


Simple and Rapid High-Performance Liquid Chromatography Method for Simultaneous Determination of Picloram and 2,4-D in Pesticide Formulations

Abstract
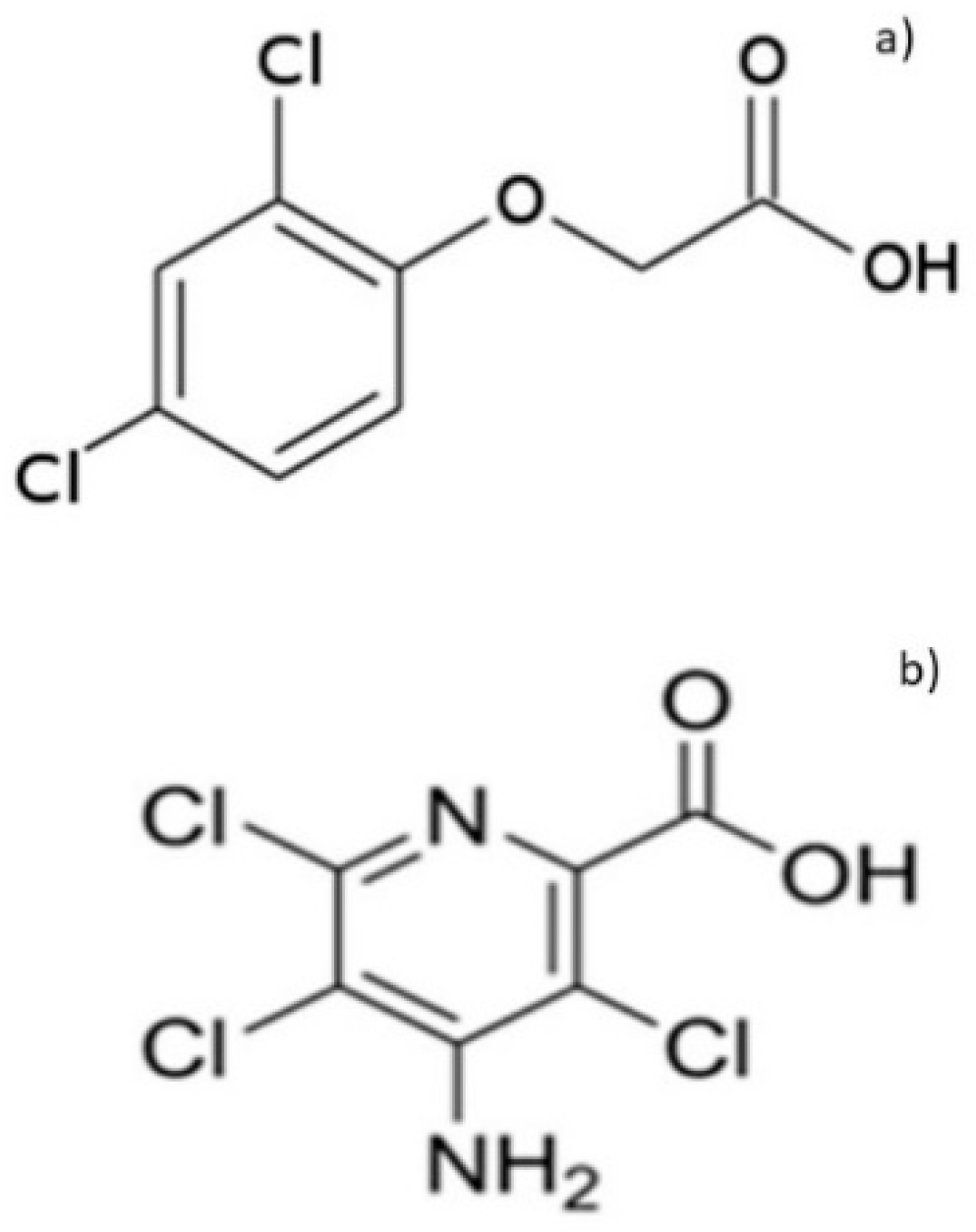
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Instrumentation
2.3. Chromatographic Procedure
2.4. Sample Preparation
2.4.1. Standard Solution
2.4.2. Samples Solutions
2.5. Method Validation
2.5.1. Trueness and Precision
2.5.2. Linearity
2.5.3. LoD and LoQ
3. Results and Discussion
3.1. Specificity
3.2. Linearity
3.3. Precision (Repeatability)
3.4. Reproducibility
3.5. Accuracy
3.6. LoD and LoQ
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tian, Y.; Liu, X.; Dong, F.; Xu, J.; Lu, C.; Kong, Z.; Zheng, Y. Simulataneous determination of Aminopyralid, Clopyralid and Picloram residues in vegetables and fruits using Ultra-performance Liquid chromatography/Tandem mass Spectrometry. J. AOAC Int. 2012, 95, 554–559. [Google Scholar] [CrossRef] [PubMed]
- De Gerónimo, E.; Botero-Coy, A.M.; Marín, J.M.; Aparicio, V.C.; Costa, J.L.; Sancho, J.V.; Hernández, F. Simple and rapid analytical methodology based on liquid chromatography–tandem mass spectrometry for monitoring pesticide residues in soils from Argentina. Anal. Methods 2015, 7, 9504–9512. [Google Scholar] [CrossRef]
- Tang, L.; Zeng, G.M.; Shen, G.L.; Li, Y.P.; Zhang, Y.; Huang, D.L. Rapid detectionof Picloram in Agricultural Field Samples Using a Disposable Immunomembrane-based electrochemical sensor. Environ. Sci. Technol. 2008, 42, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Sack, C.; Vonderbrink, J.; Smoker, M.; Smith, R.E. Determination of Acid herbicides using Modified QuEChERS with fast Switching ESI+/ESI- LC/MS/MS. J. Agric. Food Chem. 2015, 63, 9657–9665. [Google Scholar] [CrossRef]
- Li, W.K.; Zhang, J.; Wang, S.; Ma, Z.Q.; Feng, J.T.; Pei, H.W.; Liu, Y.M. Simulataneous determination of three herbicides residues in wheat flour based on the hollow fiber supported carbon dots. J. Food Compos. Anal. 2022, 108, 104426. [Google Scholar] [CrossRef]
- Rodríguez, R.; Mañes, J.; Picó, Y. Off-line Solid-phase microextraction and capillary electrophoresis mass spectrometry to determine acidic pesticides in fruits. Anal. Chem. 2003, 75, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Koesukwiwat, U.; Sanguankaew, K.; Leepipatpiboon, N. Rapid determination of phenoxy acid residues in rice by modified QuEChERS extraction and liquid chromatorgraphy-tandem mass spectrometry. Anal. Chim. Acta 2008, 626, 10–20. [Google Scholar] [CrossRef]
- Mwakalesi, A.J.; Potter, I.D. Removal of picloram herbicide from an aqueous environment using polymer inclusion membranes. J. Environ. Chem. Eng. 2020, 8, 103936. [Google Scholar] [CrossRef]
- AOAC. Official Method 976:03, Picloram and 2,4-D in Pesticide Formulations, Liquid Chromatographic Method. 1996. Available online: http://www.aoacofficialmethod.org/index.php?main_page=product_info&cPath=1&products_id=1903 (accessed on 25 October 2022).
- Swathi, R.; Ramanjulu, C.; Ramachandra, B.; Naidu, N.V. Development and validation of spectrophotometric method for the determination of Picloram. Der Pharm. Lettre. 2015, 7, 86–93. [Google Scholar]
- Abramovic, B.F.; Anderluh, V.B.; Gall, F.F.; Sojic, D.V. Derivative spectrophotometric determination of the herbicides picloram and triclopyr in mixtures. J. Serb. Chem. Soc. 2007, 72, 809–819. [Google Scholar] [CrossRef]
- Stevens, T.S. Assay of Formulations containing 2,4-D and/or Picloram by direct injection high pressure Liquid Chromatography. J. Assoc. Off. Anal. Chem. 1979, 62, 297–303. [Google Scholar] [CrossRef]
- Henriet, J.; Lovett, J.F.; Martijn, A.; Povlsen, H.H. Method MT174/SL/M/-. 2,4-D + Picloram aqueous solutions. In CIPAC Handbook “Analysis of Technical and Formulated Pesticides” Editor CIPAC; CIPAC: Harpendn, UK, 1989; Volume 1B, pp. 1757–1759. [Google Scholar]
- Kowalska, G.; Pankiewicz, U.; Kowalski, R. Estimation of pesticide residues in selected products of plant origin from Poland with the use of the HPLC-MS/MS technique. Agriculture 2020, 10, 192. [Google Scholar] [CrossRef]
- Wang, L.; Ma, P.; Chen, H.; Chang, M.; Lu, P.; Chen, N.; Zhang, X. Rapid determination of mixed pesticide residues on apple surface by surface-enhanced Raman spectroscophy. Foods 2022, 11, 1089. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.; Carvalho, T.; Sousa, A.; Neto, V.S.; Fechine, P.; Nascimento, R. Determination of insecticide Residues in vegetal fruits. Chromatogr. Res. Int. 2011, 2011, 713256. [Google Scholar] [CrossRef]
- Botero-Coy, A.M.; Marin, J.M.; Ibanez, M.; Sancho, J.V.; Hernandez, F. Multi-residue determination of pesticides in tropical fruits using liquid chromatography/tandem mass spectrometry. Anal. Bioanal. Chem. 2012, 402, 2287–2300. [Google Scholar] [CrossRef] [PubMed]
- Chamkasem, N.; Morris, C. Direct determination of 2,4-dichorophenoxyacetic acid in Soybean and corn by liquid chromatography/tandem mass spectrometry. J. Regul. Sci. 2016, 02, 9–18. [Google Scholar]
- Larson, R.D.; Houglum, J.E. Liquid-chromatography of pesticide formulations containing Dicamba, 2,4-D, and MCPP. J. AOAC 1991, 74, 679–681. [Google Scholar] [CrossRef]
- Velkoska-Markovska, L.; Petanovska-Ilievska, B. Quantitative determination of 2,4-D in pesticides Monosan Herbi and DMA-6. J. Agric. Food Environ. Sci. 2016, 67, 62–67. [Google Scholar]
- Hafeez, A.; Tawab, I.A.; Iqbal, S. Development and validation of an HPLC method for the simultaneous determination of fipronil, Clorfenapyr, and Pyriproxyfen in Insecticide formualtions. J. AOAC Int. 2016, 99, 1185–1190. [Google Scholar] [CrossRef]
- Balayiannis, G.; Karasali, H.; Ambrus, A. Rapid determination of Famoxadone and Cymoxanil in commercial pesticide Formulation by High Performance Liquid Chromatography Using a C18 Monolithic Rod Column. Bull. Environ. Contam. Toxicol. 2014, 93, 775–780. [Google Scholar] [CrossRef]
- Quintas, G.; Armenta, S.; Morales-Noé, A.; Garrigues, S.; Guardia, M. Simultaneous determination of Folpet and Metalaxyl in pesticide formualtions by flow injection Fourier transform infrered spectrometry. Anal. Chim. Acta 2003, 480, 11–21. [Google Scholar] [CrossRef]
- Karasali, H.; Kasiotis, K.; Machera, K. Rapid determination of fosetyl-aluminium in commercial pesticide formualtions by high-performance liquid chromatography. Chem. Pap. 2014, 68, 725–731. [Google Scholar] [CrossRef]
- Pose-Juan, E.; Rial-Otero, R.; Martínez-Carballo, E.; López-Periago, E.; Simal-Gándara, J. Determination of metalaxyl and identification of adjuvants in wettable powder pesticide technical formulas. Anal. Bioanal. Chem. 2009, 394, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Płonka, M.; Walorczyk, S.; Miszczyk, M.; Kronenbach-Dylong, D. Simulataneous gas chromatographic determination of Chlorpyrifos and its impurity sulfotep in liquid pesticide formualtions. J. Environ. Sci. Health Part B 2016, 51, 736–741. [Google Scholar] [CrossRef]
- Miszczyk, M.; Płonka, M.; Bober, K.; Dołowy, M.; Pyka, A.; Pszczolińska, K. Application of chemometric analysis based on physicochemical and chromatographic data for the differentiation origin of plant protection products containing chlorpyrifos. J. Environ. Sci. Health Part B 2015, 50, 744–751. [Google Scholar] [CrossRef]
- Marczewska, P.; Płonka, M.; Rolnik, J.; Sajewicz, M. Determination of azoxystrobin and its impurity in pesticide formualtions by liquid chromatography. J. Environ. Sci. Health Part B 2020, 56, 599–603. [Google Scholar] [CrossRef]
- Marczewska, P.; Miszczyk, M.; Płonka, M.; Kronenbach-Dylong, D.; Szeremeta, D.; Sajewicz, M. Application of different chromatographic techniques and chemometric analysis in authenticity testing of plant protection products containing azoxystrobin as an active substance. J. Environ. Sci. Health Part B 2019, 54, 590–597. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Repeatability Retention Time (RSD%) | Repeatability Area (RSD%) | Tailing Factor | Resolution |
|---|---|---|---|
| 2,4-D | |||
| 0.045 | 0.401 | 1.164 ± 0.005; RSD% = 0.465 | 9.84 ± 0.04 RSD% = 0.38 |
| Picloram | |||
| 0.098 | 0.191 | 1.227 ± 0.002; RSD% = 0.148 | 5.33 ± 0.03 RSD% = 0.51 |
| Prophiophenone | |||
| 0.04 | 0.249 | 1.071 ± 0.002; RSD% = 0.241 | 10.86 ± 0.03 RSD% = 0.28 |
| Picloram | 2,4-D | |||
|---|---|---|---|---|
| 1st Day | 2nd Day | 1st Day | 2nd Day | |
| Mean value (n = 5) | 31.0 | 31.0 | 114.0 | 113.8 |
| Standard deviation (SD) | 0.39 | 0.48 | 1.76 | 1.62 |
| Relative Standard Deviation (RSD%) | 1.26 | 1.54 | 1.54 | 1.42 |
| Horwitz RSDr | 2.25 | 2.25 | 1.87 | 1.87 |
| Horwitz RSDR | 3.36 | 3.36 | 2.79 | 2.79 |
| Horwitz ratio (Horrat) | 0.56 | 0.68 | 0.76 | 0.76 |
| Analyte | Label Content (g/kg) | Amount Measured (g/kg; n = 5) | Intraday Accuracy (% n = 5) | Inter Day Accuracy (% n = 10) |
|---|---|---|---|---|
| Picloram | 32.8 | 31.03 | 94.63 | 94.60 |
| 2,4-D | 109 | 114.04 | 104.62 | 104.53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santilio, A.; Girolimetti, S.; Picardo, V. Simple and Rapid High-Performance Liquid Chromatography Method for Simultaneous Determination of Picloram and 2,4-D in Pesticide Formulations. Analytica 2022, 3, 430-438. https://doi.org/10.3390/analytica3040029
Santilio A, Girolimetti S, Picardo V. Simple and Rapid High-Performance Liquid Chromatography Method for Simultaneous Determination of Picloram and 2,4-D in Pesticide Formulations. Analytica. 2022; 3(4):430-438. https://doi.org/10.3390/analytica3040029
Chicago/Turabian StyleSantilio, Angela, Silvana Girolimetti, and Valentina Picardo. 2022. "Simple and Rapid High-Performance Liquid Chromatography Method for Simultaneous Determination of Picloram and 2,4-D in Pesticide Formulations" Analytica 3, no. 4: 430-438. https://doi.org/10.3390/analytica3040029
APA StyleSantilio, A., Girolimetti, S., & Picardo, V. (2022). Simple and Rapid High-Performance Liquid Chromatography Method for Simultaneous Determination of Picloram and 2,4-D in Pesticide Formulations. Analytica, 3(4), 430-438. https://doi.org/10.3390/analytica3040029