. [Google Scholar] [CrossRef]
- Carlsson, H.; Nilsson, U.; Becker, G.; Östman, C. Organophosphate Ester Flame Retardants and Plasticizers in the Indoor Environment: Analytical Methodology and Occurrence. Environ. Sci. Technol. 1997, 31, 2931–2936. [Google Scholar] [CrossRef]
- Salamova, A.; Ma, Y.; Venier, M.; Hites, R.A. High Levels of Organophosphate Flame Retardants in the Great Lakes Atmosphere. Environ. Sci. Technol. 2014, 1, 8–14. [Google Scholar] [CrossRef]
- Zeng, X.; Wu, Y.; Liu, Z.; Gao, S.; Yu, Z. Occurrence and distribution of organophosphate ester flame retardants in indoor dust and their potential health exposure risk. Environ. Toxicol. Chem. 2018, 37, 345–352. [Google Scholar] [CrossRef]
- Zeng, Y.; Ding, N.; Wang, T.; Tian, M.; Fan, Y.; Wang, T.; Chen, S.J.; Mai, B.X. Organophosphate esters (OPEs) in fine particulate matter (PM(2.5)) in urban, e-waste, and background regions of South China. J. Hazard. Mater. 2020, 385, 121583. [Google Scholar] [CrossRef]
- Pang, L.; Liu, H.; Yang, H.; Pang, R.; Liu, J. Seasonal variation and affecting factors of organophosphate esters in particulate matter in air: A comparison between measured data and model predictions. Environ. Sci. Pollut. Res. Int. 2021, 28, 36669–36679. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.E.; Yoon, S.; Sheesley, R.J.; Usenko, S. Pressurized liquid extraction technique for the analysis of pesticides, PCBs, PBDEs, OPEs, PAHs, alkanes, hopanes, and steranes in atmospheric particulate matter. Chemosphere 2015, 137, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, P.; Li, Y.; Wang, D.; Matsiko, J.; Yang, R.; Sun, H.; Hao, Y.; Zhang, Q.; Jiang, G. Spatial and temporal distribution of organophosphate esters in the atmosphere of the Beijing-Tianjin-Hebei region, China. Environ. Pollut. 2019, 244, 182–189. [Google Scholar] [CrossRef]
- EPA. Biden-Harris Administration Finalizes Ban on Most Uses of Methylene Chloride, Protecting Workers and Communities from Fatal Exposure. 2024. Available online: https://www.epa.gov/newsreleases/biden-harris-administration-finalizes-ban-most-uses-methylene-chloride-protecting (accessed on 26 August 2024).
- EPA. Fact Sheet: Methylene Chloride or Dichloromethane (DCM). 2023. Available online: https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/fact-sheet-methylene-chloride-or-dichloromethane-dcm-0# (accessed on 13 March 2024).
- EPA. Definition and Procedure for the Determination of the Method Detection Limit, Revision 2. 2016. Available online: https://www.epa.gov/sites/default/files/2016-12/documents/mdl-procedure_rev2_12-13-2016.pdf (accessed on 12 September 2023).
- Björklund, E.; Sporring, S.; Wiberg, K.; Haglund, P.; Holst, C.v. New strategies for extraction and clean-up of persistent organic pollutants from food and feed samples using selective pressurized liquid extraction. TrAC 2006, 25, 318–325. [Google Scholar] [CrossRef]
- Giergielewicz-Możajska, H.; Dąbrowski, Ł.; Namieśnik, J. Accelerated Solvent Extraction (ASE) in the Analysis of Environmental Solid Samples—Some Aspects of Theory and Practice. Crit. Rev. Anal. Chem. 2001, 31, 149–165. [Google Scholar] [CrossRef]
- Clark, A.E.; Yoon, S.; Sheesley, R.J.; Usenko, S. Spatial and Temporal Distributions of Organophosphate Ester Concentrations from Atmospheric Particulate Matter Samples Collected across Houston, TX. Environ. Sci. Technol. 2017, 51, 4239–4247. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Exp. # | Solvent Composition | Extraction Temp. (°C) | Surrogate Recovery 1 (Average ± Standard Deviation) (%) |
|---|---|---|---|
| 1 | 2:1 DCM/ACE | 100 | 71.1 ± 23.1 2 |
| 2 | 1:1 HEX/ACE | 100 | 88.1 ± 18.8 2 |
| 3 | 1:1 HEX/ACE | 120 | 74.4 ± 18.1 2 |
| 4 | 2:1 HEX/ACE | 100 | 56.0 ± 10.8 2 |
| 5 | 2:1 HEX/ACE | 120 | 66.5 ± 19.2 2 |
| 6 | 1:1 HEX/EA | 100 | 73.8 ± 8.1 3 |
| 7 | 1:1 HEX/EA | 120 | 64.6 ± 8.6 3 |
| 8 | 2:1 HEX/EA | 100 | 56.0 ± 8.4 3 |
| 9 | 2:1 HEX/EA | 120 | 62.4 ± 10.3 3 |
| Precision and Accuracy | SRM 2585 | ||||
|---|---|---|---|---|---|
| Compound Abbr. | Percent Recovery (n = 6; %) | MDL (ppb) | Detected (pg mg−1) | Reported (pg mg−1) | % Error |
| dTBP | 86.8 ± 5.2 | --- | --- | --- | --- |
| TBP | 79.3 ± 13.3 | 51.0 | 437 ± 64 | 276 ± 14 | 58% |
| dTCEP | 100 ± 5 | --- | --- | --- | --- |
| TCEP | 94.9 ± 8.3 | 14.2 | 872 ± 21 | 925 ± 149 | 6% |
| dTCPP | 81.9 ± 4.4 | --- | --- | --- | --- |
| TCPP | 101 ± 20 | 41.1 | 1157 ± 250 | 1220 ± 350 | 5% |
| dTDCPP | 90.9 ± 6.1 | --- | --- | --- | --- |
| TDCPP | 95.9 ± 5.1 | 39.7 | 3149 ± 238 | NR 1 | --- |
| dTPP | 83.3 ± 5.2 | --- | --- | --- | --- |
| TPP | 92.4 ± 5.9 | 53.4 | 1282 ± 105 | 1190 ± 130 | 8% |
| EHDPP | 108 ± 9 | 34.3 | 1086 ± 57 | NR 1 | --- |
| TEHP | 111 ± 14 | 55.9 | 939 ± 48 | NR 1 | --- |
| 2IPPDPP | 110 ± 3 | 26.7 | 431 ± 57 | NR 1 | --- |
| TOTP | 108 ± 5 | 27.4 | 139 ± 47 | NR 1 | --- |
| 3IPPDPP | 102 ± 3 | 24.0 | --- | --- | --- |
| 2tBPDPP | 101 ± 4 | 19.0 | --- | --- | --- |
| TMTP | 118 ± 4 | 20.9 | --- | --- | --- |
| 3tBPDPP | 96.3 ± 3.2 | 19.2 | 97.6 ± 11.2 | NR 1 | --- |
| B2IPPPP | 99.4 ± 4.8 | 14.4 | 231 ± 30 | NR 1 | --- |
| 24DIPPDPP | 107 ± 5 | 18.2 | 314 ± 64 | NR 1 | --- |
| 4tBPDPP | 110 ± 4 | 19.6 | 481 ± 29 | NR 1 | --- |
| TPTP | 97.2 ± 10.0 | 16.1 | 239 ± 8 | NR 1 | --- |
| B3IPPPP | 111 ± 6 | 25.3 | --- | --- | --- |
| T2IPPP | 95.0 ± 3.2 | 19.4 | 174 ± 13 | NR 1 | --- |
| B2tBPPP | 99.0 ± 3.5 | 17.7 | --- | --- | --- |
| T35DMPP | 111 ± 5 | 21.9 | --- | --- | --- |
| 4IPPDPP | 98.7 ± 3.6 | 17.4 | --- | --- | --- |
| B3tBPPP | 105 ± 4 | 22.0 | --- | --- | --- |
| B4IPPP | 111 ± 4 | 21.7 | --- | --- | --- |
| T3IPPP | 122 ± 6 | 24.5 | --- | --- | --- |
| B24DIPPPP | 101 ± 3 | 21.5 | --- | --- | --- |
| T34DMPP | 111 ± 5 | 17.6 | --- | --- | --- |
| T3tBPP | 125 ± 9 | 18.2 | --- | --- | --- |
| B4tBPPP | 111 ± 4 | 22.9 | 294 ± 24 | NR 1 | --- |
| T4IPPP | 117 ± 4 | 20.2 | --- | --- | --- |
| T4tBPP | 128 ± 8 | 25.9 | 113 ± 19 | NR 1 | --- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gathof, A.; Bonanno, T.; Rossicone, P.; Clark, A.E. Improved and Novel Methods for Investigating Organophosphate Esters in Particulate Matter. Analytica 2024, 5, 471-480. https://doi.org/10.3390/analytica5040032
Gathof A, Bonanno T, Rossicone P, Clark AE. Improved and Novel Methods for Investigating Organophosphate Esters in Particulate Matter. Analytica. 2024; 5(4):471-480. https://doi.org/10.3390/analytica5040032
Chicago/Turabian StyleGathof, Annie, Tess Bonanno, Paige Rossicone, and Adelaide E. Clark. 2024. "Improved and Novel Methods for Investigating Organophosphate Esters in Particulate Matter" Analytica 5, no. 4: 471-480. https://doi.org/10.3390/analytica5040032
APA StyleGathof, A., Bonanno, T., Rossicone, P., & Clark, A. E. (2024). Improved and Novel Methods for Investigating Organophosphate Esters in Particulate Matter. Analytica, 5(4), 471-480. https://doi.org/10.3390/analytica5040032