
pH Effects on the Conformations of Galacturonan in Solution: Conformational Transition and Loosening, Extension and Stiffness †



Abstract
1. Introduction
1.1. Conformational Transitions: Helices and Coils
1.2. The Loose Helix
1.3. The Case of Galacturonan
1.4. Experimental Approaches to the Conformational Transition of Galacturonan
1.5. The Coupling of the Conformational Changes of Galacturonan
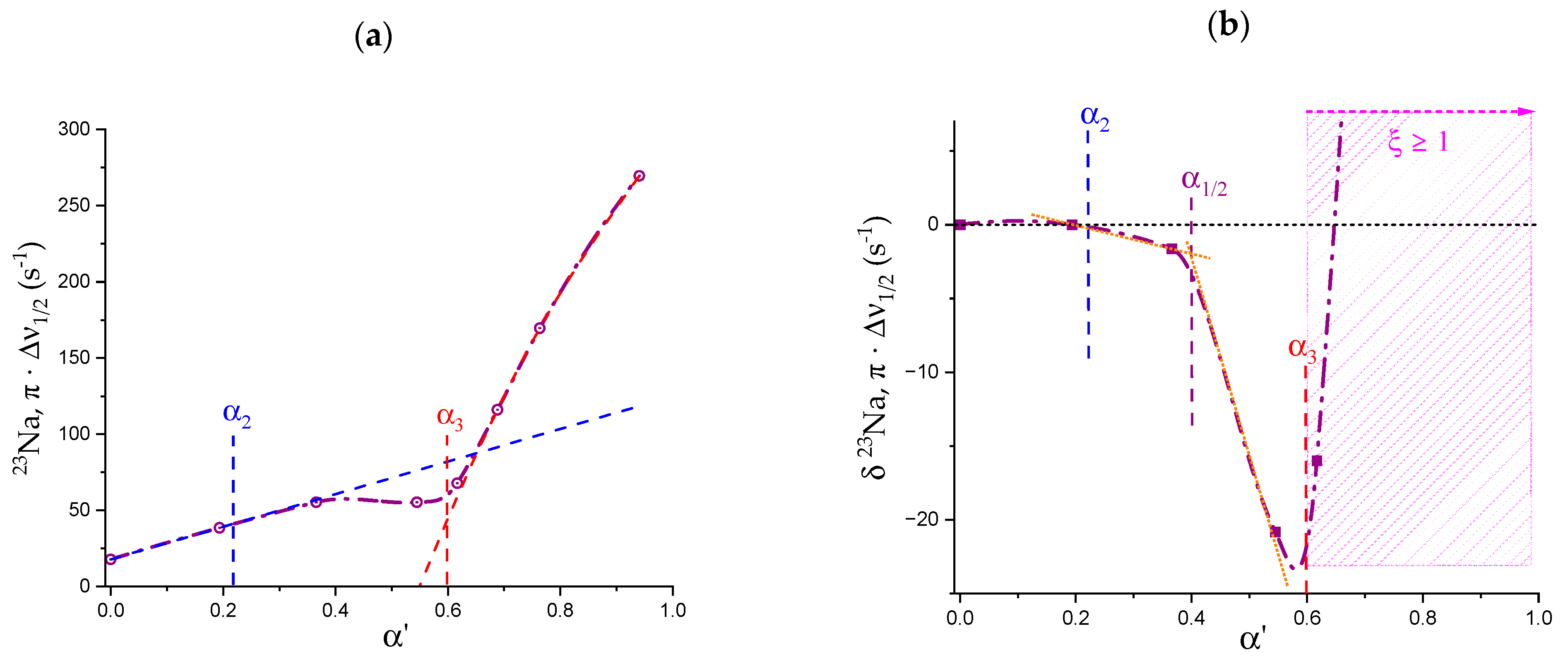
1.6. Specific Effects of Ions on the Conformation of Galacturonan
1.7. Hydration of Galacturonan
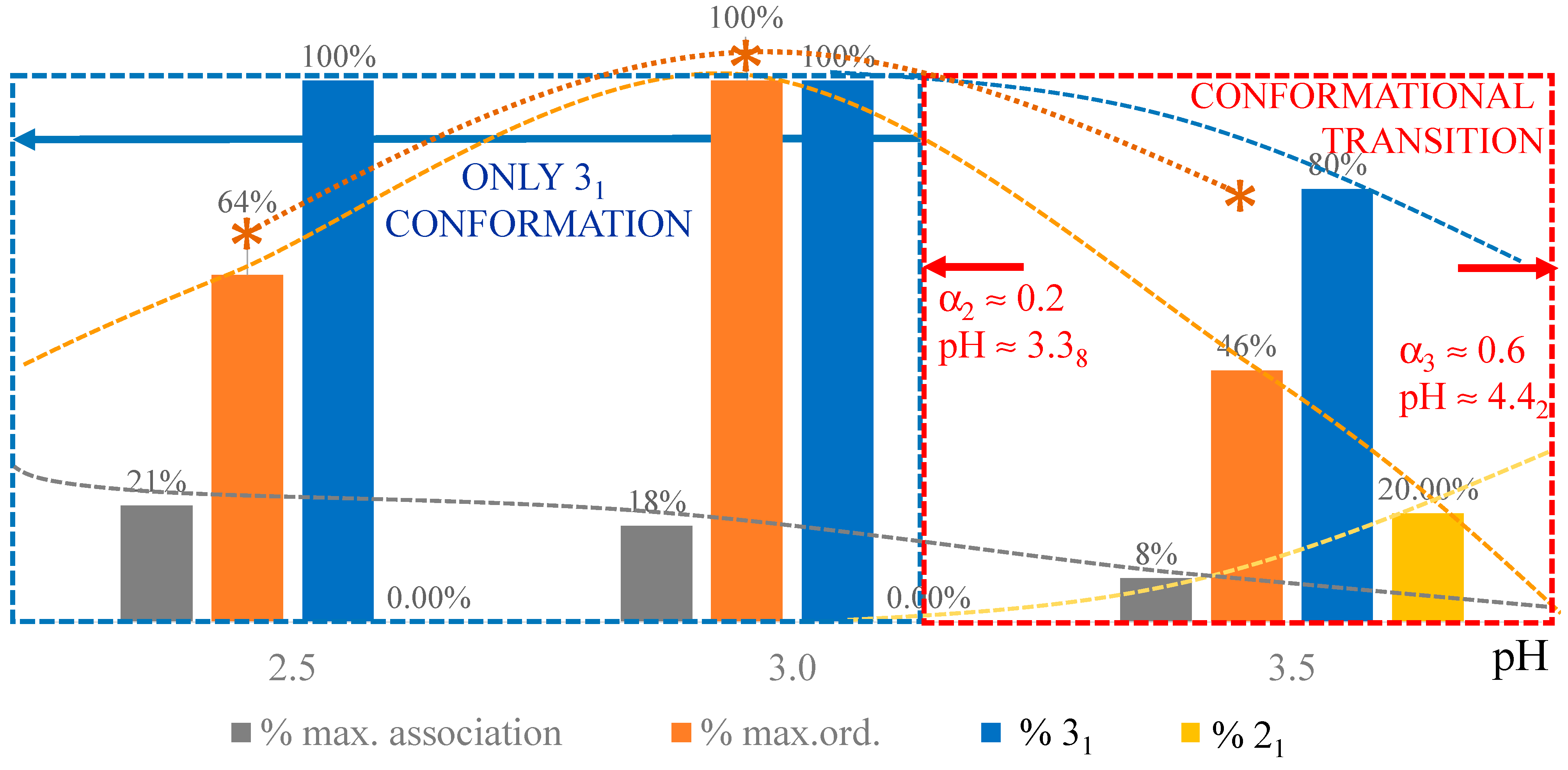
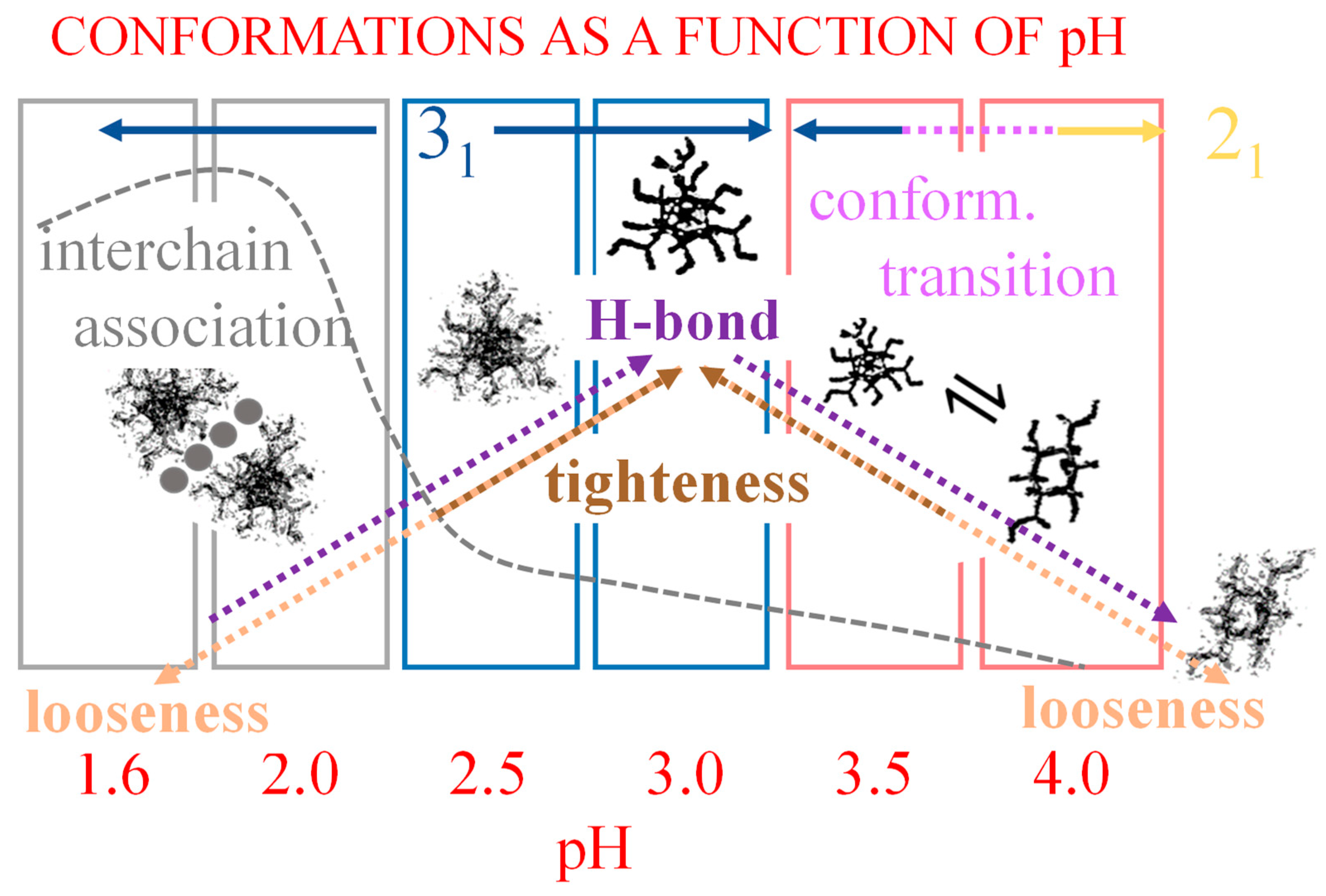
2. The Charge-Induced Conformational Transition of Galacturonan
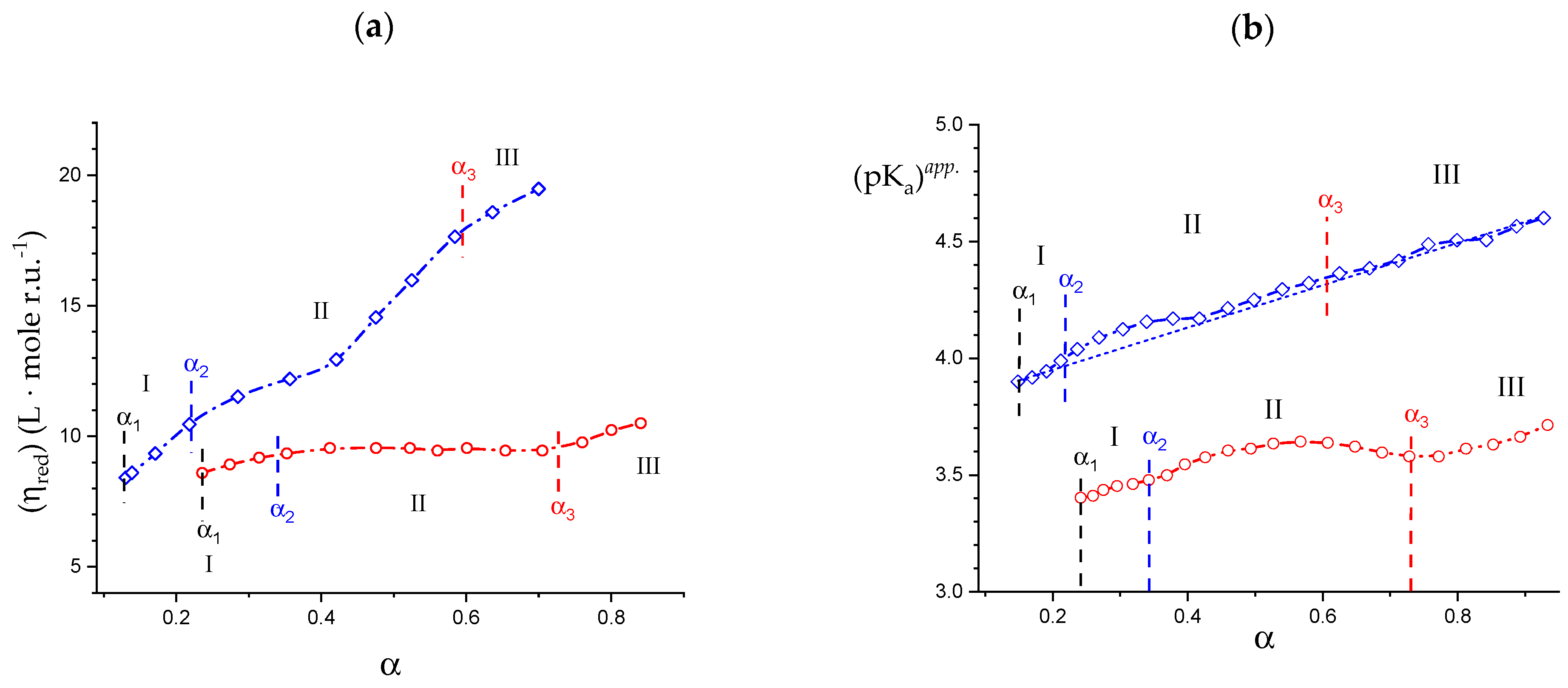
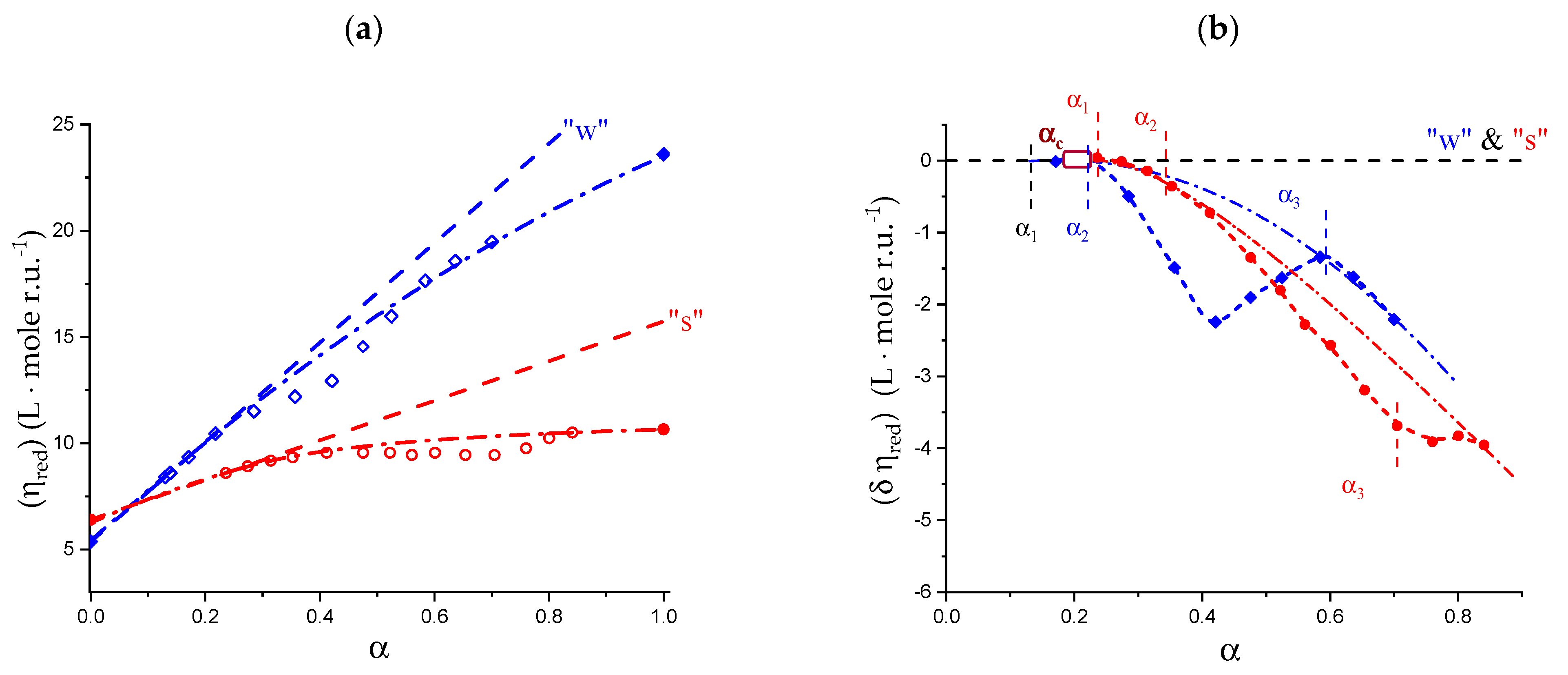
2.1. Viscometric and Potentiometric Data
2.2. Isothermal Micro-Calorimetric Data in Aqueous NaClO4
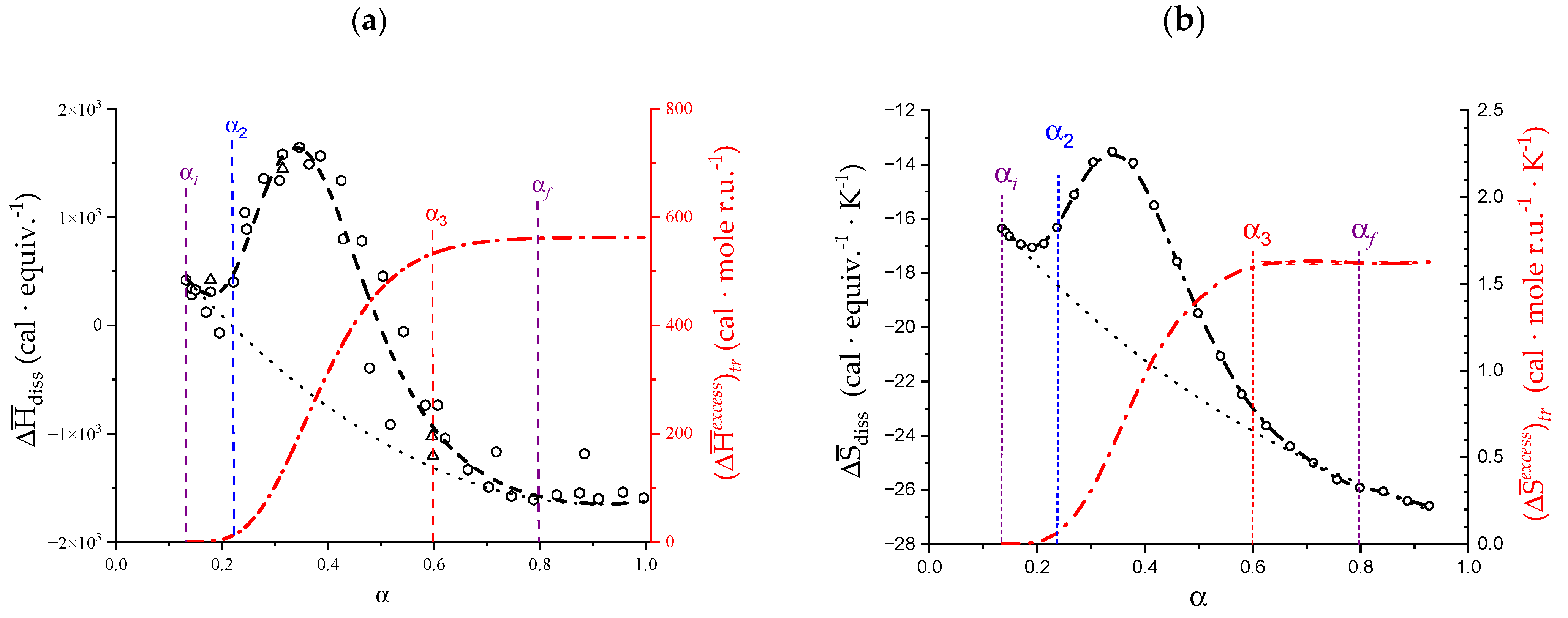
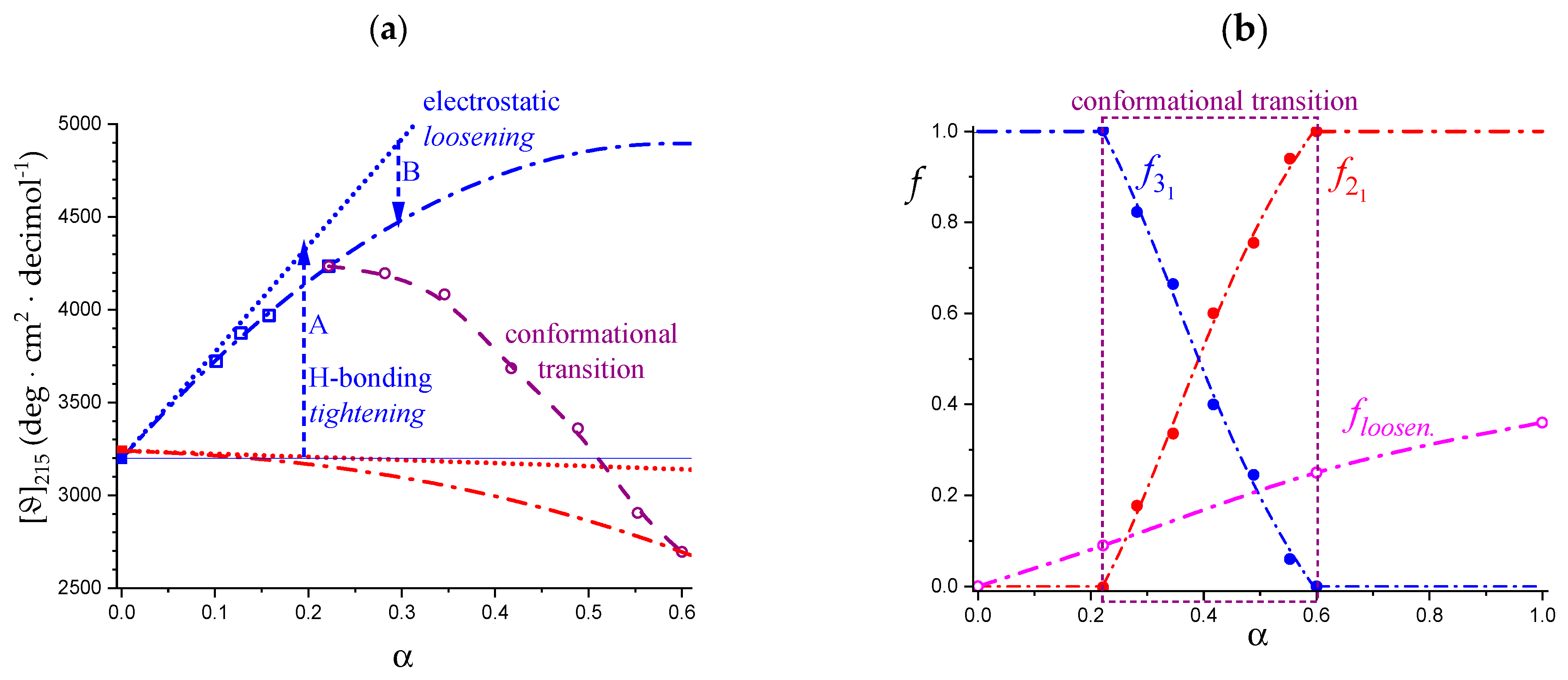
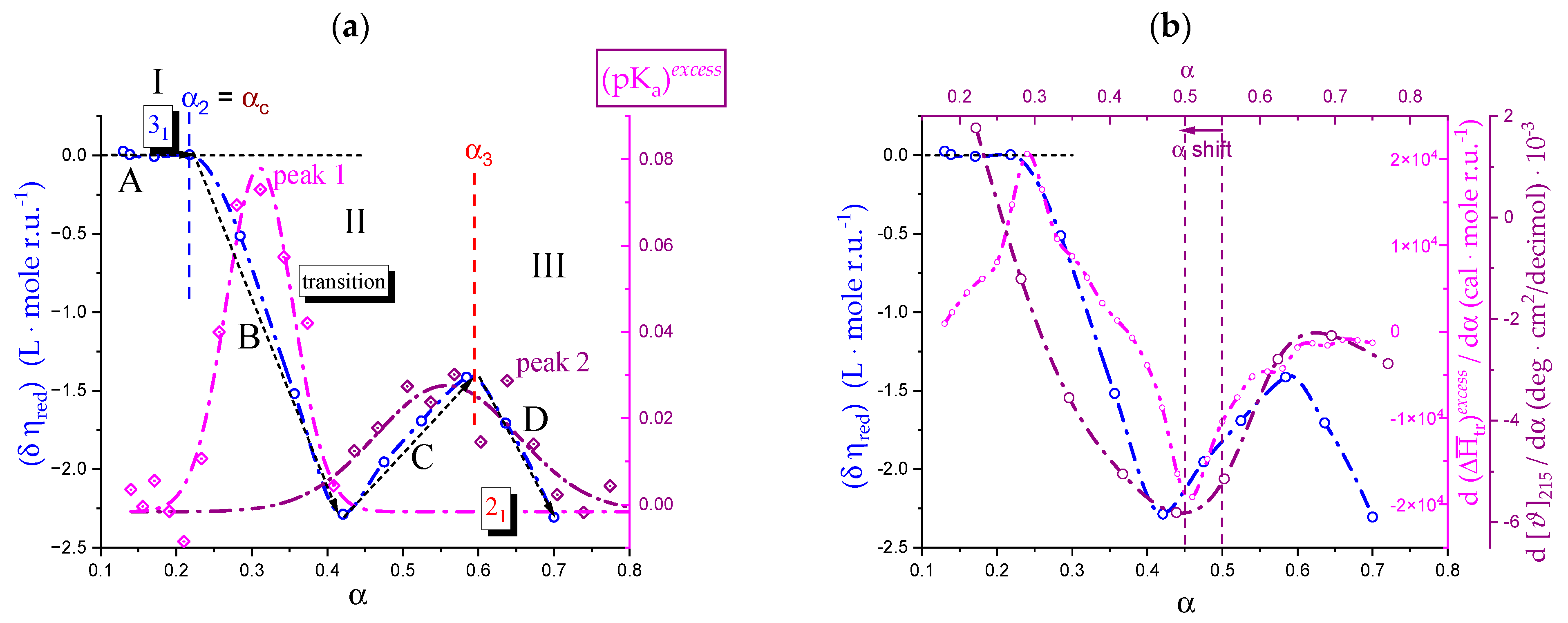
2.3. Decoupling of the Conformational Changes
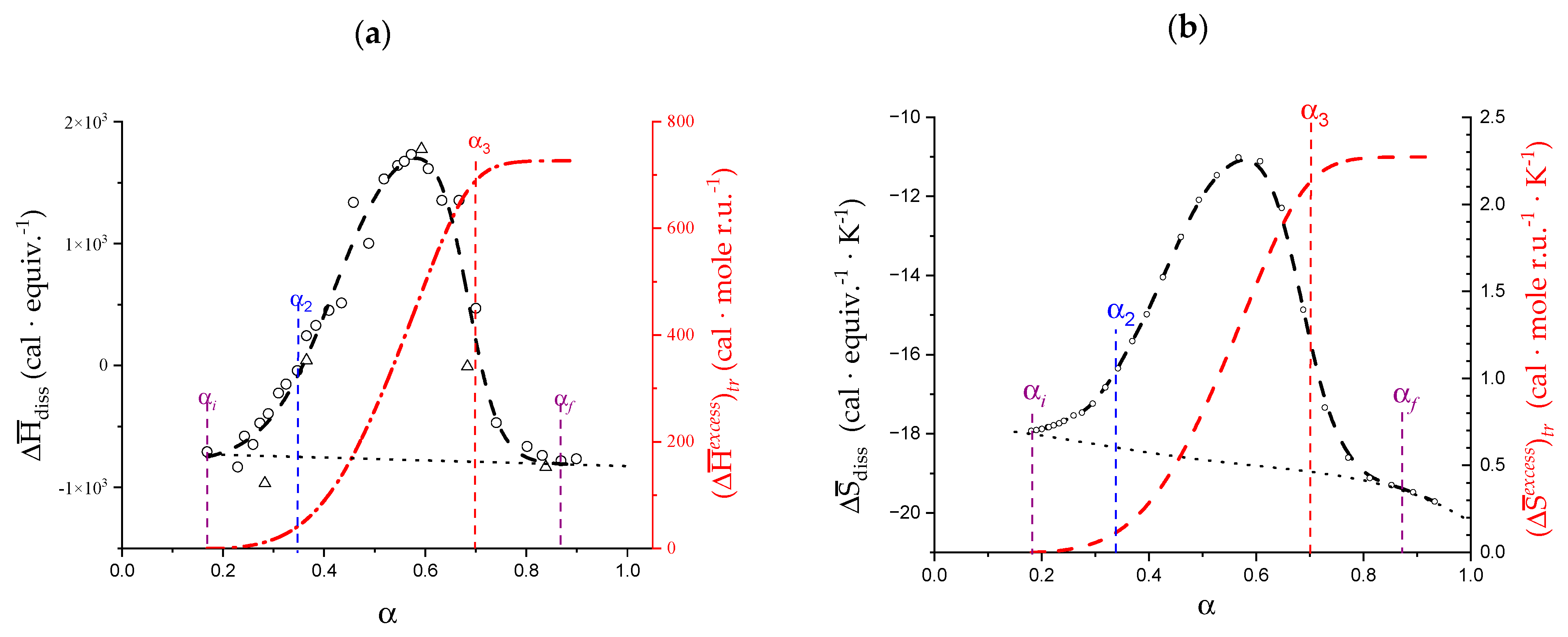
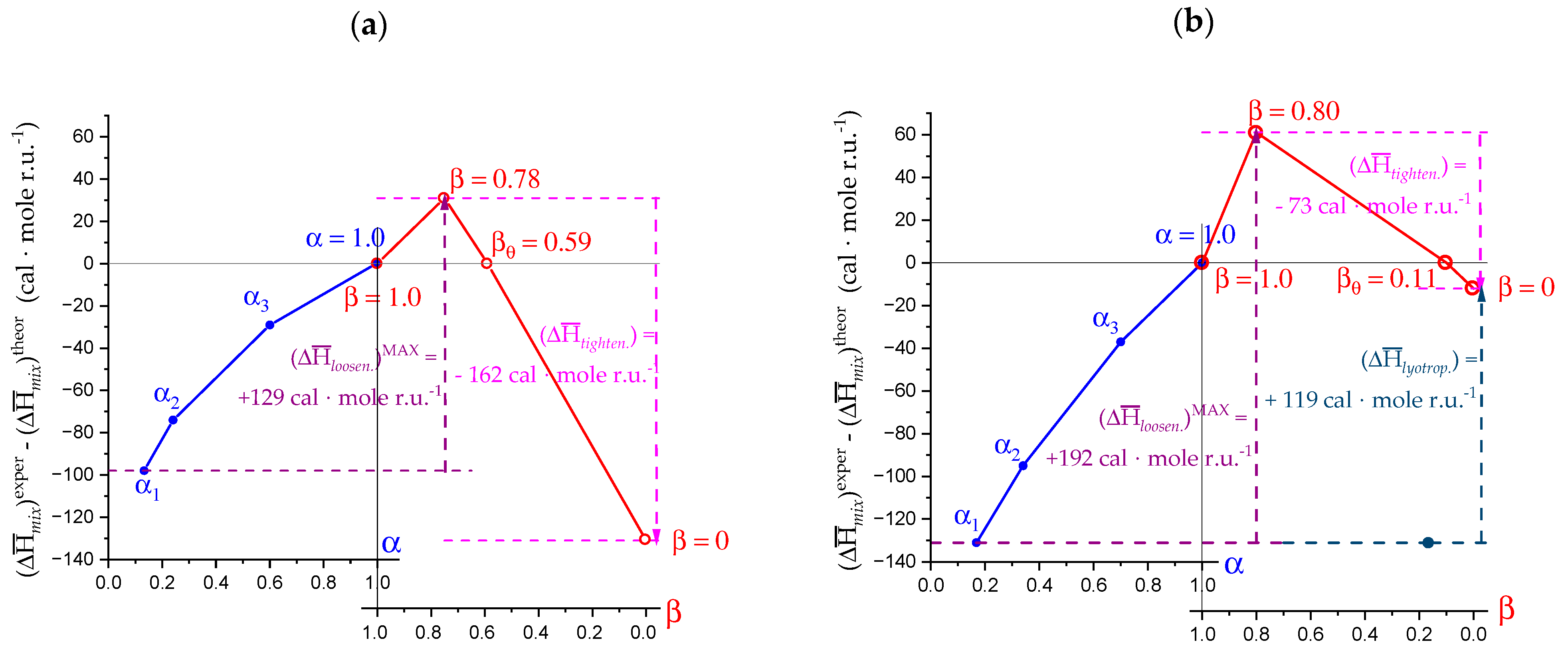
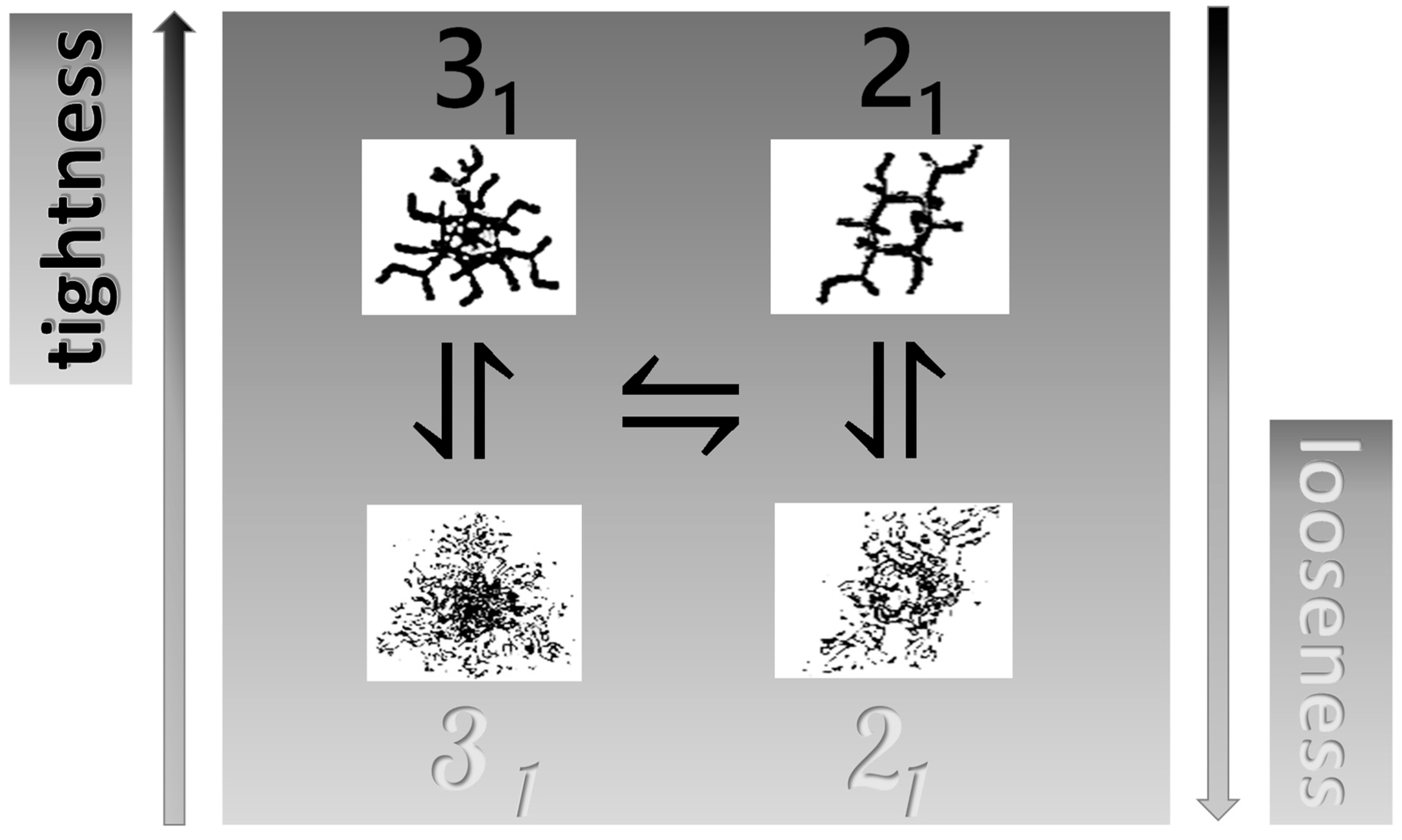
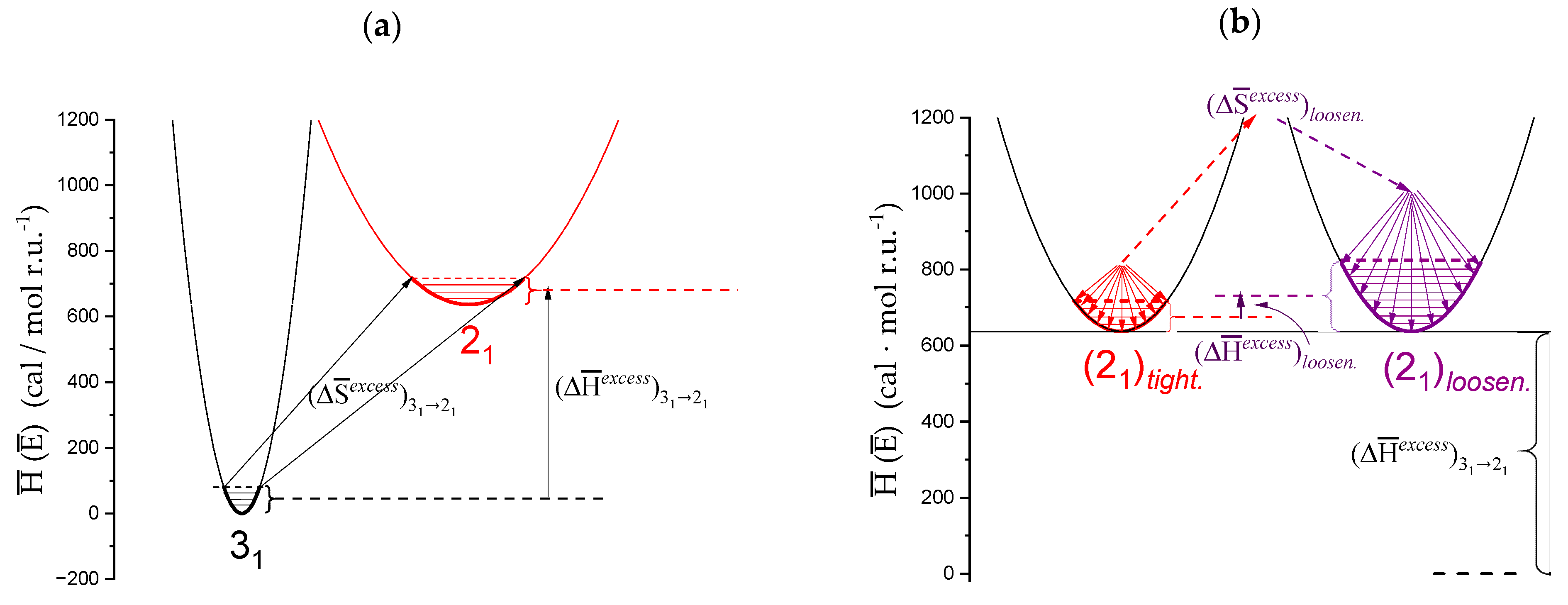
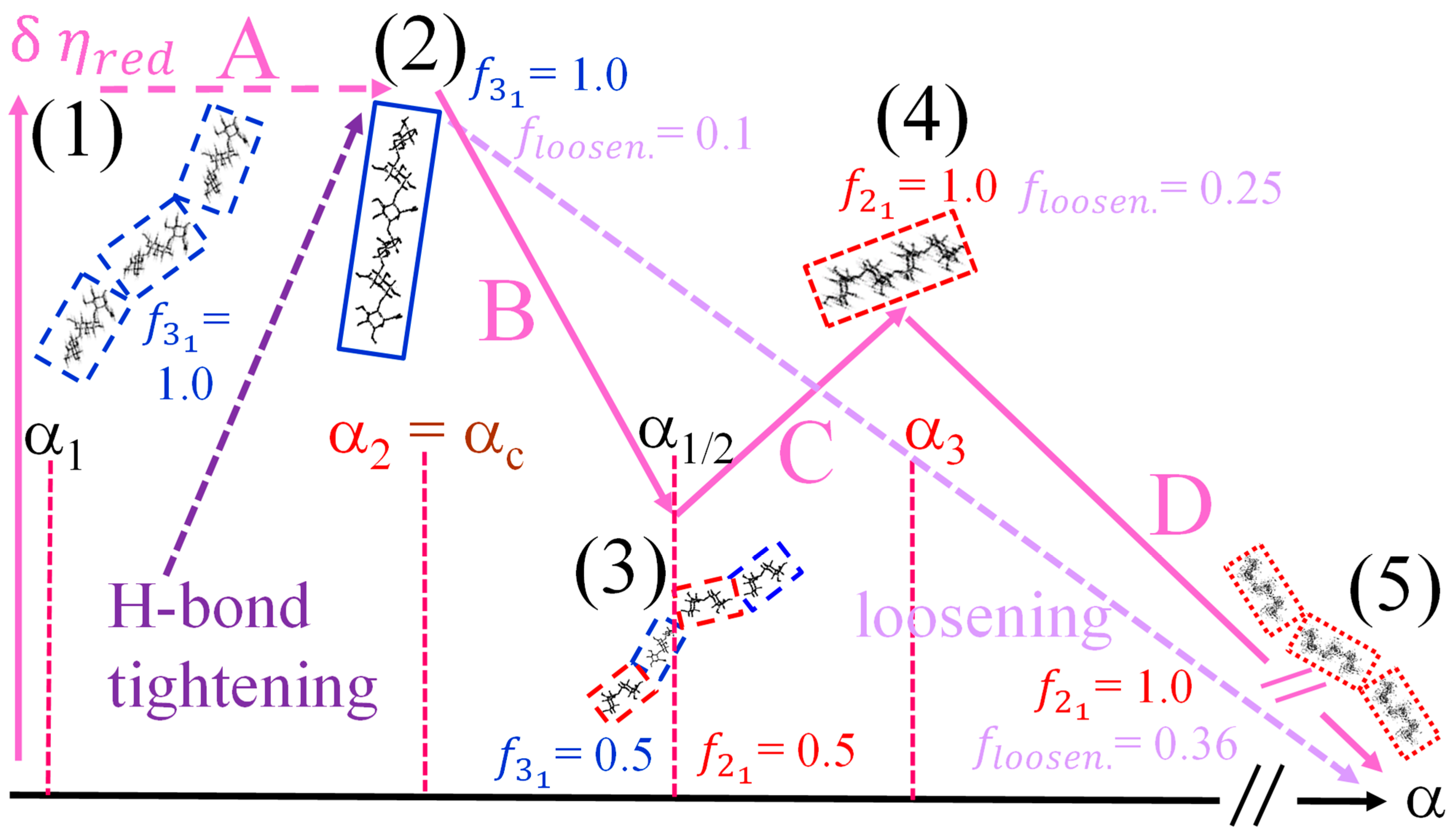
- The experimental transition () thermodynamic changes (, with = , , ) are supposed to result from the additive contribution of the inter-conformational transition term (31 → 21) and the loosening term (loosen.): + );
- In the ranges αi → α2 and α3 → αf (being αi = α1), the measured effect stems from the loosening. terms only: and . Moreover, no other excess term being detected beyond αf, the final condition (αf, R) can be substituted by (α = 1, R). For the case in “salt”, R = 10;
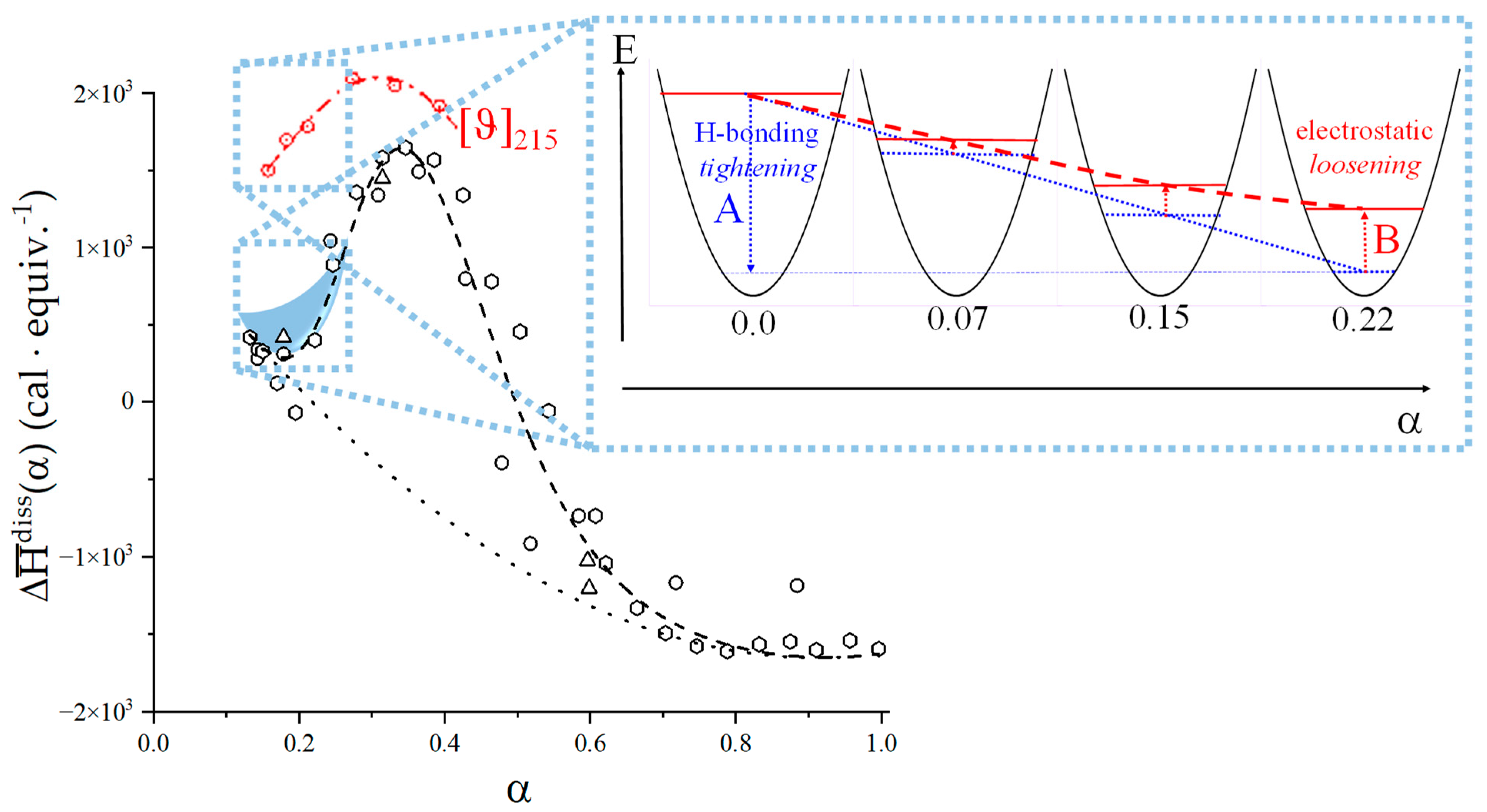
- To evaluate the loosening contribution in the transition interval α2 → α3, two methods have been devised, which are described in Supplementary paragraph 2. Briefly, they both assume that reflects the trends before α2 and beyond α3. The results are encouragingly similar, allowing for the trivial estimation of as the sum of the three contributions in the three sequential intervals of α. The results have been reported in block a. of Table 2;
- The terms have then been evaluated as the difference between and (see block b. of Table 2). The terms are assumed to be independent of α, as usual in calculating the (non-electrostatic) thermodynamic properties of the conformational transition when there is no—or a negligible—variation in charge density values between the initial and final states. This holds in the present case, where the difference in charge density between the 31 and the 21 fully-charged conformations is as low as 0.46% at full dissociation (as already mentioned in Section 1.1), which reduces to 0.16% at α2 and to 0.32% at α3. Operationally, the electrostatic terms are fully taken care of by the and baselines;
- The total excess changes from α1 to α = 1 () are then the bare sum of the two previous sets (see block c. of Table 2).
2.4. Isothermal Micro-Calorimetric Data in Salt-Free Aqueous Solution
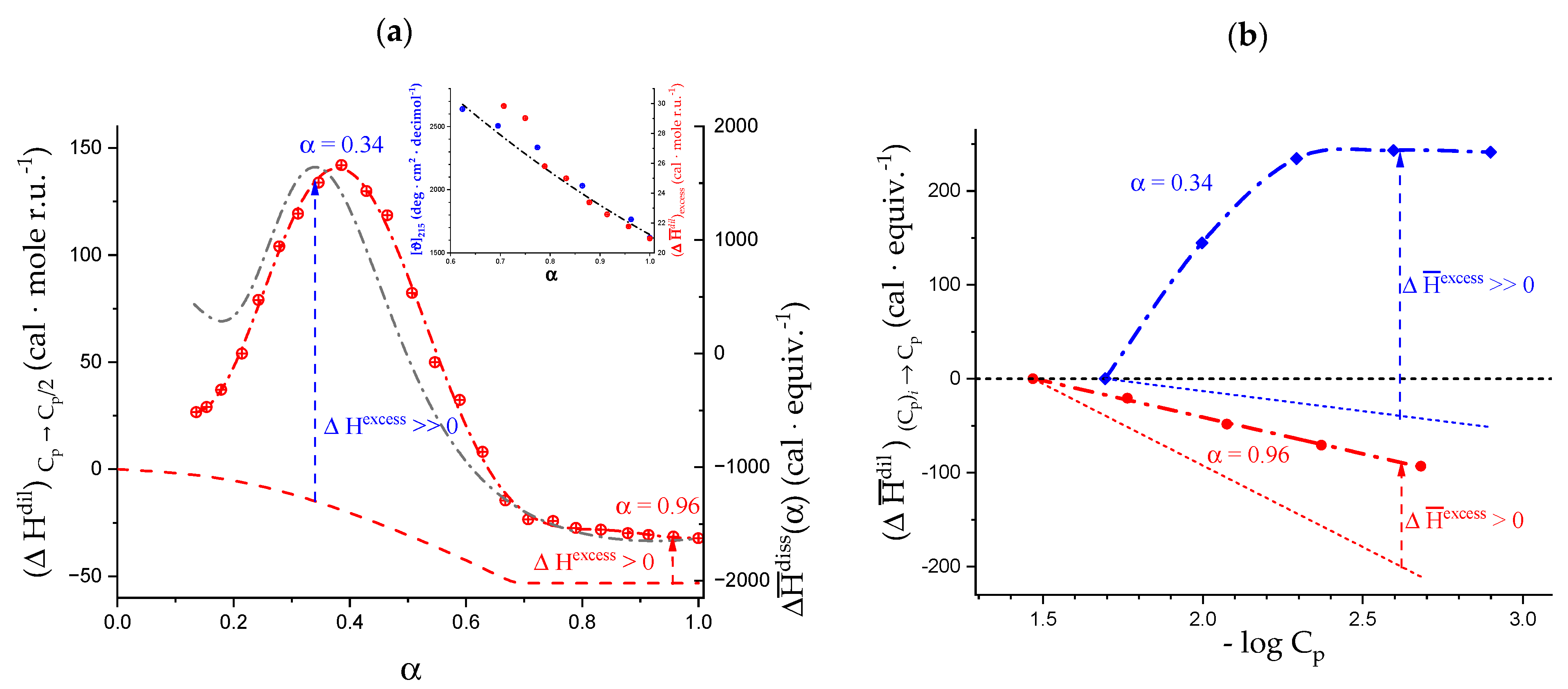
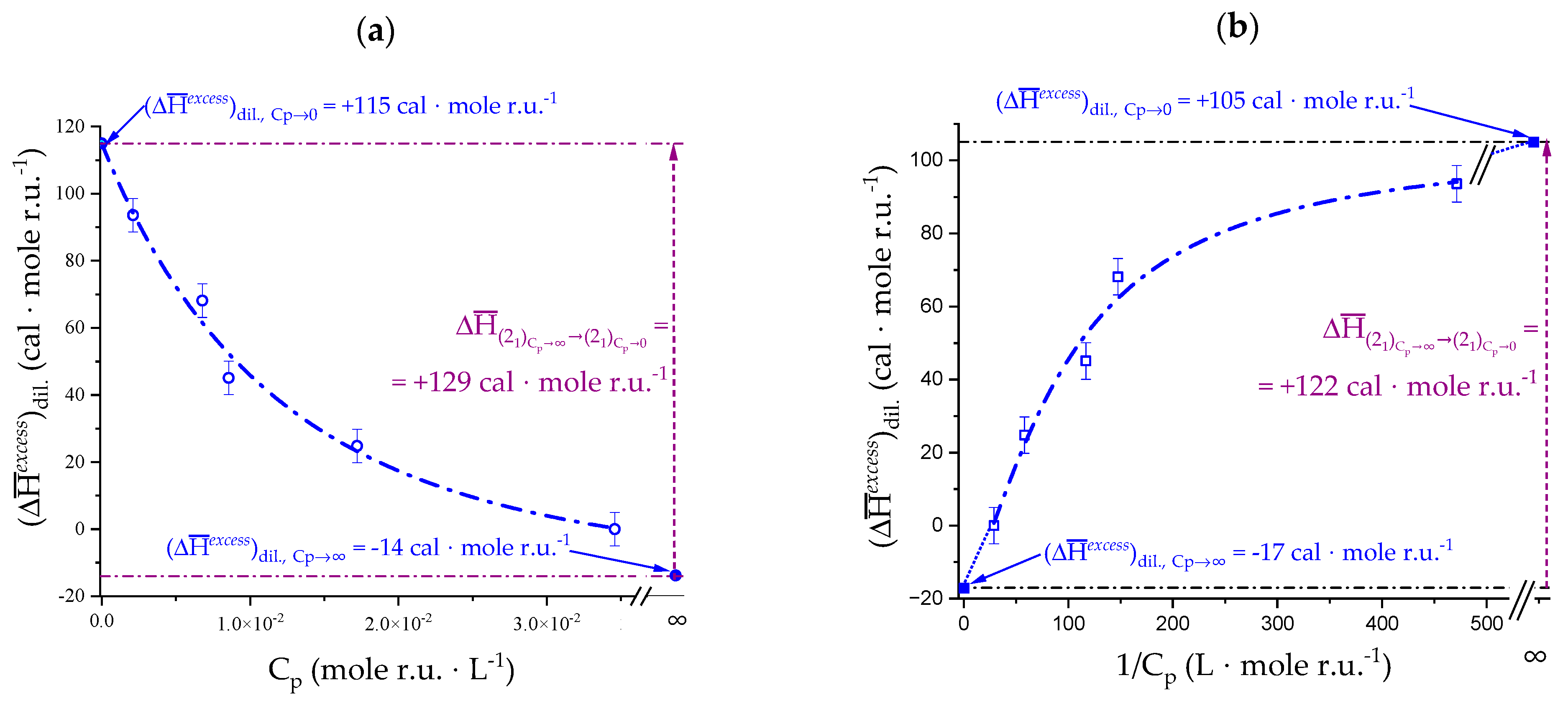
2.5. Enthalpy Changes of Dilution: Galacturonan in Water
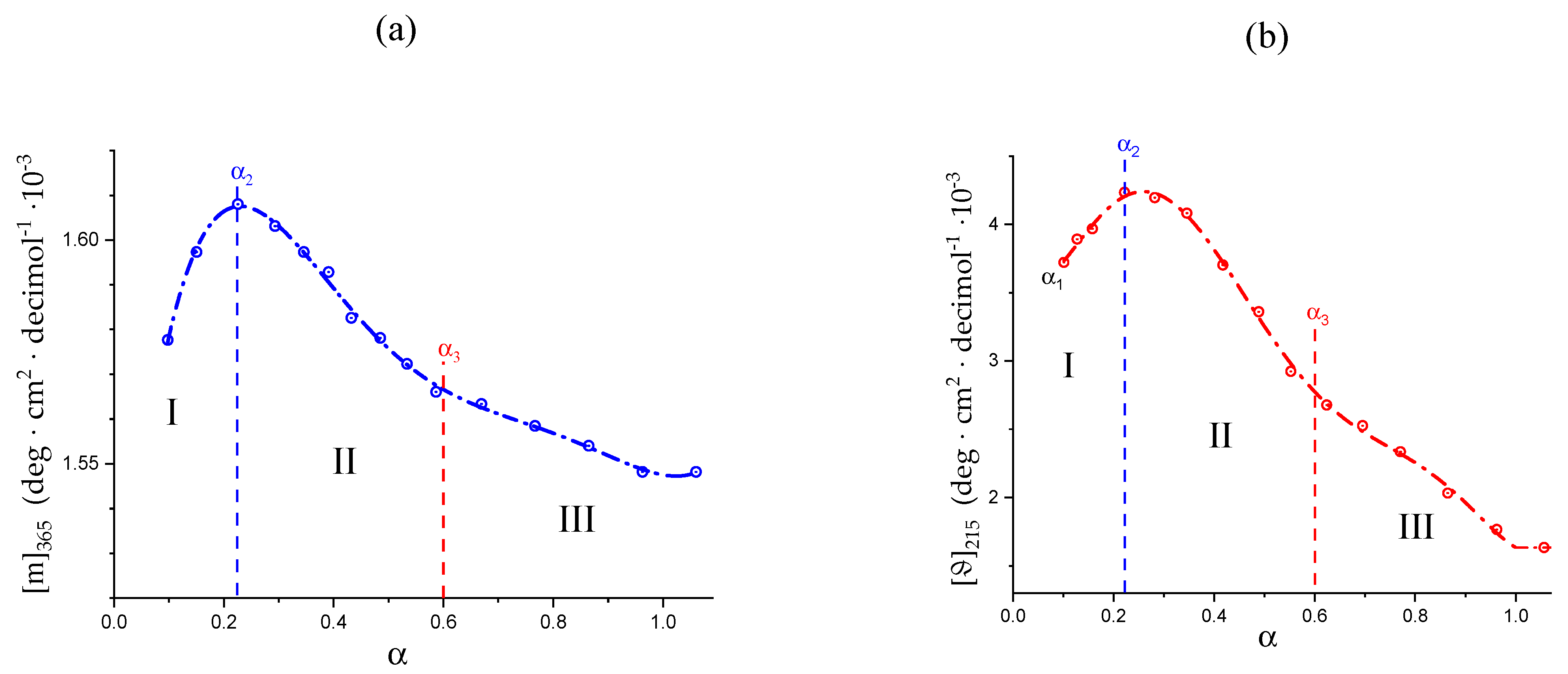
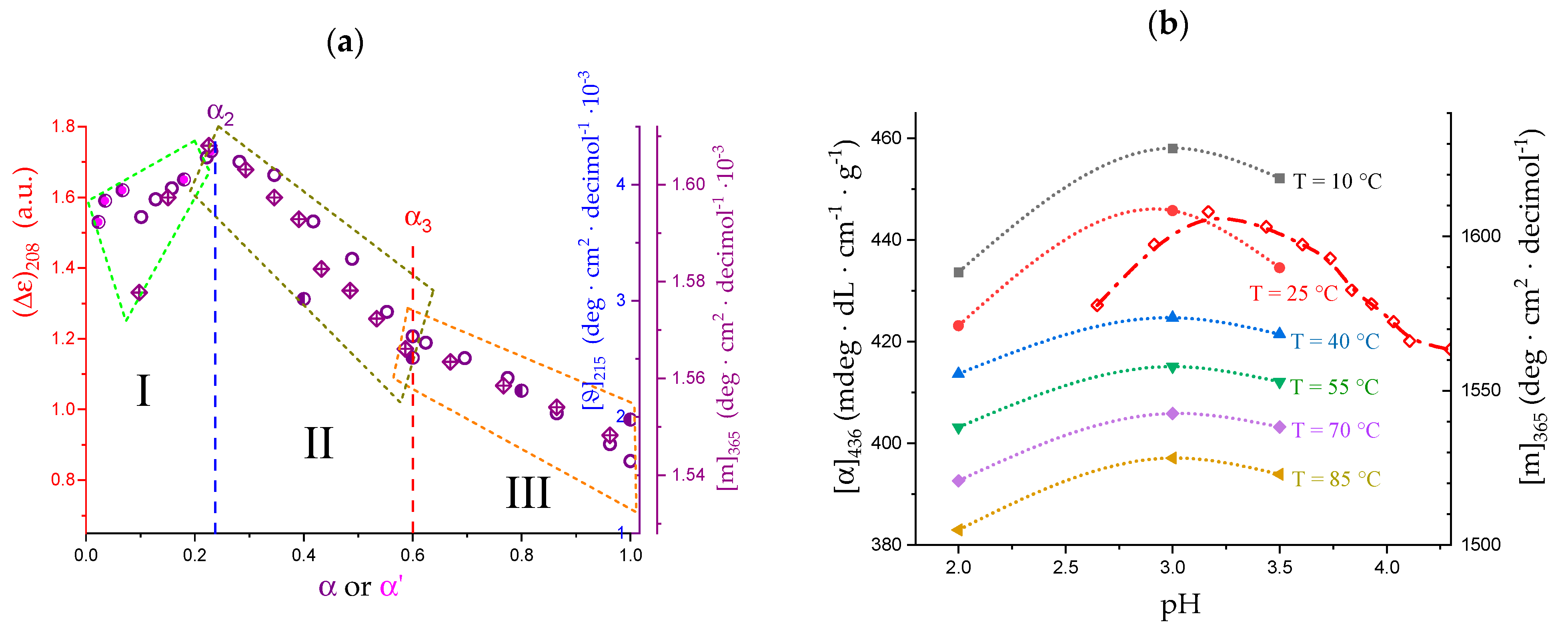
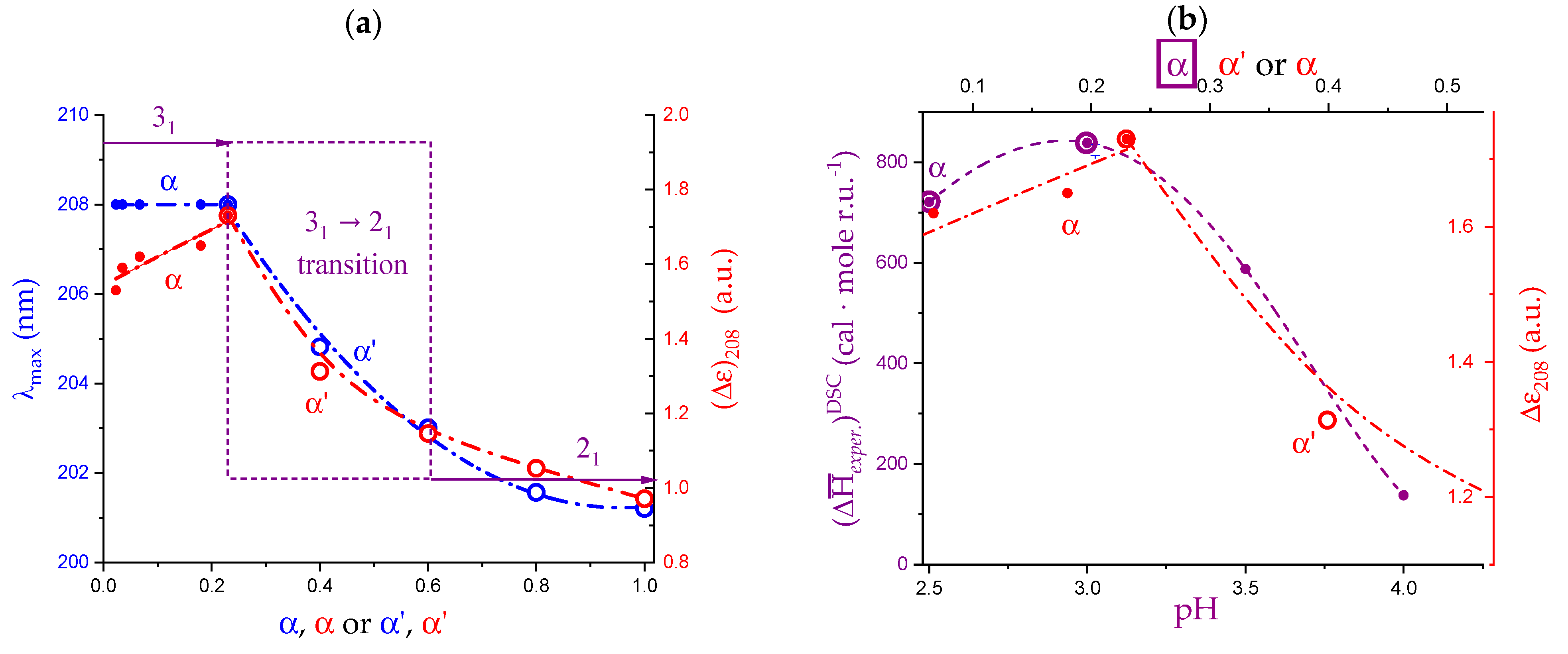
2.6. On the Peculiar Behavior of the Dependence of the Chiro-Optical Properties of Galacturonan in Water

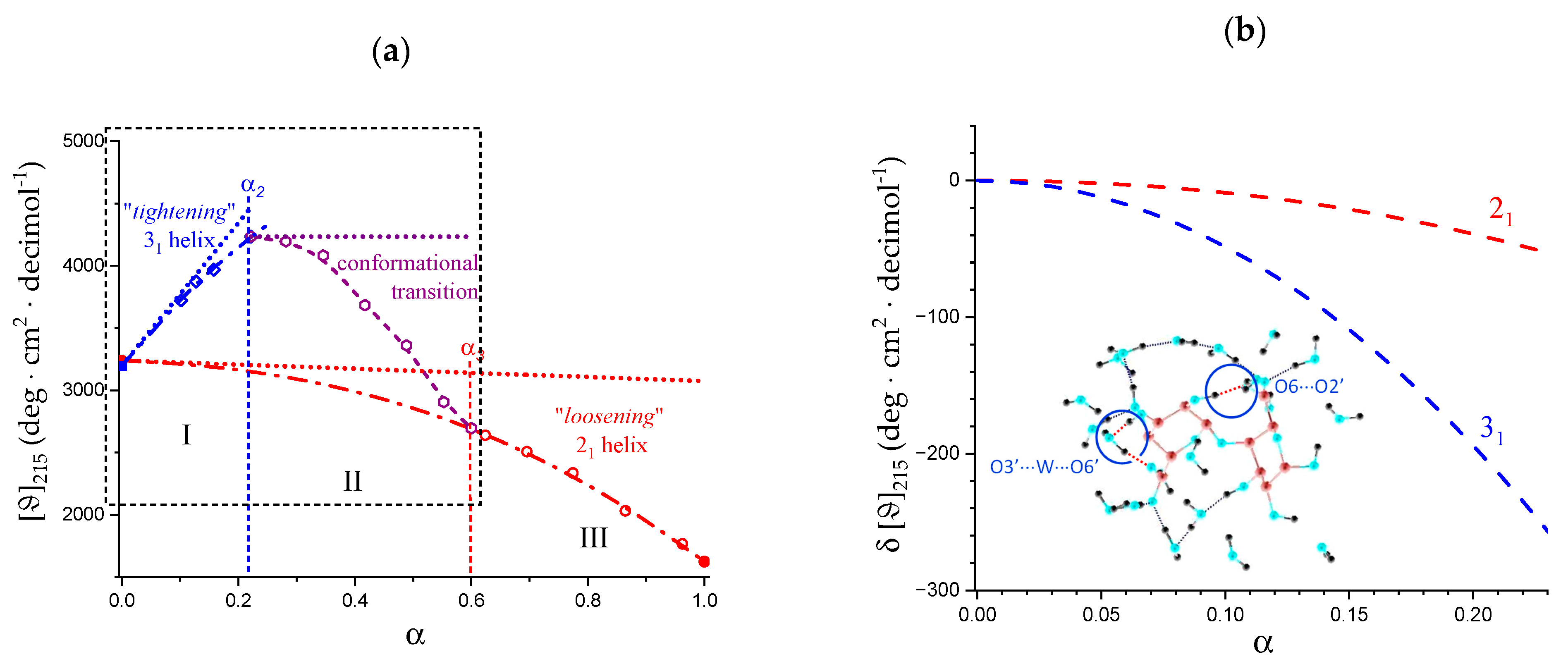
2.7. The Unique Interplay of Stiffening and Loosening of the 31 Conformation Induced by Charging: The Progress of the Conformational Transition
2.8. Opposing Enthalpic Effects upon Charging the 31 Helix
3. The Temperature-Induced Conformational Transition of Galacturonan
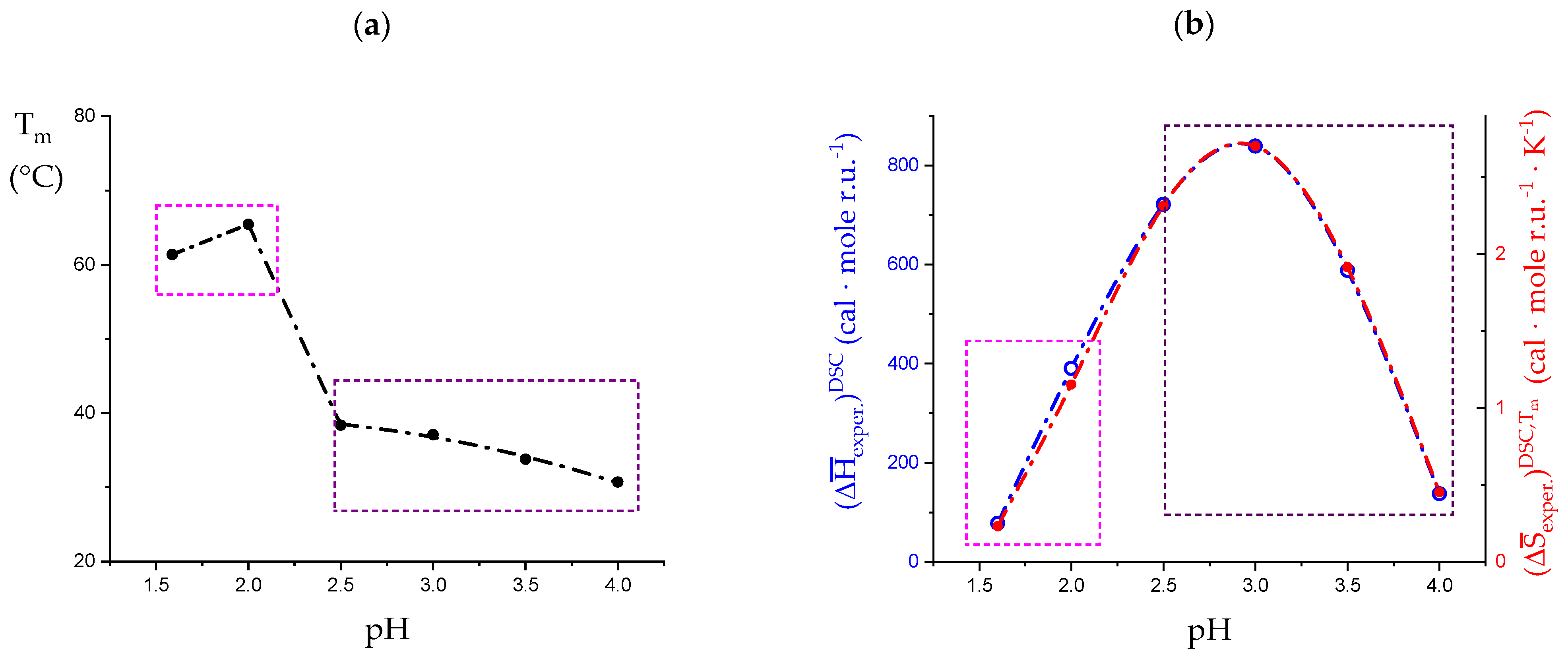
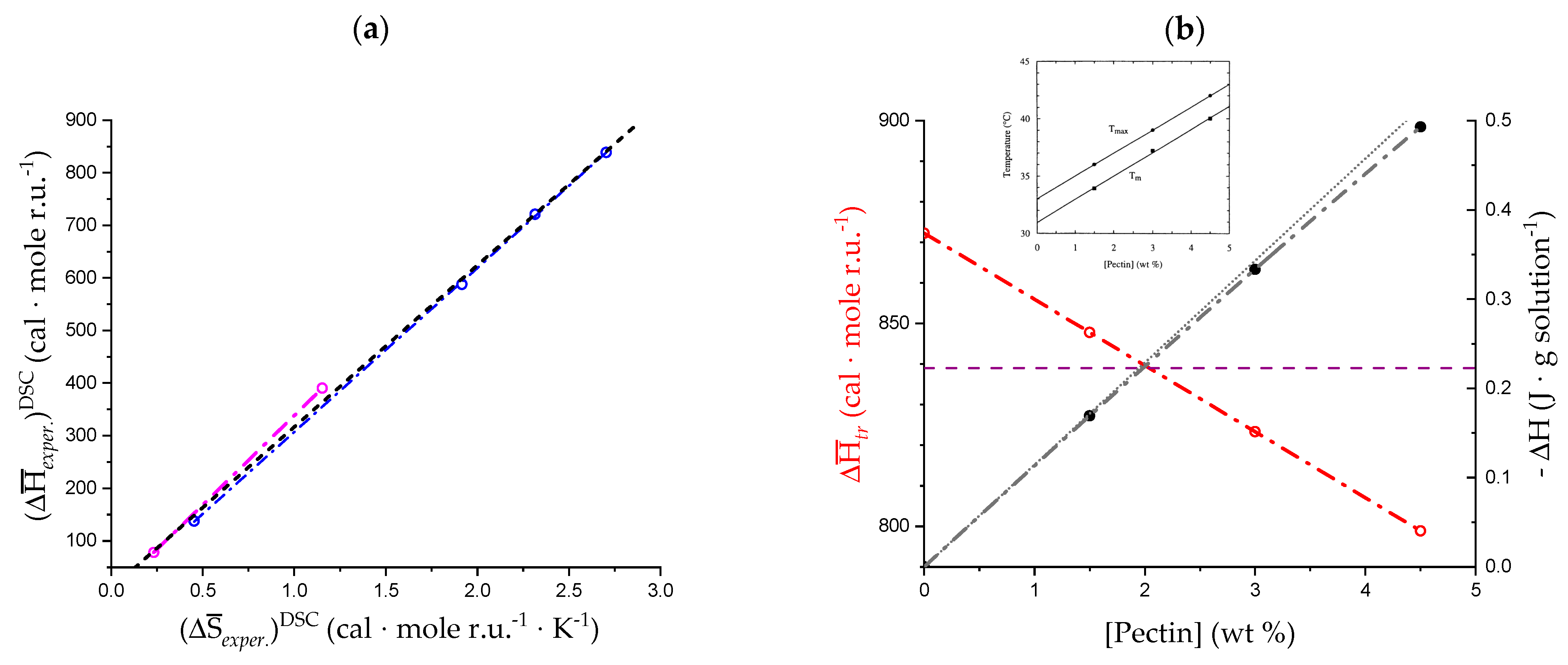
3.1. Enthalpy Changes and Melting Temperatures from DSC Data
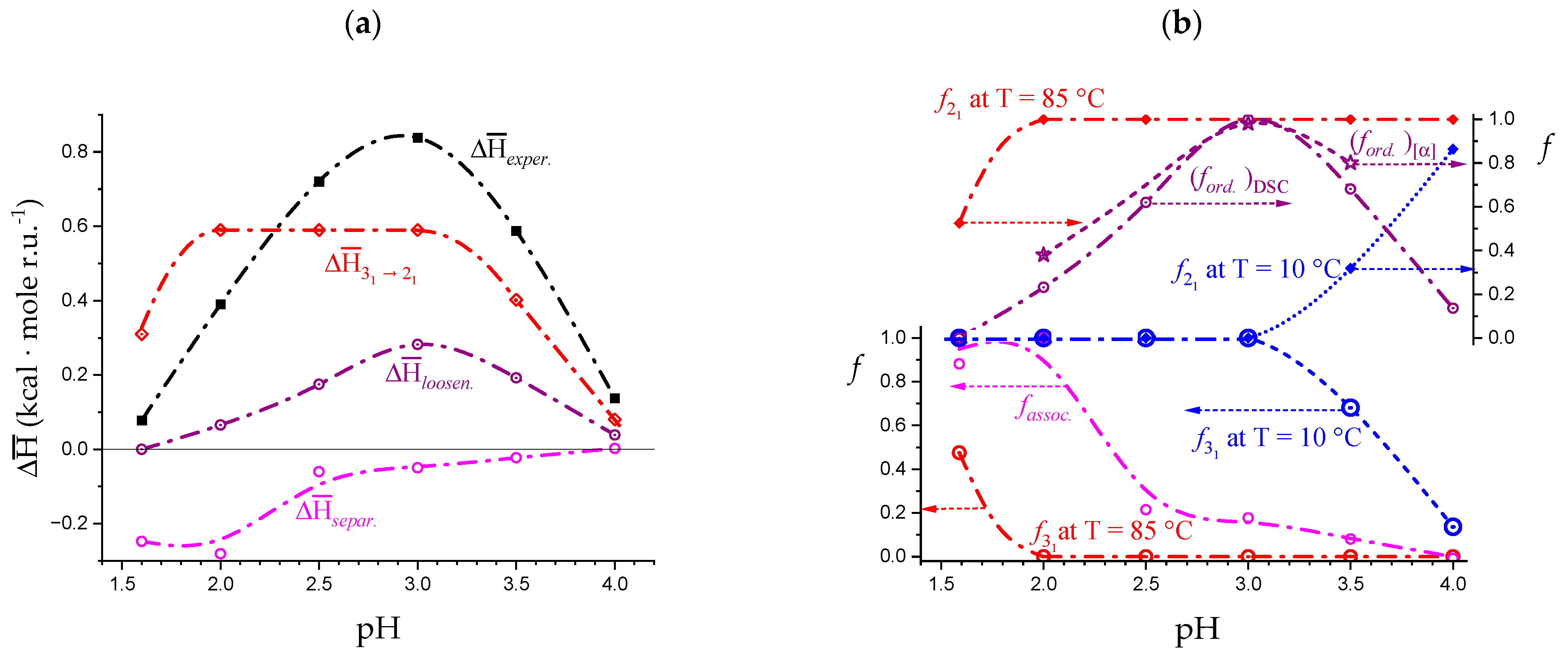
- The enthalpy change accompanying the “melting” of the associated structures. The presence of contacts between chains (association), which is a prerequisite of gel formation in LMP, was demonstrated by light-scattering experiments [38]. It is quite reasonable to suppose that the junctions are held together thanks to hydrophobic (van der Waals) interactions between the COOCH3 groups of facing chain stretches (with a correlated enthalpy change in association, ). Their melting brings about a negative (exothermic) value of , , with the opposite process being endothermic: “The formation of the hydrophobic bond is endothermic at low temperatures” [13] ( = ; separ. stands for “(chain) separation”, having restricted the word “dissociation” to the ionization process of galacturonic acid). From the experimental standpoint, negative (exothermic) values of have been observed in several associated systems held together by hydrophobic interactions [56,58,73]. In experiments of melting, like in the DSC heating experiments, one should then expect that the experimental enthalpy of melting—albeit being endothermic due to conformational transition, see points 2. and 3.—becomes increasingly less endothermic upon increasing polymer concentration, Cp. This increasing exothermic contribution stems from the obvious assumption that association if favored by an increase in Cp. Of course, in principle, other interchain interactions might also be present, like interchain hydrogen bonds. In this case, however, breaking such bonds would bring about an endothermic enthalpy change, indistinguishable from those described hereafter in 2. and 3. and, more important, it would add to those, thereby increasing the net endothermic effect;
- The enthalpy change accompanying the conformational transition from the 31 to the 21 helical symmetry, . This effect is endothermic and, for the 100% change in conformation, it coincides with the value of ( = +0.59 kcal·mole r.u.−1), reported in Table 2 for the “salt” case from ITμC experiments. Should the final 21 conformation be only partially present in the initial state, then the extent of this effect would be equal to , where is the fraction of the polymer in the 31 conformation, trivially being + = 1;
- The enthalpy change accompanying the loss of order (“loosening”) of either regular conformation that accompanies the progressive increase in temperature, . This effect is endothermic (as very well known for the full order → disorder transitions of proteins and nucleic acids) and it coincides with the entity called in Table 2; its value can vary depending on the given set of initial and final states;
- The sum of and of . can be operationally identified with , i.e., the enthalpy changes of the two processes pertaining to the transition of the isolated chain, corresponding the processes whose thermodynamic quantities have been reported in Table 2. On the other side, all terms contributing to chain–chain dissociation are collectively expressed by .
3.2. Insights into Conformational Changes from Data as a Function of Temperature
3.3. On the Difference between the Disordered Conformations of Pectin Brought about by Charging or Temperature
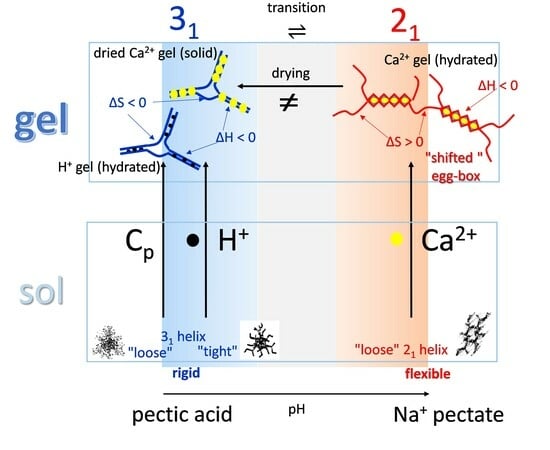
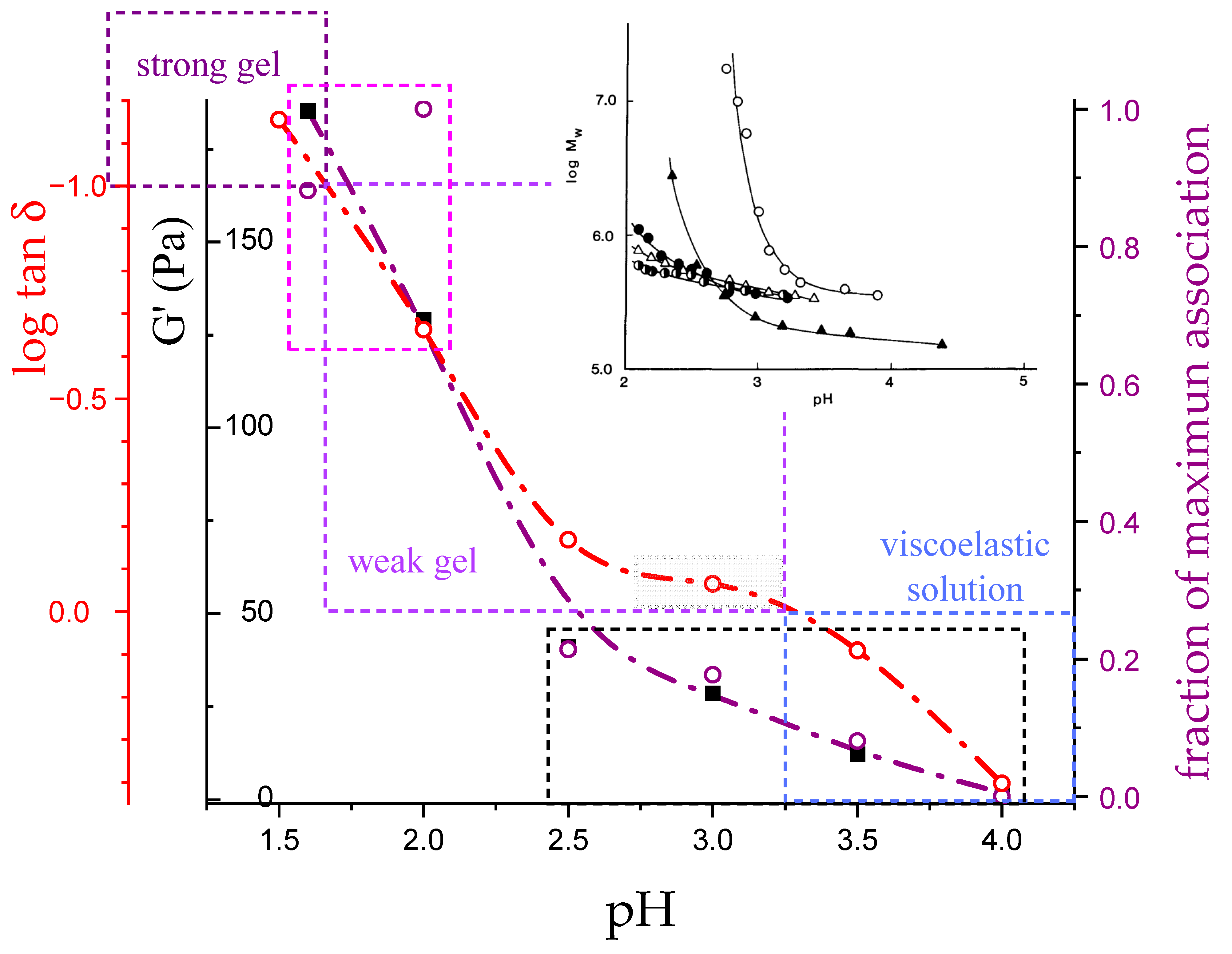
3.4. Molecular Aspects of the Acid–Gel Formation of LMP
- The 31 conformation is required to produce strong gels in very acidic conditions;
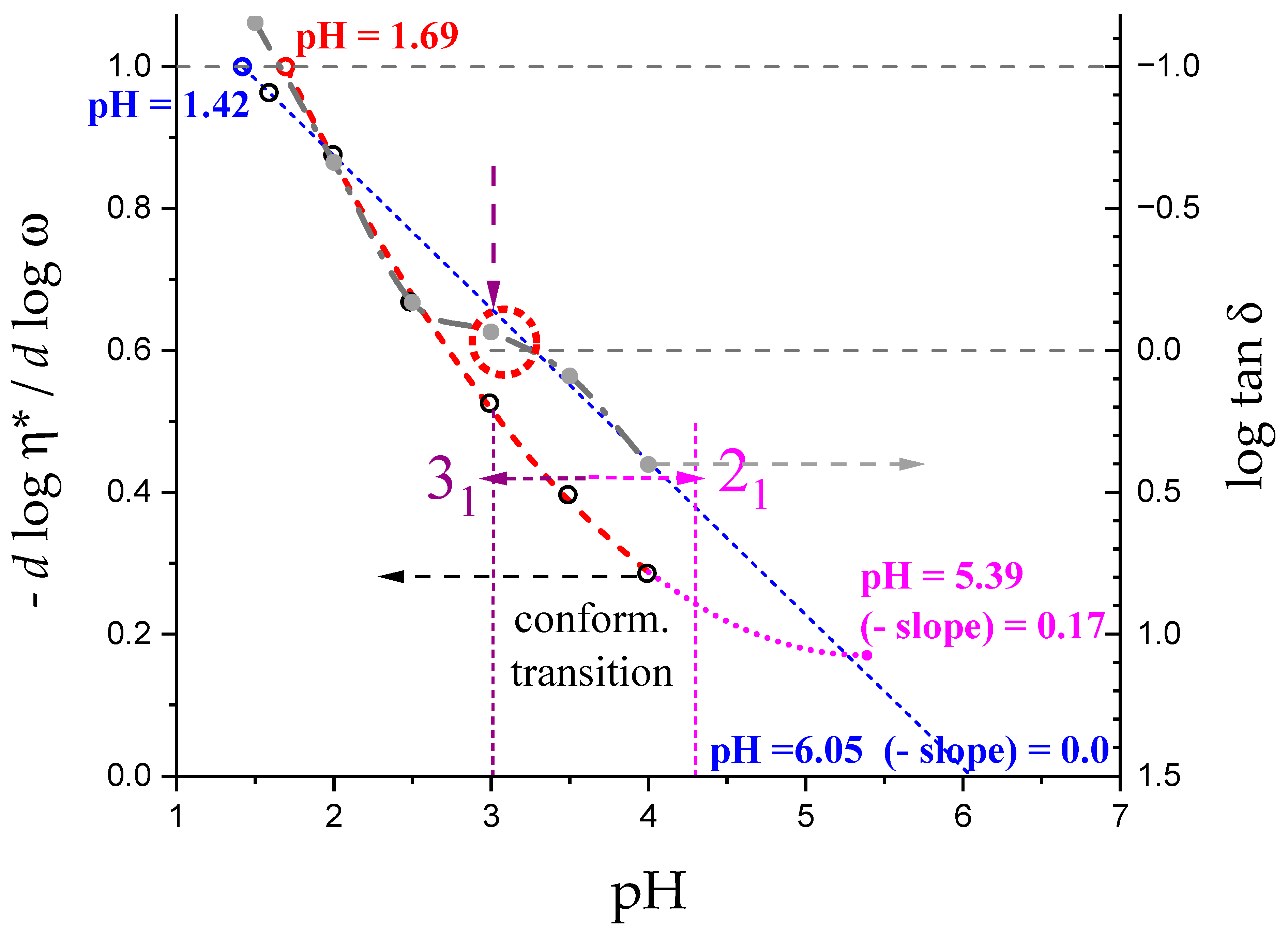
- “Even in the absence of Ca2+ and above pH 3.5 LMP associates spontaneously and forms weak gels at sufficiently high concentration that can, however, be easily broken by shear” [74]. This is documented by the values of the (−slope) function, which at pH = 3.5 (within the conformational transition range) show a value as large as 0.4. Even at the last point of the parabolic trend (i.e., at pH 5.39) the value of the (−slope) function is 0.17, pointing to a clearly viscoelastic solution (see Figure 14 and Figure 15). Interestingly, “This association is favored by increasing the temperature” [74], i.e., this is accompanied by an endothermic enthalpy change, exactly corresponding to the presently determined > 0 (see Section 3.1);
- The parabolic rise of the function (−slope) upon decreasing pH is even steeper below about pH = 3.0 (at α2), at which the 31 conformation is at its maximum tightness and in its most extended conformation (see the above discussion in Section 2.6 and Section 2.7 and in the following Section 6 on viscosity). For α < α2 (i.e., pH < 3.0), the chain gets progressively more disordered. This indicates that the increasing gel-like character—as shown by the increase in the function (−slope)—is not due to an increase in contacts between stretches in the (31) ordered conformation. Rather, the major players,—effective in producing the rheologically effective interchain links,—are as follows: (1) the polyelectrolyte charge density, which must be reduced to a negligible amount by reduction in pH; (2) the presence of attractive van der Waals (hydrophobic) interactions between COOCH3 groups on facing—almost uncharged—chains;
- For pH values lower than 3.0, the 31 conformation gets progressively looser upon decreasing pH; still, the nice parabolic trend must be viewed upon as intimately linked to a persisting fraction of tightness. Should the system proceed along that trend, the value of (−slope) = 1.0 would have been reached already at the (higher) pH value of 1.69. On the contrary, the pH value of the intercept at = 1.0 for the linear trend of the two lowest values pH data points is 1.42. It is possible to hypothesize two opposing mechanisms of gel–junction formation. The former one could be due to the formation of H-bonds between locally regular sequences of facing repeating units; the latter one could be due to interactions between the non-regularly distributed methyl-ester groups on different chains. The regularity required by the first mechanism implies the ordering and stiffening of the 31 conformation, which, moreover, would be favored by a limited increase in fractional charge on the COOH groups, contrary to the observed effect. Moreover, as addressed in Section 3.1, breaking such interchain H-bonds (if present in significant amount) would imply a major endothermic effect, at variance with the opposite experimental findings (see Figure 12a). At variance, the second mechanism benefits from an increased flexibility to favor the geometrically undefined van der Waals contacts between opposing chains—the less charged the better—with a substantial gain of configurational entropy. The observed break of the parabolic trend of the function upon decreasing pH suggests a progressive reduction in any H-bond based association mechanism (if ever present) in favor of that based on associative hydrophobic (van der Waals) interactions at lower residual fractional charging;
- That the conformation of LMP at pH ≤ 2.0 is largely disordered is supported by the intercept of the blue dashed line with the line of −slope = 0.0 (Newtonian liquid) at pH = 6.05. It means that such disordered LMP conformation, if it could be ideally kept up to neutral conditions, would behave as a pure dissipative liquid, without traces of elasticity that, on the contrary, characterize the “real” LMP solutions at a neutrality whose viscoelastic behavior (−slope = 0.17) is rooted in the—albeit “loose”—21 helical conformation.
4. Lyotropic Effect of Perchlorate on the Conformations of Galacturonan
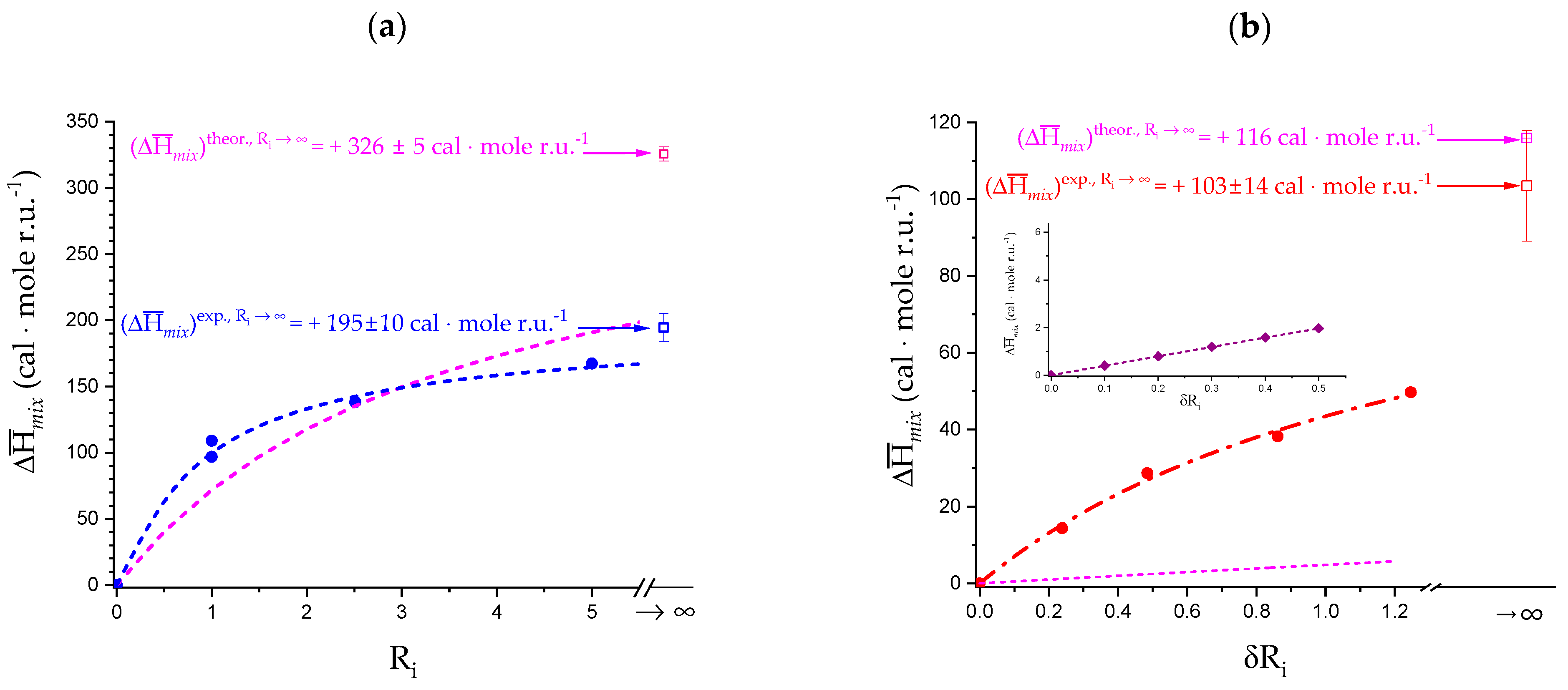
4.1. Enthalpy Changes of Mixing with Salt
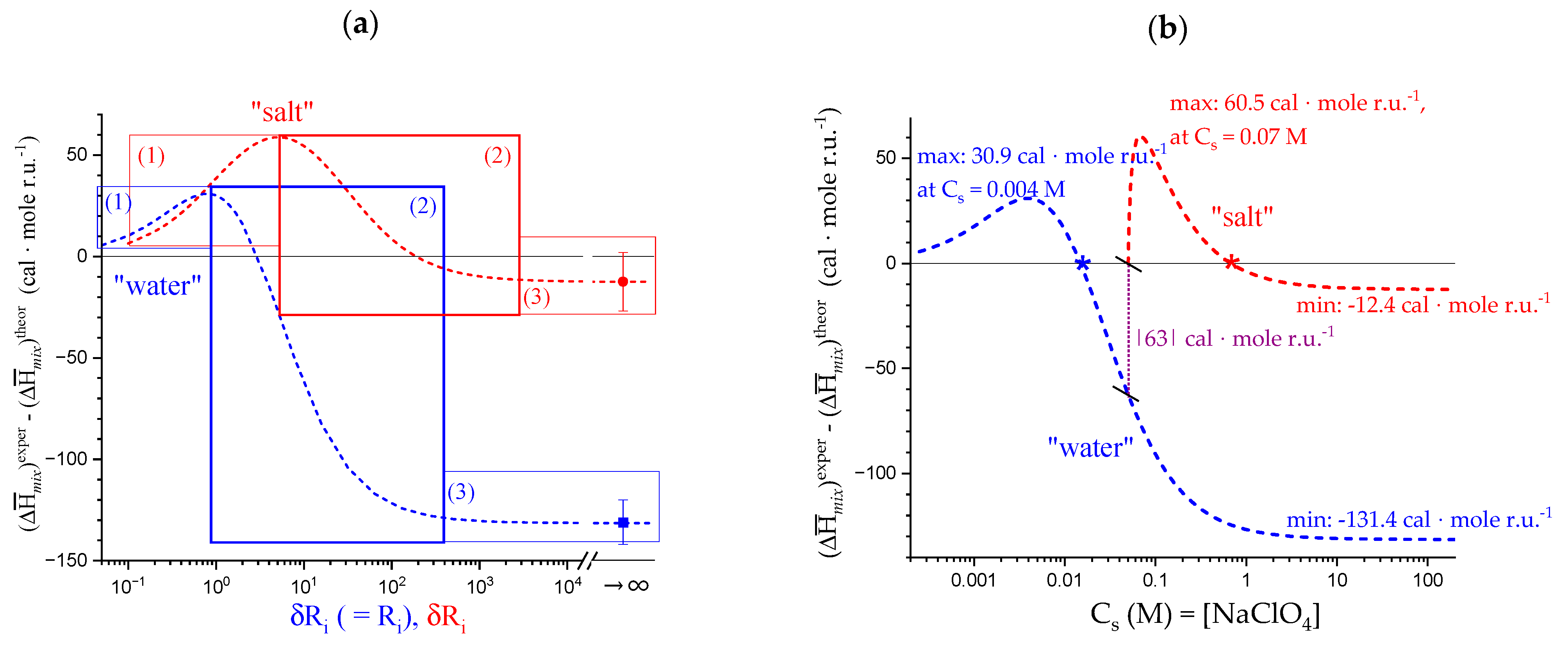
4.2. On the Difference between Infinite Ionic Strength and Infinite (Specific) Salt Concentration: Hofmeister and Lyotropic Effects
5. Whole-Chain Properties of Galacturonan
5.1. Capillary Viscosity Data

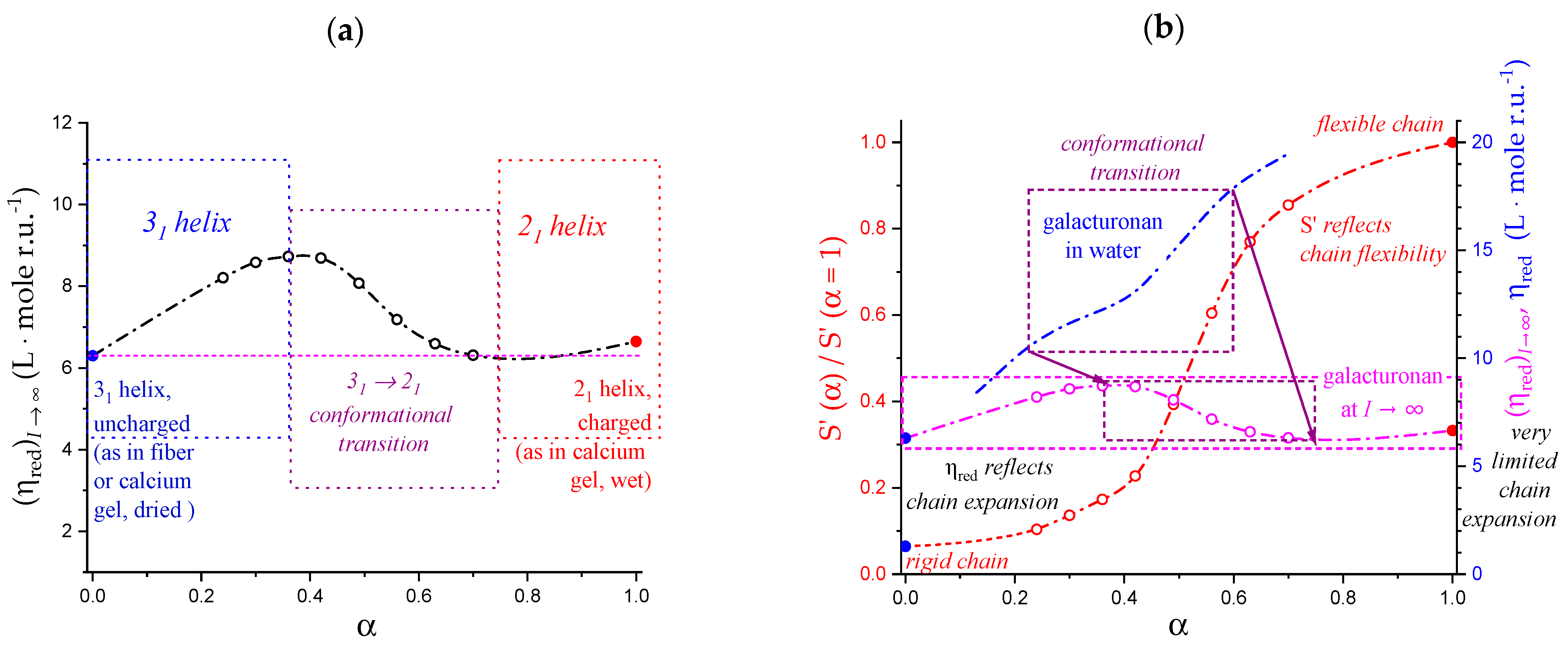
5.2. “Intrinsic” Chain Expansion and Stiffness of Pectic Acid
6. Conclusions
- Increasing the fraction of charging (as degree of dissociation, α) from 0.0 to 1.0—at 25 °C—brings about the following:
- An expansion of the 31 helical conformation—tightened by the formation of intra-molecular H-bond in H2 → O’6—with the formation of rheologically significant cooperative stretches up to a critical value of charge density, ξc (ξc = αc·ξ0);
- A conformational transition from the stiffer 31 to the more flexible 21 helical conformation; the α width of the transition is constant (δαtr = 0.38), i.e., independent of the ionic strength of the medium;
- An ever increasing—with α—loosening of either helical conformation;
- The 31 → 21 transition can also be achieved by heating LMP in water (namely, from 10 °C to 85 °C). The pH profile of the heat effects (by DSC) indicated that the condition pH = 3.0 produces the maximum value, paralleling the pH profile of the optical activity. It corresponds to the maximum tightening by the formation of intra-molecular H-bond;
- Both negative charging (by COOH dissociation) and temperature are effective in disordering (“loosening”) galacturonan helical conformations. However, the latter variable is by far more effective in doing so. Moreover, NaClO4, in due concentration, can stabilize the loosened conformation. This has been shown as the failure of an increase in the ionic strength to completely revert the loosening provoked by the increase in charge density through electrostatic shielding. Such effect is the first reported evidence of the impact of the chaotropic anion perchlorate on galacturonan (Hofmeister effect);
- For pH values ≤ 2.0, a massive association of LMP builds up, which is mostly driven by hydrophobic (van der Waals) interactions. A concerted action of highly reduced charge density, increased conformational loosening and interchain association produce interchain junctions at the root of acid–gel formation [32];
- The extrapolation of the reduced viscosity data to infinite ionic strength confirmed that the expansion of the 31 and the 21 helical conformations at α = 0.0 and 1.0, respectively, was the same (to within 1%) [31]; on the contrary, the response to ionic strength change showed a 15-fold increase in the 31 → 21 transformation, pointing to a very large accompanying increase in flexibility;
- The viscosity data confirmed that the three-fold helical conformation undergoes progressive tightening and expansion thanks to inter-residue H-bond formation up to a critical value of α, αc, beyond which a breakdown of the cooperative helix starts to produce the much more flexible two-fold helical conformation.
Supplementary Materials
- Intermediate values of at the intermediate values of α, from α1 to α = 1.0
- Calculation of the “loosen” contribution in the conformational transition interval
- More on the Thermodynamics of Association
- On the possible sources of deviation from theory of the enthalpy of mixing data (including Figure S1)
- More on Hofmeister and Lyotropic Effects in Polysaccharide Systems
- On the Coupling of the Polyelectrolyte Aspects with the Lyotropic Ones (including Figure S2)
- On the Viscometric behavior of Galacturonan as Compared to that of a “Normal” Weak Polyacid (including Figure S3)
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Zugenmaier, P. (Ed.) History of Cellulose Research BT. In Crystalline Cellulose and Derivatives: Characterization and Structures; Springer: Berlin/Heidelberg, Germany, 2008; pp. 7–51. ISBN 978-3-540-73934-0. [Google Scholar]
- Walkinshaw, M.D.; Arnott, S. Conformations and Interactions of Pectins: I. X-ray Diffraction Analyses of Sodium Pectate in Neutral and Acidified Forms. J. Mol. Biol. 1981, 153, 1055–1073. [Google Scholar] [CrossRef]
- Rees, D.A. Chain Conformation: Order versus Disorder. In Polysaccharide Shapes; Springer: Berlin/Heidelberg, Germany, 1977. [Google Scholar]
- Morris, E.R.; Powell, D.A.; Gidley, M.J.; Rees, D.A. Conformations and Interactions of Pectins: I. Polymorphism between Gel and Solid States of Calcium Polygalacturonate. J. Mol. Biol. 1982, 155, 507–516. [Google Scholar] [CrossRef]
- Palmer, K.J.; Hartzog, M.B. An X-ray Diffraction Investigation of Sodium Pectate. J. Am. Chem. Soc. 1945, 67, 2122–2127. [Google Scholar] [CrossRef]
- Sterling, C. Structure of Oriented Gels of Calcium Polyuronates. Biochim. Biophys. Acta 1957, 26, 186–197. [Google Scholar] [CrossRef]
- Ravanat, G.; Rinaudo, M. Investigation on Oligo- and Polygalacturonic Acids by Potentiometry and Circular Dichroism. Biopolymers 1980, 19, 2209–2222. [Google Scholar] [CrossRef]
- Walkinshaw, M.D.; Arnott, S. Conformations and Interactions of Pectins: II. Models for Junction Zones in Pectinic Acid and Calcium Pectate Gels. J. Mol. Biol. 1981, 153, 1075–1085. [Google Scholar] [CrossRef]
- Ruggiero, J.; Urbani, R.; Cesàro, A. Conformational Features of Galacturonans. I. Structure and Energy Minimization of Charged and Uncharged Galacturonan Dimeric Units. Int. J. Biol. Macromol. 1995, 17, 205–212. [Google Scholar] [CrossRef]
- Ruggiero, J.; Urbani, R.; Cesàro, A. Conformational Features of Galacturonans. II. Configurational Statistics of Pectic Polymers. Int. J. Biol. Macromol. 1995, 17, 213–218. [Google Scholar] [CrossRef]
- Cros, S.; du Penhoat, C.H.; Bouchemal, N.; Ohassan, H.; Imberty, A.; Pérez, S. Solution Conformation of a Pectin Fragment Disaccharide Using Molecular Modelling and Nuclear Magnetic Resonance. Int. J. Biol. Macromol. 1992, 14, 313–320. [Google Scholar] [CrossRef]
- Di Nola, A.; Fabrizi, G.; Lamba, D.; Segre, A.L. Solution Conformation of a Pectic Acid Fragment by 1H-Nmr and Molecular Dynamics. Biopolymers 1994, 34, 457–462. [Google Scholar] [CrossRef]
- Scheraga, H.A.; Némethy, G.; Steinberg, I.Z. The Contribution of Hydrophobic Bonds to the Thermal Stability of Protein Conformations. J. Biol. Chem. 1962, 237, 2506–2508. [Google Scholar] [CrossRef]
- Smith, L.J.; Fiebig, K.M.; Schwalbe, H.; Dobson, C.M. The Concept of a Random Coil: Residual Structure in Peptides and Denatured Proteins. Fold. Des. 1996, 1, R95–R106. [Google Scholar] [CrossRef]
- Inman, R.B.; Baldwin, R.L. Helix-Random Coil Transitions in DNA Homopolymer Pairs. J. Mol. Biol. 1964, 8, 452–469. [Google Scholar] [CrossRef]
- Fresco, J.R.; Doty, P. Polynucleotides. I. Molecular Properties and Configurations of Polyriboadenylic Acid in Solution1. J. Am. Chem. Soc. 1957, 79, 3928–3929. [Google Scholar] [CrossRef]
- Myer, Y.P. The PH-Induced Helix-Coil Transition of Poly-L-Lysine and Poly-L-Glutamic Acid and the 238-Mμ Dichroic Band. Macromolecules 1969, 2, 624–628. [Google Scholar] [CrossRef]
- Jordan, R.C.; Brant, D.A.; Cesàro, A. A Monte Carlo Study of the Amylosic Chain Conformation. Biopolymers 1978, 17, 2617–2632. [Google Scholar] [CrossRef]
- Gagnaire, D.; Pérez, S.; Tran, V. Configurational Statistics of Single Chains of α-Linked Glucans. Carbohydr. Polym. 1982, 2, 171–191. [Google Scholar] [CrossRef]
- Arnott, S.; Fulmer, A.; Scott, W.E.; Dea, I.C.M.; Moorhouse, R.; Rees, D.A. The Agarose Double Helix and Its Function in Agarose Gel Structure. J. Mol. Biol. 1974, 90, 269–284. [Google Scholar] [CrossRef]
- Foord, S.A.; Atkins, E.D.Y. New X-ray Diffraction Results from Agarose: Extended Single Helix Structures and Implications for Gelation Mechanism. Biopolymers 1989, 28, 1345–1365. [Google Scholar] [CrossRef]
- Guenet, J.-M.; Brûlet, A.; Rochas, C. Agarose Chain Conformation in the Sol State by Neutron Scattering. Int. J. Biol. Macromol. 1993, 15, 131–132. [Google Scholar] [CrossRef]
- Guenet, J.-M.; Rochas, C. Agarose Sols and Gels Revisited. Macromol. Symp. 2006, 242, 65–70. [Google Scholar] [CrossRef]
- Guenet, J.-M. Factors Influencing Gelation versus Crystallization in Cooling Polymer Solutions. Trends Polym. Sci. 1996, 4, 6–11. [Google Scholar]
- Donati, I.; Benegas, J.; Paoletti, S. On the Molecular Mechanism of the Calcium-Induced Gelation of Pectate. Different Steps in the Binding of Calcium Ions by Pectate. Biomacromolecules 2021, 22, 5000–5019. [Google Scholar] [CrossRef]
- Cros, S.; Garnier, C.; Axelos, M.A.V.; Imberty, A.; Pérez, S. Solution Conformations of Pectin Polysaccharides: Determination of Chain Characteristics by Small Angle Neutron Scattering, Viscometry, and Molecular Modeling. Biopolymers 1996, 39, 339–351. [Google Scholar] [CrossRef]
- Pérez, S.; Mazeau, K.; Hervé du Penhoat, C. The Three-Dimensional Structures of the Pectic Polysaccharides. Plant Physiol. Biochem. 2000, 38, 37–55. [Google Scholar] [CrossRef]
- Thibault, J.F.; Rinaudo, M. Interactions of Counteroins with Pectins Studied by Potentiometry and Circular Dichroism. In Chemistry and Function of Pectins; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1986; Volume 310, pp. 6–61. ISBN 9780841209749. [Google Scholar]
- Cesàro, A.; Ciana, A.; Delben, F.; Manzini, G.; Paoletti, S. Physicochemical Properties of Pectic Acid. I. Thermodynamic Evidence of a PH-Induced Conformational Transition in Aqueous Solution. Biopolymers 1982, 21, 431–449. [Google Scholar] [CrossRef]
- Paoletti, S.; Cesaro, A.; Delben, F.; Ciana, A. Ionic Effects on the Conformation, Equilibrium, Properties, and Rheology of Pectate in Aqueous Solutions and Gels. In Chemistry and Function of Pectins; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1986; Volume 310, pp. 7–73. ISBN 9780841209749. [Google Scholar]
- Guidugli, S.; Villegas, M.; Benegas, J.; Donati, I.; Paoletti, S. Solvation and Expansion of Neutral and Charged Chains of a Carbohydrate Polyelectrolyte: Galacturonan in Water. A Critical Revisiting. Biophys. Chem. 2023, 295, 106960. [Google Scholar] [CrossRef]
- Gilsenan, P.M.; Richardson, R.K.; Morris, E.R. Thermally Reversible Acid-Induced Gelation of Low-Methoxy Pectin. Carbohydr. Polym. 2000, 41, 339–349. [Google Scholar] [CrossRef]
- Huynh, U.T.D.; Lerbret, A.; Neiers, F.; Chambin, O.; Assifaoui, A. Binding of Divalent Cations to Polygalacturonate: A Mechanism Driven by the Hydration Water. J. Phys. Chem. B 2016, 120, 1021–1032. [Google Scholar] [CrossRef]
- Assifaoui, A.; Lerbret, A.; Uyen, H.T.D.; Neiers, F.; Chambin, O.; Loupiac, C.; Cousin, F. Structural Behaviour Differences in Low Methoxy Pectin Solutions in the Presence of Divalent Cations (Ca2+ and Zn2+): A Process Driven by the Binding Mechanism of the Cation with the Galacturonate Unit. Soft Matter 2015, 11, 551–560. [Google Scholar] [CrossRef]
- Ventura, I.; Jammal, J.; Bianco-Peled, H. Insights into the Nanostructure of Low-Methoxyl Pectin–Calcium Gels. Carbohydr. Polym. 2013, 97, 650–658. [Google Scholar] [CrossRef]
- Catoire, L.; Derouet, C.; Redon, A.-M.; Goldberg, R.; Hervédu Penhoat, C. An NMR Study of the Dynamic Single-Stranded Conformation of Sodium Pectate. Carbohydr. Res. 1997, 300, 19–29. [Google Scholar] [CrossRef]
- Thibault, J.F.; Rinaudo, M. Interactions of Mono- and Divalent Counterions with Alkali- and Enzyme-Deesterified Pectins in Salt-Free Solutions. Biopolymers 1985, 24, 2131–2143. [Google Scholar] [CrossRef]
- Sawayama, S.; Kawabata, A.; Nakahara, H.; Kamata, T. A Light Scattering Study on the Effects of PH on Pectin Aggregation in Aqueous Solution. Food Hydrocoll. 1988, 2, 31–37. [Google Scholar] [CrossRef]
- Alagna, L.; Prosperi, T.; Tomlinson, A.A.G.; Rizzo, R. Extended X-ray Absorption Fine Structure Investigation of Solid and Gel Forms of Calcium Poly(+.Alpha.-D-Galacturonate). J. Phys. Chem. 1986, 90, 6853–6857. [Google Scholar] [CrossRef]
- Schmidt, I.; Cousin, F.; Huchon, C.; Boué, F.; Axelos, M.A. V Spatial Structure and Composition of Polysaccharide−Protein Complexes from Small Angle Neutron Scattering. Biomacromolecules 2009, 10, 1346–1357. [Google Scholar] [CrossRef]
- Braccini, I.; Grasso, R.P.; Pérez, S. Conformational and Configurational Features of Acidic Polysaccharides and Their Interactions with Calcium Ions: A Molecular Modeling Investigation. Carbohydr. Res. 1999, 317, 119–130. [Google Scholar] [CrossRef]
- Ström, A.; Schuster, E.; Goh, S.M. Rheological Characterization of Acid Pectin Samples in the Absence and Presence of Monovalent Ions. Carbohydr. Polym. 2014, 113, 336–343. [Google Scholar] [CrossRef]
- Yoo, S.-H.; Lee, B.-H.; Savary, B.J.; Lee, S.; Lee, H.G.; Hotchkiss, A.T. Characteristics of Enzymatically-Deesterified Pectin Gels Produced in the Presence of Monovalent Ionic Salts. Food Hydrocoll. 2009, 23, 1926–1929. [Google Scholar] [CrossRef]
- Yoo, S.-H.; Fishman, M.L.; Savary, B.J.; Hotchkiss, A.T. Monovalent Salt-Induced Gelation of Enzymatically Deesterified Pectin. J. Agric. Food Chem. 2003, 51, 7410–7417. [Google Scholar] [CrossRef]
- Voet, A. Quantative Lyotropy. Chem. Rev. 1937, 20, 169–179. [Google Scholar] [CrossRef]
- Kunz, W.; Henle, J.; Ninham, B.W. ‘Zur Lehre von Der Wirkung Der Salze’ (about the Science of the Effect of Salts): Franz Hofmeister’s Historical Papers. Curr. Opin. Colloid Interface Sci. 2004, 9, 19–37. [Google Scholar] [CrossRef]
- Grasdalen, H.; Smidsroed, O. Iodide-Specific Formation of.Kappa.-Carrageenan Single Helixes. Iodine-127 NMR Spectroscopic Evidence for Selective Site Binding of Iodide Anions in the Ordered Conformation. Macromolecules 1981, 14, 1842–1845. [Google Scholar] [CrossRef]
- Norton, I.T.; Morris, E.R.; Rees, D.A. Lyotropic Effects of Simple Anions on the Conformation and Interactions of Kappa-Carrageenan. Carbohydr. Res. 1984, 134, 89–101. [Google Scholar] [CrossRef]
- Stanley, C.B.; Strey, H.H. Osmotically Induced Helix-Coil Transition in Poly(Glutamic Acid). Biophys. J. 2008, 94, 4427–4434. [Google Scholar] [CrossRef]
- Olsen, C.M.; Shikiya, R.; Ganugula, R.; Reiling-Steffensmeier, C.; Khutsishvili, I.; Johnson, S.E.; Marky, L.A. Application of Differential Scanning Calorimetry to Measure the Differential Binding of Ions, Water and Protons in the Unfolding of DNA Molecules. Biochim. Biophys. Acta-Gen. Subj. 2016, 1860, 990–998. [Google Scholar] [CrossRef]
- Satoh, M.; Komiyama, J.; Iijima, T. Viscometric Study of Poly(α-L-Glutamic Acid) in NaCl Solution. Colloid Polym. Sci. 1980, 258, 136–141. [Google Scholar] [CrossRef]
- Noda, I.; Tsuge, T.; Nagasawa, M. The Intrinsic Viscosity of Polyelectrolytes. J. Phys. Chem. 1970, 74, 710–719. [Google Scholar] [CrossRef]
- Leyte, J.C.; Mandel, M. Potentiometric Behavior of Polymethacrylic Acid. J. Polym. Sci. Part A Gen. Pap. 1964, 2, 1879–1891. [Google Scholar] [CrossRef]
- Nagasawa, M.; Holtzer, A. The Helix-Coil Transition in Solutions of Polyglutamic Acid. J. Am. Chem. Soc. 1964, 86, 538–543. [Google Scholar] [CrossRef]
- Pan, A.; Biswas, T.; Rakshit, A.K.; Moulik, S.P. Enthalpy–Entropy Compensation (EEC) Effect: A Revisit. J. Phys. Chem. B 2015, 119, 15876–15884. [Google Scholar] [CrossRef] [PubMed]
- Crescenzi, V.; Quadrifoglio, F.; Delben, F. Calorimetric Investigation of Poly(Methacrylic Acid) and Poly(Acrylic Acid) in Aqueous Solution. J. Polym. Sci. Part A-2 Polym. Phys. 1972, 10, 357–368. [Google Scholar] [CrossRef]
- Delben, F.; Cesaro, À.; Paoletti, S.; Crescenzi, V. Monomer Composition and Acetyl Content as Main Determinants of the Ionization Behavior of Alginates. Carbohydr. Res. 1982, 100, C46–C50. [Google Scholar] [CrossRef]
- Crescenzi, V.; Quadrifoglio, F.; Delben, F. Calorimetric Studies of Polycarboxylic Acids in Aqueous Solution. II. Maleic Acid–Butyl Vinyl Ether Copolymer. J. Polym. Sci. Part C Polym. Symp. 1972, 39, 241–246. [Google Scholar] [CrossRef]
- Abe, A.; Furuya, H. Conformational Characteristics of Polypeptide Chains with Special Focus on the α-Helix-Sense Inversion. In Modern Applications of Flory’s “Statistical Mechanics of Chain Molecules”; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2020; Volume 1356, pp. 5–63. ISBN 9780841298866. [Google Scholar]
- Watanabe, J.; Okamoto, S.; Satoh, K.; Sakajiri, K.; Furuya, H.; Abe, A. Reversible Helix−Helix Transition of Poly(β-Phenylpropyl l-Aspartate) Involving a Screw-Sense Inversion in the Solid State. Macromolecules 1996, 29, 7084–7088. [Google Scholar] [CrossRef]
- Sakajiri, K.; Satoh, K.; Yoshioka, K.; Kawauchi, S.; Watanabe, J. A Study on the Isothermal Kinetics of Helix–Helix Transformation Poly(β-Phenylpropyl l-Aspartate) Solid by DSC Analysis. J. Mol. Struct. 1999, 477, 175–179. [Google Scholar] [CrossRef]
- Braccini, I.; Pérez, S. Molecular Basis of Ca2+-Induced Gelation in Alginates and Pectins: The Egg-Box Model Revisited. Biomacromolecules 2001, 2, 1089–1096. [Google Scholar] [CrossRef]
- Perić, L.; Pereira, C.S.; Pérez, S.; Hünenberger, P.H. Conformation, Dynamics and Ion-Binding Properties of Single-Chain Polyuronates: A Molecular Dynamics Study. Mol. Simul. 2008, 34, 421–446. [Google Scholar] [CrossRef]
- Gouvion, C.; Mazeau, K.; Heyraud, A.; Taravel, F.R.; Tvaroska, I. Conformational Study of Digalacturonic Acid and Sodium Digalacturonate in Solution. Carbohydr. Res. 1994, 261, 187–202. [Google Scholar] [CrossRef]
- Lewis, G.; Johnson, A.F. Interpretation of the Heat of Dilution of Polymer Solutions. Polymer 1970, 11, 336–338. [Google Scholar] [CrossRef]
- Paoletti, S.; Delben, F.; Crescenzi, V. Enthalpies of Dilution of Partially Neutralized Maleic Acid Copolymers in Water. Correlation of Experiments with Theories. J. Phys. Chem. 1981, 85, 1413–1418. [Google Scholar] [CrossRef]
- Krasna, A.I. Changes in the Light-Scattering Properties of DNA on Denaturation. J. Colloid Interface Sci. 1972, 39, 632–646. [Google Scholar] [CrossRef]
- Colvill, A.J.E.; Jordan, D.O. The Influence of Ionic Strength on the Reversibility of the Denaturation of DNA in Dilute Solution. J. Mol. Biol. 1963, 7, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Manunza, B.; Deiana, S.; Pintore, M.; Gessa, C. Molecular Dynamics Study of Polygalacturonic Acid Chains in Aqueous Solution. Carbohydr. Res. 1997, 300, 85–88. [Google Scholar] [CrossRef]
- Noto, R.; Martorana, V.; Bulone, D.; San Biagio, P.L. Role of Charges and Solvent on the Conformational Properties of Poly(Galacturonic Acid) Chains: A Molecular Dynamics Study. Biomacromolecules 2005, 6, 2555–2562. [Google Scholar] [CrossRef]
- Guidugli, S.; Villegas, M.; Esteban, C.; Fernández Gauna, C.; Pantano, S.; Paoletti, S.; Benegas, J. Flexibilidad de Cadenas Neutras y Cargadas de Ácido Poligalacturonico En Distintos Solventes. An. AFA 2009, 21, 238–242. [Google Scholar]
- Wendler, K.; Thar, J.; Zahn, S.; Kirchner, B. Estimating the Hydrogen Bond Energy. J. Phys. Chem. A 2010, 114, 9529–9536. [Google Scholar] [CrossRef]
- Fenyo, J.C.; Delben, F.; Paoletti, S.; Crescenzi, V. Thermodynamics of Polycarboxylate Aqueous Solutions. 4. Special Features of Hydrophobic Maleic Acid Olefin Copolymers. J. Phys. Chem. 1977, 81, 1900–1905. [Google Scholar] [CrossRef]
- Capel, F.; Nicolai, T.; Durand, D.; Boulenguer, P.; Langendorff, V. Calcium and Acid Induced Gelation of (Amidated) Low Methoxyl Pectin. Food Hydrocoll. 2006, 20, 901–907. [Google Scholar] [CrossRef]
- Paoletti, S.; Cesàro, A.; Delben, F.; Crescenzi, V.; Rizzo, R. Polyelectrolytic Aspects of Conformational Transitions and Interchain Interactions in Ionic Polysaccharide Solutions: Comparison of Theory and Microcalorimetric Data. In Microdomains in Polymer Solutions; Dubin, P., Ed.; Springer: Boston, MA, MA, 1985; pp. 159–189. ISBN 978-1-4613-2123-1. [Google Scholar]
- Donati, I.; Cesàro, A.; Paoletti, S. Specific Interactions versus Counterion Condensation. 1. Nongelling Ions/Polyuronate Systems. Biomacromolecules 2006, 7, 281–287. [Google Scholar] [CrossRef]
- Eklund, L.; Hofer, T.S.; Persson, I. Structure and Water Exchange Dynamics of Hydrated Oxo Halo Ions in Aqueous Solution Using QMCF MD Simulation, Large Angle X-ray Scattering and EXAFS. Dalt. Trans. 2015, 44, 1816–1828. [Google Scholar] [CrossRef]
- Walrafen, G.E. Raman Spectral Studies of the Effects of Perchlorate Ion on Water Structure. J. Chem. Phys. 2003, 52, 4176–4198. [Google Scholar] [CrossRef]
- Mazzini, V.; Craig, V.S.J. What Is the Fundamental Ion-Specific Series for Anions and Cations? Ion Specificity in Standard Partial Molar Volumes of Electrolytes and Electrostriction in Water and Non-Aqueous Solvents. Chem. Sci. 2017, 8, 7052–7065. [Google Scholar] [CrossRef] [PubMed]
- Lo Nostro, P.; Ninham, B.W. Hofmeister Phenomena: An Update on Ion Specificity in Biology. Chem. Rev. 2012, 112, 2286–2322. [Google Scholar] [CrossRef]
- Asciutto, E.K.; General, I.J.; Xiong, K.; Asher, S.A.; Madura, J.D. Sodium Perchlorate Effects on the Helical Stability of a Mainly Alanine Peptide. Biophys. J. 2010, 98, 186–196. [Google Scholar] [CrossRef][Green Version]
- Crevenna, A.H.; Naredi-Rainer, N.; Lamb, D.C.; Wedlich-Söldner, R.; Dzubiella, J. Effects of Hofmeister Ions on the α-Helical Structure of Proteins. Biophys. J. 2012, 102, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Hu, C.; Mei, A.; Lou, X. The Influence of the Coexisting Anions on the Adsorption of Perchlorate from Water by the Modified Orange Peels. Nat. Environ. Pollut. Technol. 2016, 15, 1359–1362. [Google Scholar]
- Clarke-Sturman, A.J.; Pedley, J.B.; Sturla, P.L. Influence of Anions on the Properties of Microbial Polysaccharides in Solution. Int. J. Biol. Macromol. 1986, 8, 355–360. [Google Scholar] [CrossRef]
- Mráček, A.; Varhaníková, J.; Lehocký, M.; Gřundělová, L.; Pokopcová, A.; Velebný, V. The Influence of Hofmeister Series Ions on Hyaluronan Swelling and Viscosity. Molecules 2008, 13, 1025–1034. [Google Scholar] [CrossRef]
- Tatini, D.; Sarri, F.; Maltoni, P.; Ambrosi, M.; Carretti, E.; Ninham, B.W.; Lo Nostro, P. Specific Ion Effects in Polysaccharide Dispersions. Carbohydr. Polym. 2017, 173, 344–352. [Google Scholar] [CrossRef]
- Fishman, M.L.; Chau, H.K.; Kolpak, F.; Brady, J. Solvent Effects on the Molecular Properties of Pectins. J. Agric. Food Chem. 2001, 49, 4494–4501. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Ratcliffe, I.; Williams, P.A.; Luo, S.; Chen, J.; Liu, C. The Influence of PH and Monovalent Ions on the Gelation of Pectin from the Fruit Seeds of the Creeping Fig Plant. Food Hydrocoll. 2021, 111, 106219. [Google Scholar] [CrossRef]
- Benegas, J.C.; Cesàro, A.; Rizzo, R.; Paoletti, S. Conformational Stability of Biological Polyelectrolytes: Evaluation of Enthalpy and Entropy Changes of Conformational Transitions. Biopolymers 1998, 45, 203–216. [Google Scholar] [CrossRef]
- Engel, J.; Schwarz, G. Cooperative Conformational Transitions of Linear Biopolymers. Angew. Chem. Int. Ed. Engl. 1970, 9, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Poland, D.; Scheraga, H.A. Theory of Helix-Coil Transitions in Biopolymers. In Statistical Mechanical Theory of Order-Disorder Transitions in Biological Macromolecules; Academic Press: Cambridge, MA, USA, 1970. [Google Scholar]
- Zimm, B.H.; Doty, P.; Iso, K. Determination of the Parameters for Helix Formation in Poly-γ-Benzyl-L-Glutamate. Proc. Natl. Acad. Sci. USA 1959, 45, 1601–1607. [Google Scholar] [CrossRef] [PubMed]
- Kidera, A.; Nakajima, A. Light Scattering from Poly(L-Glutamic Acid) in Aqueous Solution in the Helix-Coil Transition Region. Macromolecules 1984, 17, 659–663. [Google Scholar] [CrossRef]
- Smidsrød, O.; Haug, A. Estimation of the Relative Stiffness of the Molecular Chain in Polyelectrolytes from Measurements of Viscosity at Different Ionic Strengths. Biopolymers 1971, 10, 1213–1227. [Google Scholar] [CrossRef]
- Manning, G.S. The Persistence Length of DNA Is Reached from the Persistence Length of Its Null Isomer through an Internal Electrostatic Stretching Force. Biophys. J. 2006, 91, 3607–3616. [Google Scholar] [CrossRef]
- Malovikova, A.; Milas, M.; Rinaudo, M.; Borsali, R. Viscometric Behavior of Sodium Polygalacturonate in the Presence of Low Salt Content. In Macro-Ion Characterization; ACS Symposium Series; American Chemical Society: Washingtong, DC, USA, 1993; Volume 548, pp. 24–315. ISBN 9780841227705. [Google Scholar]
- Powell, D.A.; Morris, E.R.; Gidley, M.J.; Rees, D.A. Conformations and Interactions of Pectins: II. Influence of Residue Sequence on Chain Association in Calcium Pectate Gels. J. Mol. Biol. 1982, 155, 517–531. [Google Scholar] [CrossRef]
- Grasdalen, H.; Kvam, B.J. Sodium-23 NMR in Aqueous Solutions of Sodium Polyuronates. Counterion Binding and Conformational Conditions. Macromolecules 1986, 19, 1913–1920. [Google Scholar] [CrossRef]
- Minin, K.A.; Zhmurov, A.; Marx, K.A.; Purohit, P.K.; Barsegov, V. Dynamic Transition from α-Helices to β-Sheets in Polypeptide Coiled-Coil Motifs. J. Am. Chem. Soc. 2017, 139, 16168–16177. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Luo, Y.; Ma, B.; Nussinov, R.; Wei, G. Conformational Distribution and α-Helix to β-Sheet Transition of Human Amylin Fragment Dimer. Biomacromolecules 2014, 15, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Donati, I.; Asaro, F.; Paoletti, S. Experimental Evidence of Counterion Affinity in Alginates: The Case of Nongelling Ion Mg2+. J. Phys. Chem. B 2009, 113, 12877–12886. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.S. Limiting Laws and Counterion Condensation in Polyelectrolyte Solutions: IV. The Approach to the Limit and the Extraordinary Stability of the Charge Fraction. Biophys. Chem. 1977, 7, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.S. Limiting Laws and Counterion Condensation in Polyelectrolyte Solutions: V. Further Development of the Chemical Model. Biophys. Chem. 1978, 9, 65–70. [Google Scholar] [CrossRef]
- Vanderzee, C.E.; Swanson, J.A. Heats of Dilution and Relative Apparent Molal Enthalpies of Aqueous Sodium Perchlorate and Perchloric Acid. J. Phys. Chem. 1963, 67, 285–291. [Google Scholar] [CrossRef]
- Messikomer, E.E.; Wood, R.H. The Enthalpy of Dilution of Aqueous Sodium Chloride at 298.15 to 373.15 K, Measured with a Flow Calorimeter. J. Chem. Thermodyn. 1975, 7, 119–130. [Google Scholar] [CrossRef]
- Andreeva, D.; Hartmann, H.; Taneva, S.G.; Krastev, R. Regulation of the growth, morphology, mechanical properties and biocompatibility of natural polysaccharide-based multilayers by Hofmeister anions. J. Mater. Chem. B 2016, 4, 7092–7100. [Google Scholar] [CrossRef]
- Sangeetha, P.; Selvakumari, T.M.; Selvasekarapandian, S. Preparation of primary magnesium battery based on kappa-carrageenan with magnesium perchlorate and its application to electrochemical devices. Polym. Bull. 2023. [Google Scholar] [CrossRef]
- Seale, R.; Morris, E.R.; Rees, D.A. Interactions of alginates with univalent cations. Carbohydr. Res. 1982, 110, 101–112. [Google Scholar] [CrossRef]
- Rochas, C.; Rinaudo, M. Activity Coefficients of Counterions and Conformation in kappa-Carrageenan Systems. Biopolymers 1980, 19, 1675–1687. [Google Scholar] [CrossRef]
- Zhang, W.; Piculell, L.; Nilsson, S. Salt Dependence and Ion Specificity of the Coil-Helix Transition of Furcellaran. Biopolymers 1991, 31, 1727–1736. [Google Scholar] [CrossRef]
- Zhang, W.; Piculell, L.; Nilsson, S. Effects of Specific Anion Binding on the Helix-Coil Transition of Lower Charged Carrageenans. NMR Data and Conformational Equilibria Analyzed within the Poisson-Boltzmann Cell Model. Macromolecules 1992, 25, 6165–6172. [Google Scholar] [CrossRef]
- Yin, W.; Zhang, H.; Huang, L.; Nishinari, K. Effects of the lyotropic series salts on the gelation of konjac glucomannan in aqueous solutions. Carbohydr. Polym. 2008, 74, 68–78. [Google Scholar] [CrossRef]
- Singh, T.; Meena, R.; Kumar, A. Effect of sodium sulfate on the gelling behavior of agarose and water structure inside the gel networks. J. Phys. Chem. B 2009, 113, 2519–2525. [Google Scholar] [CrossRef] [PubMed]



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Transition | |||
|---|---|---|---|---|
| kcal·mol−1 | cal·mol−1·K−1 | kcal·mol−1 | ||
| Reference [29] | αi → αf, s a | +0.70 ± 0.10 b | +2.2 ± 0.3 b | +0.04 ± 0.13 b |
| αi → αf, w a | +0.50 ± 0.10 b | +1.6 ± 0.3 b | +0.02 ± 0.13 b | |
| This work | αi → αf, s a | +0.727 ± 0.05 c | +2.27 ± 0.01 c | +0.05 ± 0.05 d |
| αi → αf, w a | +0.563 ± 0.05 c | +1.62 ± 0.01 c | +0.08 ± 0.05 d |
| a. | |||||||
| condition | Cp equiv.·L−1 | Cs M | I M | kcal⋅mole r.u.−1 | cal⋅mole r.u.−1·K−1 | kcal⋅mole r.u.−1 | Tm K |
| T = 25 °C, α = 1 | |||||||
| salt (R = 10) | 0.005 | 0.05 | 0.05152 | 0.13 ± 0.00 | 0.44 ± 0.01 | 0.001 ± 0.001 | 300 ± 27 |
| water (R = 0) | 0.0064 | 0 | 0.00194 | 0.10 ± 0.01 | 0.19 ± 0.04 | 0.04 ± 0.00 | 511 ± 123 |
| b. | |||||||
| condition | Cp equiv.·L−1 | Cs M | I M | kcal⋅mole r.u.−1 | cal⋅mole r.u.−1·K−1 | kcal⋅mole r.u.−1 | Tm K |
| T = 25 °C, α = 1 | |||||||
| salt (R = 10) | 0.005 | 0.05 | 0.05152 | 0.59 ± 0.00 | 1.84 ± 0.01 | 0.05 ± 0.00 | 324 ± 2 |
| water (R = 0) | 0.0064 | 0 | 0.00194 | 0.46 ± 0.01 | 1.43 ± 0.04 | 0.04 ± 0.02 | 324 ± 12 |
| c. | |||||||
| condition | Cp equiv.·L−1 | Cs M | I M | kcal⋅mole r.u.−1 | cal⋅mole r.u.−1·K−1 | kcal⋅mole r.u.−1 | Tm K |
| T = 25 °C, α = 1 | |||||||
| salt (R = 10) | 0.005 | 0.05 | 0.05152 | 0.73 ± 0.00 | 2.28 ± 0.01 | 0.05 ± 0.00 | 319 ± 2 |
| water (R = 0) | 0.0064 | 0 | 0.00194 | 0.56 ± 0.02 | 1.63 ± 0.06 | 0.08 ± 0.02 | 346 ± 16 |
| Source | Condition | State | ||||
|---|---|---|---|---|---|---|
| kcal·mole r.u.−1 | ||||||
| this work | water a | initial | final | b | c | d |
| α = 1, Cs = 0, Cp = ∞ | α = 1, Cs = 0, Cp = 0 | 0.13 ± 0.06 | 0.12 ± 0.06 | 0.13 ± 0.09 | ||
| Isothermal (25 °C), pH-Induced Transition. Final State: α = 1.0 | Iso—pH (3.0), T-Induced Transition. Final State: T = 85 °C | RATIO | ||||
|---|---|---|---|---|---|---|
| Δ[ϑ]215 (deg·cm2·decimole−1) | 1. | Δ[α]436 (100 deg·dm−1·(% wt)−1) | 2. | 2./1. | ||
| A. 31 → 21 | 1080 a | A. 31 → 21 | 15.63 c | |||
| B. loosening | 1449 b | B. loosening | 50.0 d | |||
| Chiro-optical ratio, B./A. | 1.34 | Chiro-optical ratio, B./A. | 3.20 | 2.4 | ||
| (kcal·mole r.u.−1) | 1. | (kcal·mole r.u.−1) | 2. | 2./1. | ||
| C. 31 → 21 | 0.59 e | C. 31 → 21 | 0.59 e | |||
| D. loosening | 0.13 f | D. loosening | 0.28 g | |||
| Calorimetric ratio, D./C. | 0.22 | Calorimetric ratio, D./C. | 0.46 | 2.1 | ||
| (kcal·mole r.u.−1·K−1) | 1. | (kcal·mole r.u.−1·K−1) | 2. | 2./1. | ||
| E. 31 → 21 | 1.84 h | E. 31 → 21 | 1.84 h | |||
| F. loosening | 0.44 i | F. loosening | 1.03 j | |||
| Entropic ratio, F./E. | 0.24 | Entropic ratio, F./E. | 0.56 | 2.3 | ||
| Source | Transition | |||
|---|---|---|---|---|
| kcal ·mole r.u.−1 | ||||
| [75] | Cs = 0 → Cs = ∞, wa | +0.20 ± 0.01 | +0.33 ± 0.01 | −0.13 ± 0.01 |
| [30] | Cs = 0.05 M → Cs = ∞, sc | +0.10 ± 0.01 | +0.12 ± 0.01 | −0.01 ± 0.01 |
| a. | |||||
| condition | state | ( = 0; “as if” α = 0) | |||
| initial | final | kcal·mol−1 | |||
| water a | α1, Cs = 0 (R = 0) | α = 1, RMAX (=0.8) | 0.10 ± 0.01 | 0.03 | 0.13 ± 0.01 |
| salt b | α1, Cs = 0.05 M (R = 10) | α = 1, RMAX (=64) | 0.13 ± 0.00 | 0.06 | 0.19 ± 0.00 |
| b. | |||||
| α = 1 | c | * | † | c | |
| A | B | A + B | |||
| kcal·mol−1 | |||||
| water a | 0.03 | −0.16 | 0.13 ± 0.01 * | 0.13 ± 0.09 † | −0.03 ± 0.02 e |
| salt b | 0.06 | −0.07 | 0.19 ± 0.00 * | +0.12 ± 0.01 e | |
| α = 1 | d | c,d | d | c,d | |
| A | B | A + B | |||
| cal·mol−1·K−1 | |||||
| water a | 0.06 | −0.32 | 0.25 | −0.06 ± 0.05 e | |
| salt b | 0.20 | −0.24 | 0.64 | +0.40 ± 0.01 e | |
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paoletti, S.; Donati, I. pH Effects on the Conformations of Galacturonan in Solution: Conformational Transition and Loosening, Extension and Stiffness. Polysaccharides 2023, 4, 271-324. https://doi.org/10.3390/polysaccharides4030018
Paoletti S, Donati I. pH Effects on the Conformations of Galacturonan in Solution: Conformational Transition and Loosening, Extension and Stiffness. Polysaccharides. 2023; 4(3):271-324. https://doi.org/10.3390/polysaccharides4030018
Chicago/Turabian StylePaoletti, Sergio, and Ivan Donati. 2023. "pH Effects on the Conformations of Galacturonan in Solution: Conformational Transition and Loosening, Extension and Stiffness" Polysaccharides 4, no. 3: 271-324. https://doi.org/10.3390/polysaccharides4030018
APA StylePaoletti, S., & Donati, I. (2023). pH Effects on the Conformations of Galacturonan in Solution: Conformational Transition and Loosening, Extension and Stiffness. Polysaccharides, 4(3), 271-324. https://doi.org/10.3390/polysaccharides4030018