
Alagille Syndrome and Its Clinical and Laboratory Features: A Case Report

 ,
,  ,
,  and
and 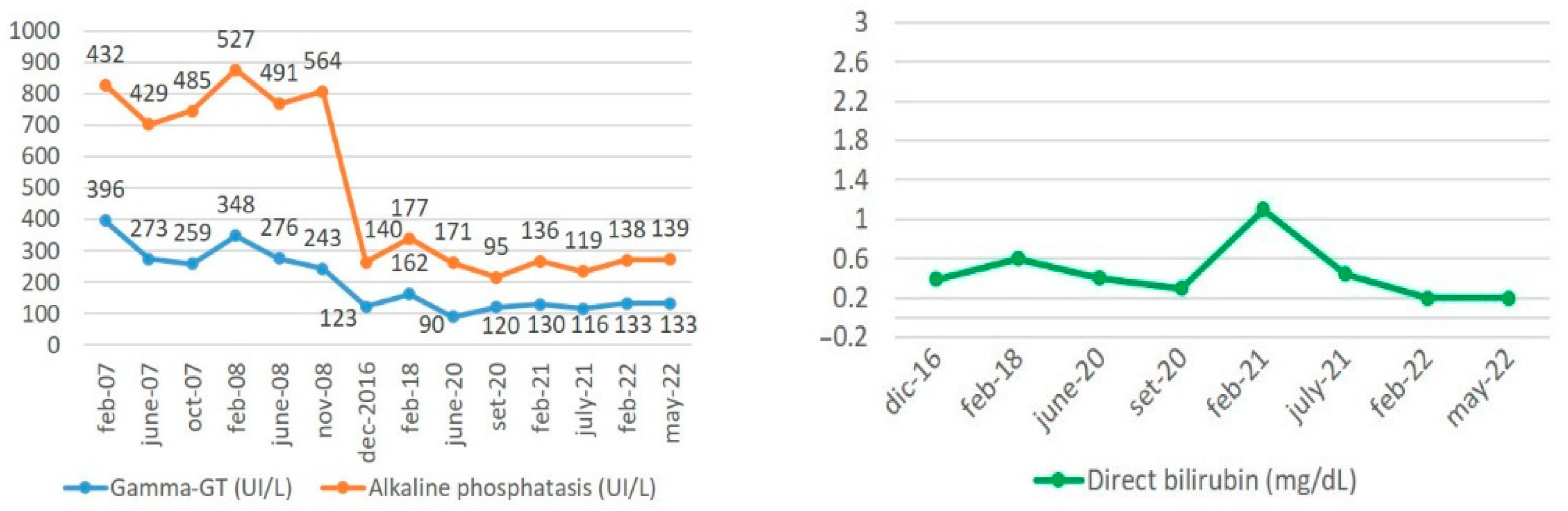{kind=link}
Abstract
1. Introduction
2. Case Report
- -
- Forty-eight hours after birth: overt jaundice developed. The newborn was treated with fluorescent blue-light phototherapy, which led to the amelioration of jaundice.
- -
- After hospital discharge: the newborn showed reduced appetite, irritability, low weight gain, and peri-oral cyanosis during breastfeeding.
- -
- Ten days after birth: she was re-hospitalized to assess the scarce weight gain along with suspected congenital heart disease.
- -
- Twentieth day of life: cholestatic jaundice developed (total bilirubin 8 mg/dL, >60% conjugated), with hypocholic stools. After excluding hepatomegaly, a therapy with phenobarbital was initiated but no significant clinical response was noted.
- -
- Two months after birth, the infant underwent heart ultrasonography which demonstrated the presence of a moderate ventricular septal defect (VSD), with left–right shunt and peripheric pulmonary trunk stenosis. An ophthalmologic evaluation showed no abnormalities. Given the neonatal episode of jaundice and the cardiovascular issues, in light of a possible congenital bile duct abnormality, a liver biopsy was performed, showing bile duct hypoplasia, compatible with the diagnosis of ALGS. The patient was then discharged with a therapy based on ursodeoxycholic acid (UDCA).
- -
- At six months of age, the patient underwent another ophthalmologic assessment which detected a posterior embryotoxon in the right eye, while it was almost absent in the left one. The patient continued the therapy with UDCA.
- -
- At the age of 3, she was hospitalized again due to growth retardation and the presence of severe itching, not responding to treatment with phenobarbital, cholestyramine, UDCA, and rifampicin. In this circumstance, percutaneous biliary drainage was attempted without success and UDCA dosage was then increased.
- -
- In subsequent years, the levels of bile acids fluctuated between 50 and 200 mmol/l, while alpha-fetoprotein remained in the range of normality.
- -
- At ten years of age, the child was evaluated with magnetic resonance imaging (MRI): intra- and extrahepatic bile ducts showed no pathological enlargements. An inhomogeneous hypointense area (6 cm of diameter) between IV and V liver segments was found. The gallbladder, spleen, kidneys, and pancreas were normal.
- -
- At the age of 12, an ultrasonographic exam performed showed important hypertrophy of the caudate lobe, and the presence of the same inhomogeneous area (now 5 cm wide) reported two years before between IV and V liver segments, not pushing nor compressing any adjacent vascular structures, was addressed as angioma or focal steatosis. The gallbladder was still normal, while the spleen volume was increased (11.5 cm) with a homogenous ultrasonographic structure. The portal vein had normal caliber and flow.
- -
- The last evaluation at the pediatric center (February 2008) included heart ultrasonography whose results showed hypoplasia of the left branch of the pulmonary trunk, with normal pressure gradient. Short stature and elevated thyroid-stimulating hormone (TSH) levels were observed, along with the presence of anti-thyroglobulin (anti-TBG) and anti-thyroperoxidase (anti-TPO) antibodies, pointing out a diagnosis of autoimmune thyroiditis, in the absence of thyroid nodules. Hepatomegaly and splenomegaly were evident at clinical palpation. Radiography of the trunk demonstrated the presence of left-convex scoliosis, with augmented lumbar lordosis, without the presence of “butterfly vertebrae”.
- -
- At the age of 13 years, the patient reported general clinical well-being, no jaundice, normal coagulation parameters, inflammatory markers, and serum protein levels, and normal kidney and pancreas functionality. She maintained her therapy with UDCA (600 mg/day), rifampicin, liposoluble vitamin supplementation, and ranitidine (150 mg/day). A genetic assessment was then performed, ruling out JAG1 pathogenic variants and parental screening was negative for other ALGS-related mutations. ALGS with characteristic facies, pulmonary trunk hypoplasia, and chronic cholestatic liver disease was pointed out. The main complaint of the patient remained itching. From 11 to 13 years old (October 2008), AP levels slightly fluctuated between 420 and 550 U/L, while GGT fluctuated between 250 and 400 U/L; total cholesterol levels remained steadily and slightly above 200 mg/dL, while total bilirubin levels did not exceed the value of 2.3 mg/dL, with a mean value of 1.4 mg/dL.
- -
- The patient missed the follow-up evaluations from 2013 to 2016, reporting general well-being except for occasional itching flares that were treated with cholestyramine 4–8 g/day with a fair control of the symptom.
- -
- In 2016, the patient came to the attention of our gastroenterology unit for the first time. The laboratory examination reported: total bilirubin 1.19 mg/dL, conjugated bilirubin 0.37 mg/dL, alanine transaminase (ALT) 36 U/L, AP 140 U/L, GGT 123 U/L, total amylase 111 U/L, pancreatic amylase 65 U/L. Inflammation markers, C-reactive protein (CRP), and alpha-fetoprotein were all normal. Vitamin D levels were in the range of normality (29.7 ng/mL). TSH was slightly elevated (5.280 uU/mL) while FT3 and FT4 levels were in the normal range. In December 2016, a re-assessment conducted by abdomen ultrasonography detected a right lobe hepatic variation (Riedel’s accessory lobe), with a normal ultrasonographic structure, normal gallbladder, without signs of gallstones, and a slightly enlarged spleen (longitudinal diameter of 125 mm). Intra- and extrahepatic bile ducts and portal vein were normal in caliber, without enlargements. Assessment of bone-mass density with dual energy X-ray absorptiometry (DEXA) reported an increased risk of fractures within the femoral neck, while a normal index was detected at the lumbar level. An ophthalmological re-assessment stated the presence of posterior embryotoxon and normal range of visus, without other pathological findings. The patient was discharged with a therapeutic protocol of UDCA 1650 mg/day, liposoluble vitamin supplementation, and rifampicin.
- -
- From 2016 to 2018, the patient reported general well-being except for flares of itching at the verge of tolerability that were treated with cholestyramine 4–8 g/day, which led to amelioration of the symptom.
- -
- At 23 years of age (2018), she was re-admitted to our unit after finding altered laboratory values from another center. Our evaluation reported normal direct bilirubin, ALT 51 U/L, aspartate transaminase (AST) 42 U/L, AP 177 U/L, GGT 162 U/L, total amylase 125 U/L, pancreatic amylase 67 U/L, anti-TPO 3704 U/mL, anti-TGB 343 U/mL, normal TSH, FT3, and FT4 levels. Heart ultrasonography was conducted, showing normal left ventricular function values (ejection fraction 60%), flow acceleration in the left pulmonary artery branch, not hemodynamically significant, a small pulmonary regurgitation, and a small tricuspidal insufficiency jet. The patient was discharged with UDCA 1800 mg/day, cholestyramine 4g ×2/day, rifampicin 300 mg on demand, and vitamin supplementation.
- -
- At a new clinical evaluation in July 2020, the patient reported general well-being, except for occasional itching flares, treated with cholestyramine.
- -
- In July 2022, at 26 years and 11 months of age, the patient reported only rare flares of pruritus, which responded positively to the treatment with cholestyramine. She took her therapy with compliance and maintained her clinical follow-up regularly with laboratory and imaging checks. The last ultrasonography (January 2022) showed a homogenous hepatic structure, Riedel’s accessory lobe, normal gallbladder without gallstones, and normal spleen with a longitudinal diameter of 12 cm. The patient is working and socially active and overall satisfied with her life quality. Laboratory results collected during this recent period are shown in Figure 1. As expected and reported in the literature, the cholestasis levels of our patient progressively decreased over time, until they reached the actual steady state, without UDCA dosage increase during the last 4 years.
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mitchell, E.; Gilbert, M.; Loomes, K.M. Alagille Syndrome. Clin. Liver Dis. 2018, 22, 625–641. [Google Scholar] [CrossRef]
- Kohut, T.J.; Gilbert, M.A.; Loomes, K.M. Alagille Syndrome: A Focused Review on Clinical Features, Genetics, and Treatment. Semin. Liver Dis. 2021, 41, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Turnpenny, P.D.; Ellard, S. Alagille syndrome: Pathogenesis, diagnosis and management. Eur. J. Hum. Genet. 2012, 20, 251–257. [Google Scholar] [CrossRef]
- Jesina, D. Alagille Syndrome: An Overview. Neonatal Netw. 2017, 36, 343–347. [Google Scholar] [CrossRef]
- Schindler, E.A.; Gilbert, M.A.; Piccoli, D.A.; Spinner, N.B.; Krantz, I.D.; Loomes, K.M. Alagille syndrome and risk for hepatocellular carcinoma: Need for increased surveillance in adults with mild liver phenotypes. Am. J. Med. Genet. 2020, 185, 719–731. [Google Scholar] [CrossRef]
- Shirley, M. Maralixibat: First Approval. Drugs 2021, 82, 71–76. [Google Scholar] [CrossRef]
- Kamath, B.M.; Ye, W.; Goodrich, N.P.; Loomes, K.M.; Romero, R.; Heubi, J.E.; Leung, D.H.; Spinner, N.B.; Piccoli, D.A.; Alonso, E.M.; et al. Outcomes of Childhood Cholestasis in Alagille Syndrome: Results of a Multicenter Observational Study. Hepatol. Commun. 2020, 4, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Tuset, E.; Ribera, J.M.; Domeenech, E.; Vaquetro, M.; Oller, B.; Armengol, M.; Planas, R.; Navarro, J.T.; Feliu, E. Pseudotumorous hyperplasia of the caudate lobe of the liver in a patient with Alagille syndrome. Med. Clin. 1995, 104, 420–422. [Google Scholar]
- Kim, B.; Park, S.-H.; Yang, H.R.; Seo, J.K.; Kim, W.S.; Chi, J.G. Hepatocellular carcinoma occurring in Alagille syndrome. Pathol. Res. Pract. 2005, 201, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Bresnahan, J.J.; Winthrop, Z.A.; Salman, R.; Majeed, S. Alagille Syndrome: A Case Report Highlighting Dysmorphic Facies, Chronic Illness, and Depression. Case Rep. Psychiatry 2016, 2016, 1657691. [Google Scholar] [CrossRef] [PubMed]
- Pati, G.K.; Singh, A.; Nath, P.; Narayan, J.; Padhi, P.K.; Parida, P.K.; Pattnaik, K.; Panda, C.; Singh, S.P. A 10-year-old child presenting with syndromic paucity of bile ducts (Alagille syndrome): A case report. J. Med. Case Rep. 2016, 10, 342. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abenavoli, L.; Boccuto, L.; Corea, A.; Gambardella, M.; Spagnuolo, R.; Luzza, F. Alagille Syndrome and Its Clinical and Laboratory Features: A Case Report. Livers 2022, 2, 258-263. https://doi.org/10.3390/livers2040021
Abenavoli L, Boccuto L, Corea A, Gambardella M, Spagnuolo R, Luzza F. Alagille Syndrome and Its Clinical and Laboratory Features: A Case Report. Livers. 2022; 2(4):258-263. https://doi.org/10.3390/livers2040021
Chicago/Turabian StyleAbenavoli, Ludovico, Luigi Boccuto, Alessandro Corea, Marialuisa Gambardella, Rocco Spagnuolo, and Francesco Luzza. 2022. "Alagille Syndrome and Its Clinical and Laboratory Features: A Case Report" Livers 2, no. 4: 258-263. https://doi.org/10.3390/livers2040021
APA StyleAbenavoli, L., Boccuto, L., Corea, A., Gambardella, M., Spagnuolo, R., & Luzza, F. (2022). Alagille Syndrome and Its Clinical and Laboratory Features: A Case Report. Livers, 2(4), 258-263. https://doi.org/10.3390/livers2040021