The presence of contaminants in food and the environment has led to the need for the development of increasingly sensitive, rapid and economical analytical techniques for the detection of these substances both in food matrices and in environmental matrices. An example is represented by herbicides, characterized by an important environmental persistence such as the case of glyphosate (Gly). Furthermore, analytical methodologies are indispensable for the evaluation of secondary products originating from treatments applied to foods, such as heterocyclic aromatic amines (HAAs). In fact, they develop because of heat treatments of foods such as meats. However, analytical techniques are also necessary for the study of compounds useful for the preservation of food matrices, as in the case of phenolic and polyphenolic compounds.
The first examples of such coupling were present in two papers published by Welch et al. in 1989 [
34] and by Mögele et al. in 1992 [
35]. In the first paper, the authors proposed an anion-exchange amino acid separation followed by pulsed coulometric detection (PSD) or by potential-sweep pulsed coulometric detection (PS-PCD). They introduced the gold electrodes. Until then, platinum electrodes were used for the pulsed amperometric detection (PAD): the analyte response in terms of reproducibility and repeatability was better because, using the non-degassed solutions, the oxygen reduction wave was well-solved from the cathodic stripping peak of gold oxide, in contrast with occurring with the Pt electrode. Under these conditions a limit of detection (LOD, defined as 3 × standard deviation of the blank, whereas Limit of quantification, LOQ, is defined as 10 × standard deviation of the blank [
36]) for lysine was 40 ppb, whereas the LODs of the other primary amino acids were within ±3 times that for lysine. It should be underlined that the analysis was carried out on underivatized amino acids. The second paper reported the determination of organic acids (i.e., oxalic acid, lactic acid, ascorbic acid), amino acids and sugars (i.e., glucose, galactose, sucrose, maltose, fructose) in foodstuffs (e.g., pear juice, soft cheese, etc.). The authors used HPLC analysis (specifically, reversed phase, RP, or flow injection analysis, FIA) followed by a post-column enzyme reaction with amperometric detection. The approach was very easy but very effective: from the oxidase-enzyme reaction hydrogen peroxide was produced, an amperometric detector was used for measuring H
+ at +0.6 V on a platinum electrode (H
2O
2 → O
2 + 2H
+ + 2e
−). The authors managed to detect such compounds with LODs ranging between nM and pM with a relative standard deviation (RSD) between 2 and 5%. Finally, in 2000 Arias de Fuentes et al. proposed a polymeric membrane ion-selective electrode (ISE) as HPLC detector for determining cholic acids in pharmaceutical preparations [
37]. The authors tested such ISE, selective for cholic acid (specifically, deoxycholic DCA, chenodeoxycholic CDCA, ursodeoxycholic UDCA, taurocholic TCA sodium salts), both in batch and in flow injection analysis (FIA), using a wall jet type of flow cell: response times < 8 s, correlation coefficients above 0.9943, average slopes (as ΔmV/ΔlogC) of 59.6 in the concentration range between 5.0 × 10
−5 and 5 × 10
−2 and satisfactory LODs (ranging between 1.6 and 3.3 µg vs. 0.2–1.8 determined by HPLC-UV) were achieved under the investigated conditions (i.e., pH 7.0–9.2, at lower pH value the ISE response drastically decreased; different solvent tested; isocratic elution, whereas gradient elution had to be carried out in further studies). This study represents an important step in the FIA because it highlighted the importance of the wall jet cell that helped to solve some problems in the flow analysis.
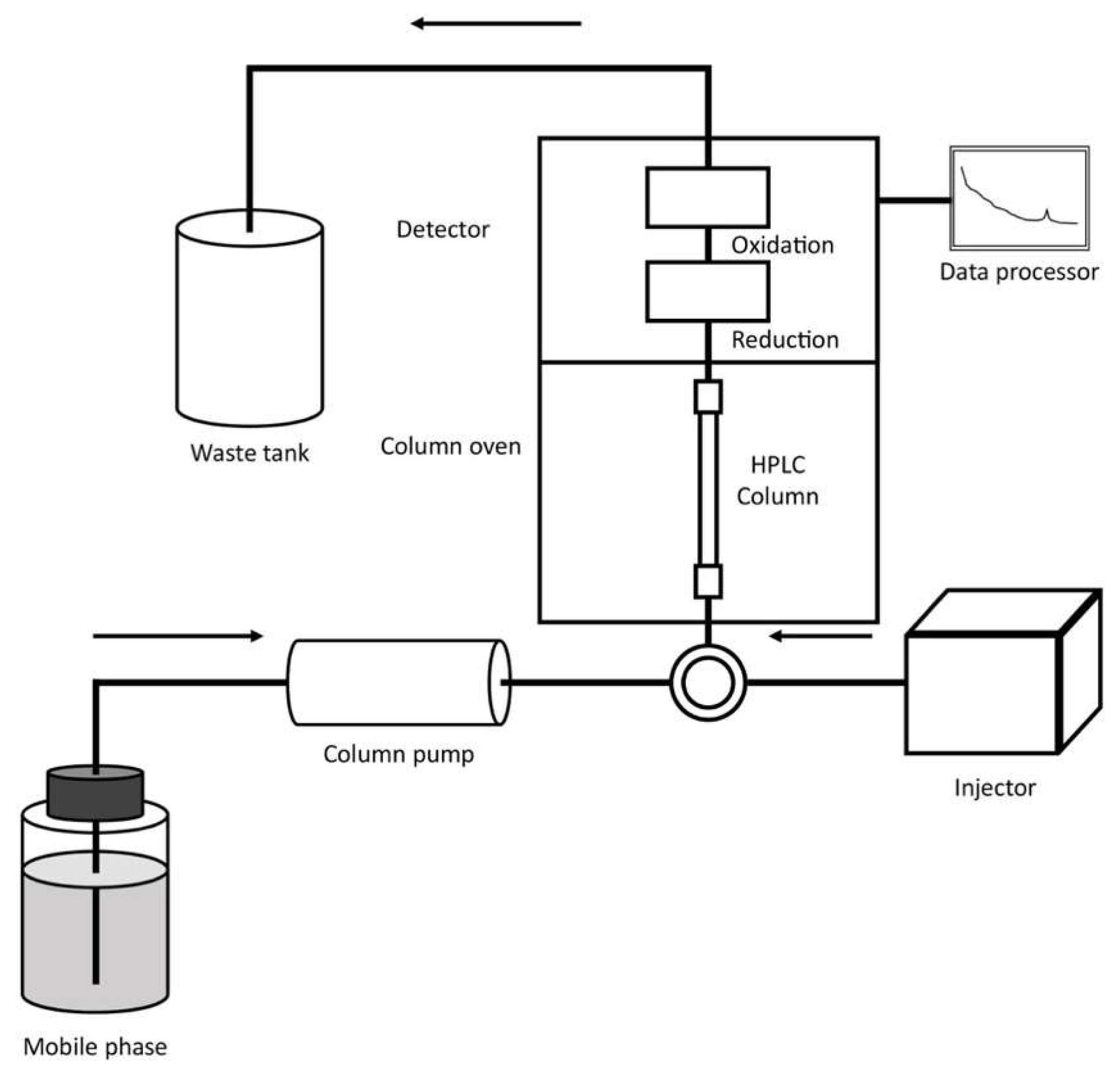
During the past four decades, the coupling between HPLC and the electrochemical detection (as amperometric, voltametric, coulometric and potentiometric methods) has been extensively used to determine different organic compounds in different alimentary (foods and beverages) and environmental samples.
3.1. Cereals, Rice and Wheat
Cereals, rice and wheat are the first samples to be presented.
Table 1 resumes all the data reported in this section.
The first paper concerning HPLC and electrochemical detection for determining altertoxins (ATX-I and ATX-II), was by Visconti et al. [
38]. They investigated the content of such toxic metabolites produced by
Alternaria alternata, a fungus colonizing a wide variety of agricultural commodities in the field or during storage, at sub-ppm levels (LOD 30 pg, RSD 3.3–5.8%) in rice, maize and tomato samples by means of an isocratic elution (methanol-water, 60:40,
v/
v) followed by dual in-series electrodes detection operating in the “redox” mode (generator electrode +1.0 V, indicator electrode −0.1 V). Another toxin compound was the citrinin, a mycotoxin produced by several fungal species including
Penicillium and
Aspergillus [
42]. Wiplinger et al. set up a procedure for analyzing such mycotoxin in different matrices (e.g., cereals, cereal-based foods, rice, wheat, etc.) by means of reversed-phase (RP-)HPLC in isocratic elution and detection with coulometric array (eight electrodes) [
39] reaching LODs between 1 and 28.5 µg kg
−1 (depending on the matrix) at detector V +600 mV. A toxic xenobiotic species, 4-nonylphenol (4-NP), was analyzed by HPLC with coulometric electrode array detection (CEAD) [
40]. The authors studied the 4-NP migration from PVC film to food (cooked rice reheated in microwave oven) wrapped with such material. The detector V rose to +670 mV for the presence of the phenolic group. Finally, in 2008, Polish authors described a RP-HPLC method with coulometric electrochemical detection for the determination of folic acid (FA) in fortified cereal products and fruit juices [
41]. FA, simply vitamin B
9, is a cofactor essential for the synthesis of purines and pyrimidines and in the production of methionine from homocysteine [
43]: it is required in the diet because humans cannot synthetize it, so it is present in fortified-folate alimentary food. The authors developed an HPLC method for analyzing it: an isocratic elution allowed to separate FA from folate with low detection limits (1.3 and 4.5 ng mL
−1, respectively), high correlation coefficients (r ≥ 0.998), good recoveries (98.0% and 103.6%, respectively), and resolution within a minimum analysis time of 16 min. For achieving good analysis, high potentials (potentials for the guard cell, electrode E1, and electrode E2 were set at +0.85, +0.20, and +0.80 V, respectively) were used for avoiding the detection of oxidized compounds and for increasing the sensitivity.
3.2. Meat and Fish
Meat and fish are two matrices where HPLC elution is largely applied for studying nutritional or toxic compounds. On some occasions, different authors have proposed the coupling with an electrochemical detector instead of ultraviolet/fluorescence/mass spectrometry detection.
Table 2 resumes all the data collected by the different papers.
First, in 1989 Vuoreal et al. described a RPHPLC method with coulometric dual-electrode detection (RPHPLC-CDD) for analyzing tyramine, a naturally trace amine derived from tyrosine, in fish and offal [
44]. The dual electrode allowed the authors to remove the interfering compounds in the first cell (thanks to their oxidation) and to detect tyramine in the second in just 5 min for the overall procedure. Five years later, Turberg et al. published a paper [
45] showing the determination of ractopamine hydrochloride, an animal feed additive administered to animals for meat production [
54], banned in some countries. Extracts from swine, cattle and turkey feed samples were analyzed by RP-HPLC in isocratic elution with CDD as detector (no analytical parameters were reported). One year later, two interesting papers reported the determination of tyrosine and coenzymes in food materials. Particularly, Bernwieser et al. [
46] determined
o and
m-tyrosine in chicken extracts. They performed an isocratic elution on a reversed phase column, after they took advantage of the oxidative/reductive detection mode for the two isomers. The coulometric array detection was carried out at V +900 mV for the reductive form and at −200 mV for the oxidative one. In this way the authors reached high selectivity and sensitivity. Further, this approach was applied by Krach et al. for analyzing the same compounds in shrimps [
47] reaching similar LODs (1.9 µg L
−1). A paper published in 2000 reported the coupling HPLC with CEAD for investigating the coenzymes Q
9 and Q
10 in different meat foods (e.g., beef steak, beef heart, herring fillet) [
48]. The authors reached good LODs (20 and 30 µg L
−1, respectively) and obtained results quite close to those obtained by HPLC-diode array detector (HPLC-DAD).
HPLC-ED has also been successfully used for HAAs determination. They are the product of the combustion reaction of creatine, creatinine, amino acids and sugars: these compounds were formed during the exposure of meat, poultry and fish to high temperatures with a risk to human health [
55]. The IARC has classified HAAs as possible or probable carcinogens (group 2A or 2B) [
56]. Krach et al. applied a HPLC method in isocratic elution coupled with coulometric array detection for determining some HAAs (i.e., 2-amino-3-methylimidazo [4,5-
f]quinoline (IQ); 2-amino-3,8-dimethylimidazo[4,5-I]quinoxaline (MeIQx); 2-amino-3,4,8-trimethyl-imidazo[4,5-
f]quinoxaline (4,8-DiMeIQx); 2-amino-3,7,8-trimethyl-imidazo[4,5-
f]quinoxaline (7,8-DiMeIQx) in soup cubes [
49]. The role of the different cells was exploited by authors for reaching high sensitivity (LODs between 2.5 and 27.5 µg L
−1). In fact, some interfering compounds were removed in the first cell because HAAs were oxidized from the second cell to the last one. In 2004 Gerbl et al. proposed an HAAs analytical determination based on HPLC-ED in meat samples [
50]. In particular, the multichannel coulometric detector consisted of eight working electrodes in two cell blocks. The samples examined were, in addition to a reference standard solution, a standardized meat extract and real samples purchased at a local fast-food restaurant such as sausage, pork knuckle and ground beef. The reference standard solution consists of a solution of five polar HAAs and five less polar HAAs in concentrations ranging between 0.8 and 2 μg g
−1 in methanol. The same compounds were added to the standardized meat extract in concentrations between 50 and 100 ng g
−1. The polar amines examined were: 2-amino-1,6-dimethylimidazo[4,5-b]pyridine (DMIP); IQ; 2-amino-3,4-dimethylimidazo[4,5-f]quinoline (MeIQ); MeIQx; 4,8-DiMeIQx; 7,8-DiMeIQx. The least polar HAAs added were: 2-amino-9H-pyrido[2,3-b]indole (AαC); 2-amino-3-methyl-9H-pyrido[2,3-b]indole (MeAαC); 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP); 3-amino-1,4-dimethyl-5H-pyrido[4,3-b]indole (Trp-P-1); 3-amino-1-methyl-5H-pyrido[4,3-b]indole (Trp-P-2). The standard solution was analyzed with a mobile phase of acetonitrile/acetic acid buffer/distilled water (15:10:75,
v/
v/
v) and with working potentials established at +280, +320, +360, +400, +440, +480, +520 and +560 mV. For the standardized meat extract, the potentials of the eight electrodes were set at values +190, +260, +330, +400, +470, +540, +610 +680 mV. In both cases, the less polar HAAs were not detected due to interference and were therefore discarded from the study. The LODs for DMIP, IQ, MeIQ, MeIQx and 4,8-DiMeIQx were 0.8, 1.4, 2.1, 1.5 and 2.5 ng g
−1, respectively. The technique was successfully applied to the real samples mentioned above. In the case of the pork knuckle sample, DMIP concentrations could not be detected due to matrix interference. In general, the highest concentrations were found in the pork knuckle.
Interesting applications regard the determination of electroactive compounds such as cysteine (Cys), L-glutathione (GSH), oxidized glutathione (GSSG) and GSH-oligomers (phytochelatins PCs). Their analysis is possible because these compounds present -SH and -SS- groups in their formula. Potesil et al. managed to separate them by means of RP-HPLC with CEAD (up to 12 cells) [
51]: in particular, they found out that GSSG and PCs were detected at V +780 mV whereas Cys and GSH at +680 mV and the LOD were between 3.9 and 27.3 µg L
−1. This method was used by authors for assessing the effect of acute toxic contamination on fish tissues due to silver ions, for instance. On the other hand, Bayram et al. followed a similar approach for studying the GSH level in HepG
2 cells [
52]: a C
18 column was used to isocratically elute GSH. Four coulometric cells were used for the detection; the maximum response was obtained at +600 mV with LOD of 0.46 µg L
−1.
An analyzed phenolic compound is rosmarinic acid. It shows an excellent antioxidant activity, and it is generally used as an additive in meat products [
57]: its determination is possible due to the ortho-hydroxyl groups positioned on the two phenolic rings present in its structure. Juric et al. developed a technique that involves the coupling of HPLC with coulometric detector (Coulochem III) [
53]. The latter consists of two electrolytic cells in series, both having a porous graphite working electrode. The matrices used were a reference sample of minced meat heat treated at 200 °C for 15 min, fresh minced meat matured for five days at 4 °C and sausages matured for three days at 4 °C. The working potentials of the two electrolytic cells were chosen. Linear voltammograms with a V ranging between +250 and +650 mV were then recorded: they showed that the oxidative I signal for rosmarinic acid reached its maximum intensity at +500 mV, whereas it reached a plateau between +200 and +500 mV. In the first electrolytic cell the V (E1) was therefore set at +200 mV, whereas in the second the V (E2) was set at +500 mV. The LODs and LOQs obtained for the methodology were 0.33 and 1 ppb, respectively. The samples were analyzed before and after the ripening period: the results obtained show a reduction in the concentration of rosmarinic acid from 20%, in the case of sausage, to 60%, in the case of minced meat.
3.3. Oils, Seeds and Vegetables
These three matrices have been largely studied in different papers where different HPLC-ED procedures have been developed for determining electroactive compounds.
Table 3 resumes the data reported in this section.
HPLC coupled with coulometric electrode array detection (CEAD) with 16 working electrodes was used for determining phenolic compounds (i.e.,
p-hydroxybenzaldehyde,
p-coumaric acid, ferulic acid, rutin, and naringenin) in tomatoes (LOD 6 µg L
−1) [
58] and for analyzing carotenoids (i.e.,
α-carotene,
β-carotene, and lycopene) in salad samples (highest response between +380 and +500 mV) [
59]. Phenolic carotenoid and tocopherol compounds were the objects of some studies carried out in the oil matrix [
60,
61,
62,
63]. In all cases, the authors focused their attention on the detector response: they found both that the response was linear over three orders of magnitude and that the ED performance was much better than the UV detection. Following the analysis of these papers, the authors agreed that the phenolic investigation could be used for determining the oil adulteration; further, they stated that the HPLC-ED procedure was valid, sensitive and effective for this aim and it could be used as routine method.
Finally, a very recent paper regarded the determination of six strobilurin fungicides residues (azoxystrobin, kresoxim-methyl, dimoxystrobin, picoxystrobin, pyraclostrobin and trifloxystrobin), a group of natural products and mostly contact fungicides with a long half time, in bean [
64]: the authors developed an analytical method based on a boron-doped diamond (BDD) electrode with amperometric detection homemade coupled to HPLC with UV-Vis detection. The BDD use gave them some advantages such as low background currents, a wide electrochemical potential window, favorable electron-transfer kinetics, and surface inertness. The amperometric detection was optimized by studying the cyclic voltammetry of each compound investigated in the range 1.0 and 2.0 V: finally, the V was set up at +1.9 V, which was the most sensitive one for the simultaneous determination of such chemicals. With the amperometric detection approach, the authors achieved regression equations for each strobilurin with R-squared (R
2) above 0.9972, LODs ranging between 1.37 and 1.57 µg kg
−1 and LOQs between 4.15 and 4.77 µg kg
−1, recoveries between 62.3 and 95.2% with RSDs between 2.3 and 9.5% and precision below 4.5%.
3.4. Fruits and Fruit Juices
In these matrices, the HPLC-ED is basically used for determining nutritional compounds;
Table 4 shows the data. Two interesting examples are from Lebiedzinska’s group: in the first paper the authors compared a developed HPLC-CDD method with a well-known HPLC-UV procedure for determining pyridoxamine, vitamins B
1, B
6 and B
12 in fruit juices [
65] and seafood, whereas in the second one, cited above, they developed an HPLC-CDD method for analyzing folic acid (FA) and 5-formyltetrahydrofolate (5-CHO-THF) in fortified fruit juices [
66]. In both cases the interfering compounds were oxidized at the first working electrode whereas the target compounds were analyzed at the second electrode. The authors suggested the method for FA and 5-CHO-THF to be valid for routine analysis. Gazdik et al. reported a method for analyzing vitamin C in orange and apples with a LOD of 15.9 µg L
−1 [
67].
Phenols and polyphenols are a large class of compounds that can be “easily” detected by electrochemical detection. For instance, 36 flavonoids and 27 phenols were analyzed by Achilli et al. by means of gradient elution RP-HPLC coupled with 16 electrodes for an array detection [
68,
69]. No information on LODs was reported for flavonoids, whereas a LOD of 0.0003 µg L
−1 was reported for phenols: the only consideration reported in both papers regarded the suggestion to perform electrochemical cleaning after the analysis for avoiding a reduction in the electrode efficiency. Similar determination was also made by Gamache et al. in fruit and juice reaching LODs ranging between 0.001 and 0.2 µg L
−1 [
70].
Another matrix in which the coupling between HPLC and CEAD was effective was strawberry. Some authors developed such methods for studying the oxidative degradation of flavonoids, and especially the unknown compound generated during this process [
71]. The authors identified the presence of two electro-active groups at +460 and +620 mV, respectively. On this basis, they proposed the first to be a hydroxy or a methoxy substituent on an aromatic ring, and the second to be a compound similar to
p-hydroxybenzoic acid. Further, they used HPLC-DAD and HPLC-MS to confirm the compound present in 6 kGy-irradiated food, namely hydroxybenzofuranones.
An Italian group published a paper regarding the determination of some flavonoids such as eriocitrin, hesperidin, quercetin, naringin and narirutin in orange juice by HPLC-CDD [
72]. The authors reported that the higher potential values could cause a background I increase because of electrooxidation of possible impurities present, resulting in a decrease in the signal-to-noise ratio (LODs ranging between 11.8 and 793 mg kg
−1). It should be noted that the authors worked hard comparing the HPLC-CDD method with the HPLC-UV and HPLC-MS analyses: they stated that these latter methods were more precise and sensitive than the HPLC-CDD one and that the selectivity of HPLC-CDD analysis was comparable to that HPLC-MS and higher than that HPLC-UV.
Finally, an HPLC-CEAD method was carried out for investigating the effect of a pear juice adulteration [
73]. In particular, the authors investigated the content of phloretin glycoside, a bioactive compound naturally present in pear and apple fruits and in juices as consequence of the processing conditions [
74].
Table 4.
HPLC-ED analysis performance in the analysis of different compounds in fruit and juice fruit matrices. The term “N/A” means the information is absent in the paper.
Table 4.
HPLC-ED analysis performance in the analysis of different compounds in fruit and juice fruit matrices. The term “N/A” means the information is absent in the paper.
| Matrix | Analyte | Column | Potential 1 | Linearity Range | LOD 2 | Recovery 3 | RSD 4 | Ref. |
|---|
| fruit juices | vitamins B1, B6, B12 | C18 | +0.85 V | 2–2800 ng mL−1 | 0.0021–9.2 ng mL−1 | 96.3–101.3 | 1.3–7.3 | [65] |
| fruit juices | folic acid | Eurospher 100 C18 | +0.8 V | 0.06–2.0 mg L−1 | 0.04 mg L−1 | 78–97 | 1.2–1.7 | [66] |
| apple and pear juice | vitamin C | Metachem Polaris C18A RP | +0.1 V | N/A | 10 ng | N/A | N/A | [67] |
| fermented beverages, fruit juices | phenolic compounds | HR 80 C18 | +0.18 < V < +0.78 V | N/A | N/A | N/A | 0.70–5.90 | [68] |
| tap water, mineral water | phenols, herbicides | HR 80 C18 | +0.08 < V < +1.2 V | 0.1–10,000 µg L−1 | 0.08–0.45 ng L−1 | 96–105 | 1.2–5.5 | [69] |
| juice fruits | phenols, flavonoids | M.S. Gel C18 | +0.06 < V < +0.90 V | N/A | 1–200 ng mL−1 | N/A | 1–5 | [70] |
| strawberries | phenols | Hypersil ODS | +0.46, +0.62 V | N/A | N/A | N/A | N/A | [71] |
| orange juice | flavonoids, flavones, flavonols | C18 | −0.5, +0.7, +0.8 V | 2–250 µg mL−1 | 0.13–1.84 µg mL−1 | 94–98 | 0.4–10 | [71] |
| juices | phloretin glycosides | Hicarbosphere 3ODS | +0.60 V | N/A | N/A | N/A | N/A | [73] |
| apple, fruit, purées, juices | phloretin glycosides | Hicarbosphere 3ODS | +0.60 V | N/A | N/A | N/A | N/A | [74] |
3.5. Beverages
The beverage matrix is well suited to the determination by means of HPLC-ED. During the last three decades, different papers concerned the use of such coupling for determining nutritional or toxic compound in water [
75,
76], milk [
77,
78,
79], tea [
80,
81], beer [
82,
83], wine [
84,
85], alcoholic drinks [
86] and brandy [
87] samples.
Table 5 shows the analytical parameters reported in these papers.
Among the different papers related to the water matrix, two papers deserved to be mentioned. In the first paper [
75], toxic compounds such as nitro-pesticides (4-nitrophenol, 3-methyl-4-nitrophenol, parathion-methyl, fenitrothion and parathion-ethyl) were analyzed by means of the HPLC-CDD method in water samples without being subjected to a preconcentration step. The interest in this paper regards the nitrophenols’ behavior. In fact, their direct determination was not possible due to the high negative potential needed for the compound reduction. A cell was devoted to the oxygen elimination from the mobile phase because it interfered in the detection, and the second cell allowed the oxidation and the related compound determination. By this method, the authors were able to reach LODs at ppb levels in river and drinking water samples. On the other hand, the second paper [
76] focused the attention on the determination of some xenoestrogens such as bisphenol A (BP-A), 2,4-dichlorophenol (2,4-DCP), 4-
tert-butylphenol (BP), 4-
n-pentylphenol (PP), 4-
n-hexylphenol (HexP), 4-
n-heptylphenol (HepP), 4-octylphenol (OP), and 4-nonylphenol (NP), by means of HPLC-CEAD preceded by a solid phase extraction (SPE) step as clean-up. Actually, the method was developed by Braunrath et al. for analyzing canned foods [
88] but it was adapted by Inoue et al. to drinking water samples; they reached LODs ranging between 0.01 and 0.02 µg L
−1.
Milk is considered a very hard matrix to analyze [
89] because it is strongly subjected to exogenous compounds. Japanese authors determined nitroxynil in cow milk by HPLC with dual electrode detection [
78]. This compound shows a nitro and a phenolic group in its formula: these groups require opposite “high” potential for their reduction/oxidation (−700/+800 mV). Further, the authors used two cells and they combined the guard cell with the first electrode for minimizing the interferences. By this procedure they obtained LOD of 0.7 µg L
−1 in commercial milk samples. The same research group analyzed another anthelmintic compound, bromofenofos, by HPLC in isocratic conditions and coulometric dual electrode detection [
77]. The compound was determined as its electroactive form, dephosphate bromofenofos, in the second cell of the detector. The authors reached a very good LOD, 0.2 µg L
−1 in raw milk samples, and high specificity so that they proposed such method as the routine protocol. An important class of contaminants in milk are antibiotics. An example is the determination of different tetracyclines (TCs) by RP-HPLC-CEAD method [
79]. The coulometric array detection is based on four detector cells: the first cell is necessary for removing interferences whereas TCs (i.e., methacycline MTC, tetracycline TC, doxycycline DC, chlortetracycline CTC, oxytetracycline OTC) were analyzed in the other three cells (LODs ranging between 10 and 25 µg L
−1).
Tea polyphenols and isoflavones are a class of compounds with high electrochemical behavior (aromatic group and phenolic group). Wu et al. set up a method based on HPLC with an eight-cell detector for analyzing such compounds in different foods [
80]. The separation occurred by gradient elution, whereas the detection was carried out by increasing potentials strictly depending on the hydroxy groups present in the molecules (e.g., +350 mV for tri-hydroxyphenols, +400 mV for
o- and
p-diphenols, and +700 mV for
m-diphenol). Another class of phenol investigated were the catechins and their related epimers, a type of natural phenol and antioxidant. Some of them, namely (−)-epigallocatechin gallate (EGCG), (−)-epigallocatechin (EGC), (−)-epicatechingallate (ECG), (−)-epicatechin (EC) and (+)-catechin hydrate (C), were analyzed by Novak et al. in green and black tea samples: RP-HPLC-ED measures were compared with measures performed by square-wave voltammetry (SWV) [
81]. The paper focused its attention on the development of the novel method, SWV: the well-validated method RP-HPLC with electrochemical detection (represented by a wall-jet type flow cell consisted of glassy-carbon working electrode at V of +0.7 V) was used as comparison for testing. The authors obtained good LODs and LOQs ranging between 23.23 nM (C) and 100.7 nM (ECG) and 77.43 nM (C) and 335.8 nM (ECG), respectively, as well as recoveries ranging between 83.7 and 105.2% (low matrix effect).
Among the different alcoholic beverages present in the market, beer shows low alcoholic grade (between 0.5% and 20% alcohol in volume ABV, normally present ranging 4–6% ABV). This matrix is a good starter for alcoholic drinks. Coumaroyl-glycerol, indicating the maize and sorghum addition, was determined in beer samples for detecting adulteration: after the SPE clean-up, the sample was analyzed by HPLC in isocratic elution followed by electrode array detector [
82]. Phenolic compounds were also analyzed in beer: the authors compared 11 columns with different stationary phases optimizing the best analytical conditions [
83].
Wine is another important drink at medium alcoholic grade. Different sensors have been set up during previous decades for analyzing different analytical parameters or flavors [
90,
91,
92]. This review would like to highlight two papers dealing HPLC analysis followed by electrochemical detection [
84]. In particular, Larcher et al. determined some phenolic compounds (i.e., 4-ethylphenol, 4-ethylguaiacol, 4-vinylphenol, and 4-vinylguaiacol) due to their presence being a potential cause of bad taste. The authors used an isocratic elution and a coulometric array detection, reaching a LOD of 1.34 µg L
−1. The other paper investigated the lignans content in red and white wines by HPLC-CEAD method [
93]. The authors determined the best potentials using the hydrodynamic voltammograms and reached LODs ranging between 0.2 and 1.2 µg L
−1.
Finally, two papers were published on the determination of different compounds in high-alcoholic-grade drinks, in particular some phenolic acids and aldehydes in distilled alcoholic beverages [
86] and fifteen benzoic and cinnamic acid derivatives in brandy samples [
87] by means of HPLC followed by coulometric detection. In the first paper, Friedrich and Sontag used dual cells for oxidative/reductive (compounds oxidized at +0.65 V and reduced at 0.0 V, respectively) modes for analyzing 3,4-dihydroxybenzaldehyde, 4-hydroxy-3-methoxy benzoic acid, 4-hydroxy-3,5-dimethoxybenzoic acid, 4-hydroxy-3-methoxy benzaldehyde, and 4-hydoxy-3,5-dimethoxy benzaldehyde in whisky, bourbon, cognac, brandy, rum and calvados samples [
86]. They reached LODs ranging between 0.01 and 1 ng with an RSD ranging between 1.2% and 11.2%. The method did not require any sample preparation, whereas the authors stated that only electrode cleaning was needed when the calibration curve slope decreased. The other paper regarded the determination of some phenolic acids (i.e., salicylic acid, benzoic acid, cinnamic acid, veratric acid, gentisic acid, vanillic acid,4-hydroxybenzoic acid, o-coumaric acid, p-coumaric acid, ferulic acid, protocatechuic acid, syringic acid, caffeic acid, sinapic acid and gallic acid) in a brandy sample [
87]. The authors compared the performance of a RP-HPLC-ED method with HPLC-UV and HPLC with particle-beam electron-impact mass spectrometry (PB-EI-MS) procedures. Hydrodynamic voltammograms were investigated for each compound for determining the peak potentials (from +260 to 335 mV for the cathodic peak and from +510 to +1450 mV for the anodic peak) for the oxidation and the reduction and oxidation of each phenolic acid. They found very good linearity (0.999 in all the cases) LODs ranging between 3.1 and 140.0 µg L
−1; these values are quite similar to those determined by HPLC-PB-EI-MS, 6.0–152.1 µg L
−1 (it should be considered that caffeic acid,
p-coumaric, acid gallic and acid 4-hydroxybenzoicacid were not analyzed by the MS technique). The authors stated that LODs obtained by LC-PB-EI-MS were about 1000-fold higher than those determined using ED, whereas HPLC-UV was not effective due to the presence of numerous interferences and inadequate sensitivity.
3.6. Honey
Honey is a complex matrix which requires different analytical steps (e.g., extraction processes, liquid–liquid separation, clean-up procedures, etc.) for analyzing its nutritional composition or the contaminants present. On the other hand, the use of ED as a detector for HPLC analysis could be a reasonable opportunity when electroactive compounds are analyzed. In this section, the main papers regarding this determination are reported and in
Table 6 the analytical parameters are highlighted. Two examples of this occurrence are described by Fuchs et al. [
94] and Jörg et al. [
95]. Both researcher groups set up an analytical procedure based on HPLC separation and CDD for analyzing phenolic compounds in honeys. The methodology was the same for both papers: the first cell oxidized the compounds for removing the interferences, whereas the second cell allowed the determination. Fuchs et al. reached LODs between 0.3 and 9.2 ppm, whereas Jörg et al. obtained LODs ranging from 5 to 80 µg L
−1. Further, Jörg et al. applied the same procedure for determining methyl-
p-hydroxybenzoate, methyl vanillate, methyl syringate,
trans-
p-methyl coumarate and
trans-methyl ferulate, still in honey samples (LODs between 0.1 and 2 µg L
−1) [
96].
Finally, a paper related to the determination of thiamethoxam residues in honeybees [
97] deserved to be cited. In this publication the authors determined such insecticide, class of neonicotinoids, which is highly toxic to honeybees by contact or ingestion, by means of RP-HPLC with a CoulArray electrochemical detector with four electrodes (three set at 100, 125 and 150 mV, and the fourth at 880 mV). The sensitivity detection was improved by a photochemical reaction, otherwise the LOD would have been too high (680 mg L
−1). After finding the best voltages (see above), a calibration curve in the range 0.01358–1.358 mg L
−1 was obtained with a slope of 0.0014, intercept of 2.1044 and a linear coefficient of 0.9998, whereas LOD, calculated at 2.1 ng g
−1, and the recovery, 67.6%, were considered suitable for detecting low thiamethoxam levels in such matrix.
3.7. Aromatic Plants and Other Food Matrices
An example of application in aromatic plants is the study by Cantalapiedra et al. [
98]. The authors described the determination of vanillin, eugenol, carvacrol and thymol in aromatic plants (basil, peppermint, cloves and vanilla). The paper focused its attention on the comparison between three types of detectors, an amperometric detector, a coulometric detector and a UV spectrophotometer, coupled to HPLC. The amperometric sensor consisted of a working electrode in glassy carbon with a surface of 7 mm
2, a reference electrode in Ag/AgCl and a counter electrode in gold. The flow coulometric sensor consisted of a porous carbon working electrode, a palladium reference electrode and a platinum counter electrode. The mobile phase was chosen according to the response obtained from the chromatographic runs in a pH range between 2.5 and 7: the optimal pH was found to be 4.6 (acetic acid buffer and ammonium acetate) with the addition of methanol as an organic modifier to optimize its adsorption to the solid phase (buffer/methanol 35:65,
v/
v). The working potentials of the electrochemical cells, both amperometric and coulometric, were determined by injecting the four analytes at different potentials. The voltammograms obtained were studied: the best ratio between signal intensity and noise was reached at the V of +0.925 V for the amperometric detector, whereas it was +0.500 V for the coulometric one. The validation of the method was carried out by analyzing real samples of commercial essences of the aromatic plants mentioned above. The coulometric detector provided a more sensitive response. However, more selective results and lower background noise were obtained with both electrochemical detectors than those observed with the spectrophotometric detector. In
Table 7 it could be noted that the LOD and LOQ values achieved by means of electrochemical detectors were considerably lower than those obtained with spectrophotometric detection.
Another compound of interest is resveratrol (3,5′,4-trihydroxy-
trans-stilbene), a phytoalexin produced by plants in response to exogenous stress, important for its benefits to human health (antioxidant, anti-inflammatory, anti-aging, therapeutic and preventive properties against tumors, and prevention against cardiovascular diseases) [
99,
100]. Němcová et al. compared the performance of two detectors coupled to an HPLC system for the resveratrol determination [
101]. The first detector was an amperometric sensor, with a working electrode in carbon paste (Carbon Paste Electrode, CPE); the second was a spectrophotometer with a λ at 306 nm. Six samples of common buckwheat and two samples of tartaric buckwheat were analyzed. All samples were ground and subjected to ethanol extraction followed by filtration. The filtrate was concentrated and added with ethanol and filtered again. The solution obtained was used for the analyses. The optimal conditions for the electrochemical determination of
trans- and
cis-resveratrol were determined by the construction of linear hydrodynamic voltammograms with Britton–Robinson (BR) buffer diluted with acetonitrile (1:1,
v/
v), in the V range between 0 and 1.4 V and pH at 3, 5 and 7. The voltammograms were obtained by injection of 20 μL of solutions containing the analytes in concentrations of 1 × 10
−4 mol L
−1.
Table 8 shows the LODs of the two detectors used, both spectrophotometric and amperometric.
The sensitivity of the detectors is comparable with regard to trans-resveratrol, whereas in the case of cis-resveratrol the electrochemical detector has a better sensitivity than the spectrophotometric detector, which induces the isomerization of this compound. However, the relevant difference is found in the selectivity: the CPE was more selective because other electrochemically active substances were absent. Finally, the authors analyzed the trans-resveratrol content in tartaric buckwheat grains (3.43–3.50 mg kg−1) and in common buckwheat grains (0.98–1.68 mg kg−1), and in tartaric leaves (0.19–0.20 mg kg−1) and in those of common buckwheat (1.81–1.82 mg kg−1).
Two officinalis plats were the matrices of Skriniar group’s paper. They determined carnosic acid in rosemary (
Rosmarinus officinalis) and sage (
Salvia officinalis). This compound, naturally present in these two plants (their dried leaves contain 1.5–2.5% of carnosic acid), is a benzenediol abietane diterpene and is used as an antioxidant preservative in food and nonfood products (extracts of rosemary, E392) [
102]. The authors exploited the presence of the benzenediol group for the electrochemical detection, particularly the CDD, and developed a HPLC-CDD method (LOD 1.2–1.4 µg L
−1). The same authors made a comparison between this method and the HPLC-UV method: they stated that the electrochemical detection was more effective that the other one
Coenzyme Q
10 (CoQ
10H
2, ubiquinol) in dietary supplements was the target of Tang [
103]: he applied a methodology previously set up for analyzing such compounds in human breast milk [
104]. He exploited the ubiquinone/ubiquinol reversible redox and the related oxidative reaction at the electrode (LOD was 5 µg L
−1). A more detailed paper was published by Kubo et al. in 2008 dealing the determination of ubiquinol-10 and ubiquinone-10 in the Japanese diet by means of HPLC-ED [
105]. These two forms (ubiquinol-10 is reduced form and ubiquinone-10 is oxidized form) of the coenzyme Q
10, extracted directly with 2-propanol, were analyzed in seventy foodstuffs (i.e., meat, different types of fish and shellfish, fruits, pulses, potatoes, dairy products). The detection was carried out by means of an amperometric electrode at V of 600 mV (vs. Ag/AgCl). Even if the paper was focused on the determination of such forms in the different matrices and the relative calculation of average intake per day, the authors determined the analytical parameters for achieving the best performances. Under the analytical conditions studied (pH, extraction solvent, mobile phase and column), the method was simple and reliable with very interesting data: correlation coefficient (0.999 for both compounds) in the range 0.08–100 µg g
−1; LOD of 0.07 µg g
−1; recoveries ranging between 87–112% with coefficients of variation (CVs) between 2.2 and 21%, and 97–106% with CVs between 0.9–4.9% for ubiquinol-10 and ubiquinone-10, respectively; good within-day (101–109% and 0.3–9.9%; 106–115% and 0.6–10.7%, respectively) and between-day (101–110% and 2.4–15.4%; 106–114% and 3.0–13.8%, respectively) accuracy and precision.
A HPLC-CEAD method was developed for analyzing some compounds responsible of the beer taste, namely α-acids, β-acids and xanthohumol in hop samples. Kac et al. determined the best electrode potentials for the analysis [
106]: below +300 mV no oxidation occurred whereas at +300, +500 and +700 mV the oxidation of the three classes of compounds, α-acids, β-acids and xanthohumol, respectively, occurred with a sensitivity much higher (between nine and twenty times) than this by HPLC-UV method.
Nel 2008 Benová et al. dealt the determination of stilbenes in three varieties of
Polygonum cuspidatum [
107]. They analyzed the content of resveratrol and its analogs, namely astringin and piceatannol and piceid, by extraction with aqueous ethanol followed by reversed-phase-(RP-)HPLC with coulometric detection. An eight-channel coulometric array detector working at potentials of 200, 300, 400, 500, 600, 700, 800, and 900 mV was used. At each V a chromatogram was obtained: piceatannol was oxidized at 500 mV, astringin and piceid at 600 mV and resveratrol at 700 mV. Under these conditions the authors achieved very good correlation coefficients (r
2 > 0.9975) and recoveries (between 90.2 and 95.3% for all the compounds) as well as LODs and LOQs, ranging between 0.008 (resveratrol) and 0.025 mg L
−1 (astringin) and between 0.029 (resveratrol) and 0.083 mg L
−1 (astringin) respectively, were lower than those obtained by means of HPLC coupled with diode array detection (DAD) (i.e., LOD 0.02 mg L
−1 for resveratrol and piceid and 0.32 mg L
−1 for astringin). Finally, the authors applied this methodology to different
Polygonum cuspidatum varieties (i.e., var.
japonica, var.
sachalinensis, var.
bohemica) for determining the content of the four stilbenes.
Another very interesting paper concerns the determination of the azo dyes, in particular the Sudan dyes. Although the use of Sudan in foods is now banned in many countries, because Sudan dyes are classified as category 3 carcinogens (not classifiable as to its carcinogenicity to humans) by IARC, they are extensively used as powder for giving a natural red appearance to the food and have been found in some products sold in the European countries. The EU fixed a limit at 0.5–1 mg kg
−1. Basically, different analytical methodologies are available and reported in literature: recently, multi-wall carbon nanotubes (MWNTs) were used in an ionic liquid-gel-modified electrode for detecting Sudan I, Sudan II, Sudan III, and Sudan IV separated by flow injection analysis (FIA) and HPLC, in soft drinks [
108]. Particularly, a glassy carbon electrode was modified by application of a thin gel film of MWNTs. The auxiliary and reference electrodes were Pt wire and Ag/AgCl, respectively. The electrochemical reaction was followed by cyclic voltammetry: the linear regression yield r was >0.99 in the range 10–0.25 ppm, the max peak V of the four compounds were determined at 0.95 V, LODs and LOQs ranged between 0.001–0.025 ppm and 0.005–0.10 ppm, respectively, with recoveries between 93.91 and 104.09%. Finally, the authors reported the determination of the four different Sudan dyes in soft drinks: their conclusions regarded that the amperometric signal was well-defined and peaks well-separated, with no interference from the matrix.
Theisen et al. described a method for analyzing sulfite in food by means of HPLC analysis coupled with a plant sulfite oxidase biosensor (immobilized enzyme reactor, IMER) [
109]. This coupling, which was very easy but really interesting, allowed the authors to achieve good analytical parameters for analyzing sulfites in 15 commercially purchased juices such as red grapes and white grapes and fruit juices based on lemon, tomato and peach. This biosensor was compared with a similar one using animal sulfite oxidase: the detection limit (0.01 mg L
−1 SO
2), linear range (0.04–20 mg L
−1 SO
2) and stability were superior. Simultaneously, the authors also evaluated the performance of the entire methodology (HPLC-IMER): this was better than that carried out by the International Federation of Fruit Juice Producers (IFU) official method [
110], IFU 7a, which was based on the Monier-Williams method optimized for fruit juices, in terms of speed (8 vs. 30 min), sensitivity, specificity and recoveries.
Table 9 resumes all the analytical data reported in the different studies discussed here.
3.8. A Particular Compound: Glyphosate
A compound that can be determined through the coupling of HPLC and electrochemical sensors is glyphosate (Gly), an inhibitor of the metabolic pathway essential for the biosynthesis of essential plant aromatic amino acids. It deserves a special section for the implications related to their toxicity [
111]. The use of Gly-based herbicides began around the 1970s, but following the expiration of Monsanto’s patent, several Gly-based formulations were commercialized on a large scale [
112]. Gly, widely used due to its low toxicity to humans, is among the products most found in water together with its main metabolite (aminomethylphosphonic acid, AMPA): it is characterized by a half-life in the soil of between 76 and 958 days. However, its use has led to a serious problem of environmental contamination. It has recently been included in group 2A by the International Agency for Research on Cancer (IARC), classifying it as a probable human carcinogen [
113]. Due to their chemical structure, Gly and AMPA are available for electrochemical determination. The studies by Coutinho et al. propose a rapid and selective analytical technique that allows their determination in food matrices. The research, conducted between 2007 and 2008, concerned the coupling of HPLC with a coulometric sensor [
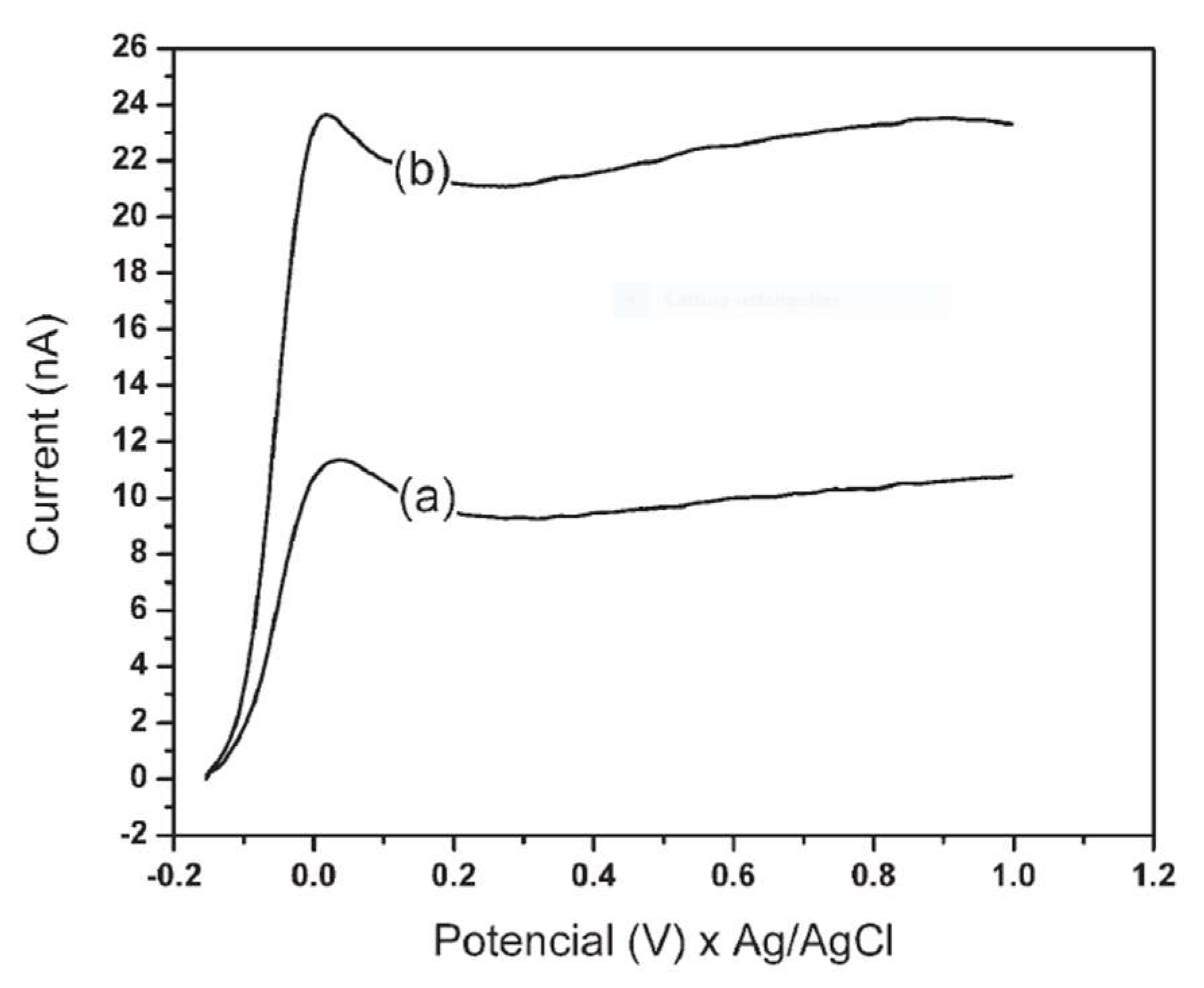
114]. The latter consists of a 60 μm diameter copper microelectrode as a working electrode and an Ag/AgCl reference electrode. This sensor allowed the Gly detection by its complexation with copper (II) thus producing an increase in the anode I, as evidenced in
Figure 2. The first step of the study was the optimization of the parameters for the electrochemical detection according to the signal obtained during the construction of voltammograms. These were recorded at a scanning rate of 0.1 V s
−1 for three types of mobile phases with three different pH values: an alkaline solution of NaOH 0.1 mol L
−1 at pH 13, a phosphate buffer at pH 6.5 and a carbonate buffer at pH 11.4.
Figure 7 shows the linear voltammograms obtained with the phosphate buffer.
The comparison between the blank and a Gly sample at known concentration highlighted that in all cases there was a linearity of the signal as a function of the concentration, but the use of phosphate buffer allowed to achieve better sensitivity and precision. The latter was therefore chosen as the mobile phase for subsequent analyzes. Methanol was then added as an organic modifier in order to regulate the elution rate and thus the peak resolution. In addition to the mobile phase, i.e., phosphate buffer 0.03 mol L
−1 and methanol (75:25,
v/
v), the working V was also experimentally determined, established as +0.5 V. Using such parameters, tomato juice was analyzed after spiking with Gly at concentration of 6.8 mg L
−1 was added: the recovery was 88%. The LOD calculated that three times the background noise was 0.1 mg L
−1. The value is satisfactory as it is lower than the maximum residual limit (MRL) of the Gly content established for food and drinks by the Environmental Protection Agency (EPA). The selectivity gives the coulometric detector a particular advantage: it is increased compared to other detectors thanks to the parameters used, specific for the Gly. Other analytes such as amino acids and organic acids, however, which have the ability to form complexes with copper (II), are not revealed. Other advantages of the technique are the simplicity of sample preparation, which does not require extraction or clean-up procedures, the cost-effectiveness of the detector, speed, as well as derivatization to be necessary [
114]. Subsequently, the same authors evaluated the analytical technique on apple juice and grape juice samples. The samples were analyzed following the addition of 6.8 mg L
−1 of Gly without clean-up or preconcentration steps: the preparation of the sample involved only centrifugation and filtration of any solid particles [
115].
Coutinho et al. continued the study in the following year [
116]. They applied the technique for the simultaneous determination of AMPA and Gly. In particular, AMPA determination is useful in order to obtain results that are more in line with the (real) total Gly content. The same technique described in the previous papers was applied with the only difference that, in this case, an electrochemical cell with three electrodes was used. In addition, a stainless-steel counter electrode was used. The working V was also evaluated by recording a voltammogram: it showed that the greatest signal for a standard mixture of Gly and AMPA, was obtained at V of +0.3 V. A sample of mineral water fortified with Gly (8.4 μg mL
−1) and AMPA (55.5 μg mL
−1) was injected: the chromatogram obtained was clean and the two peaks well resolved. The LODs were found to be 0.038 μg mL
−1 for Gly and 0.24 μg mL
−1 for AMPA. The technique, with no further preconcentration or clean-up step, was applied to real water samples: concentrations of AMPA and Gly were not revealed.
Table 10 reports the main analytical parameters found.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






