1. Introduction
Affinity binders are essential reagents and the workhorses of biological research, from academic research to clinical diagnostics and therapeutics. Traditionally, full-size IgG molecules and their fragments have been the go-to affinity binders for assays such as ELISA, flow cytometry, Western blot, and immunofluorescent staining assays [
1]. However, smaller affinity binder scaffolds have been considered for the “next-generation” of affinity reagents for their numerous advantages over conventional antibodies, including smaller size, higher thermodynamic stability, amenability to re-engineering, access to target sites, and ability to penetrate tissues and cells [
2,
3,
4]. Specifically, single domain antibody binders found in camelids, also known as nanobodies or variable domain of heavy-chain antibodies (VHHs), have recently become popular affinity reagents due to their natural availability, in addition to their abovementioned advantages [
5,
6].
The quality of any affinity binder, characterized by their specificity, affinity, and functional activity, is pivotal for the precision in their application, such as a diagnostic test or achieving their therapeutic potential. Even though VHHs are single heavy chain only fragments containing only three complementary determining regions (CDRs), compared to the six in IgGs, they have been shown to bind their targets with high affinity (K
D < nM) and specificity [
7]. This has led to rapid development of VHH-based affinity binders as potential therapeutics for diseases, including SARS-CoV2 [
8], Alzheimer’s disease [
9], and cancer [
10].
Our target in this study is K-Ras4B (hereafter referred to as K-Ras), one of the most important oncogenic proteins. K-Ras is one of the four proteins in the small Ras GTPase family [
11] and serves as upstream molecular regulators of very complex downstream kinase pathways, such as mitogen-activated protein kinases (MAP) and phosphatidylinositol-3-kinase/abstract protein kinase (PI3K/Akt) ensuring cells’ homeostasis. Since the Ras proteins play an important role in cellular signaling, their dysregulation due to mutations lead to uncontrolled cell proliferation, making it an important target of cancer diagnostics and therapeutics [
12,
13]. There is an abundance of literature on the structure, function, and molecular biology of Ras proteins [
14]. Multiple efforts have focused on the development of affinity binders to target K-Ras proteins [
15,
16]. However, K-Ras has proved to be a difficult target due to its intracellular localization, presence of multiple isoforms, and lack of druggable pockets [
17]. Recently, Amgen [
18] and Mirati [
19] therapeutics have developed covalent Ras inhibitors to the G12C mutant due to the reactivity of the cysteine mutation as a nucleophile. But this strategy cannot be extended to the other mutations, and there is still a need for developing affinity molecules to target Ras proteins.
In this work, we developed VHH binders against the most common K-Ras mutant (G12D) from a camelid-derived VHH library using phage display and identified lead hits that showed binding on phage ELISA. However, we found that most of the lead hits showed poor soluble expression in bacterial cultures limiting their characterization, functional studies, and therefore their potential as binding reagents. In this study, we evaluated the following questions: Is surface hydrophobicity of these clones playing a role in their aggregation and therefore resulting in poor solubility? If so, can we improve the soluble expression of the binders by manipulating their surface hydrophobicity through subtle changes in amino acids on their surface? Can molecular models guide us during this process?
We hypothesized that the poor solubility is due to the high surface hydrophobicity of the clones, which can be improved by hydrophilic surface mutations. To test this hypothesis, we employed a combination of random and in silico guided mutagenesis. Specifically, the in silico approach involved a computational pipeline for modeling the 3D structure of the lead hits and identifying hydrophobic hotspots on the surface of the binders. By using spatial aggregation propensity (SAP) as a measure for surface hydrophobicity, we showed that the clones with poor soluble expression contained significant hydrophobic hotspots.
Having shown correlation between the hydrophobicity and expression levels, we followed two approaches to perform mutations and produce high expression clones: (i) mutating hydrophobic amino acids centered at the hydrophobic hotspots to charged or polar ones; (ii) combining mutations that are identified through a round of random mutagenesis, which are predicted to decrease surface hydrophobicity. Both approaches led to significant increases in the soluble expression (2× to 40×) of the binders and therefore allowed characterization of their affinities to their target. While most of the binders produced by the former approach suffered from the loss of binding affinity, the latter approach resulted in high solubility without the loss of binding affinity. Therefore, based on our results, we recommend performing a round of random mutagenesis to identify mutations and using an in silico pipeline to guide the evolution of the binders by simulating several combinations of the mutations and selecting for soluble expression. Overall, our work provides a framework for rational design to improve the expression of VHH binders.
2. Materials and Methods
2.1. Homology Modeling and All-Atom MD Simulations
Modeling and simulations were performed for predictions of surface hydrophobicity of the VHH binders. The VHH binders were modeled using homology modeling, followed by energy minimization and equilibration in all-atom MD simulations in water. The Molecular Operating Environment (MOE 2022) (Montreal, QC, Canada) [
20,
21] and Iterative Threading ASSEmbly Refinement (I-TASSER 5.1) [
22] software were used for homology modeling, and NAMD parallel-computing code (NAMD 2.12) [
23] and GROningen MAchine for Computer Simulations (GROMACS-2018) [
24] were used for all-atom simulations. A 5 ns equilibration run was followed by a 100 ns production run. The MD simulations incorporated the leap-frog algorithm with a 2 fs time-step to integrate the equations of motion. The system was maintained at 300 K and 1 bar, using the velocity rescaling thermostat [
25] and Parrinello–Rahman barostat [
26], respectively. The long-ranged electrostatic interactions were calculated using the particle mesh Ewald (PME) [
27] algorithm with a real space cut-off of 1.2 nm. LJ interactions were also truncated at 1.2 nm. The TIP3P model [
28] was used to represent the water molecules, and the LINCS [
29] algorithm was used to constrain the motion of hydrogen atoms bonded to heavy atoms. Coordinates of the protein molecule were stored every 1 ps for further analysis.
2.2. SAP Calculations
Following a similar protocol as Trout and coworkers [
30], we used all-atom simulations of the VHH binders in explicit water and calculated SAP averaged over the coordinates obtained in a 100 ns simulation trajectory, as follows:
Equation (1) allows calculation of residue-specific spatial aggregation propensity (SAP
i) based on normalized solvent accessible areas (SAA) of the side chain atoms in the vicinity (defined by a distance cut-off, R) and inherent residue hydrophobicity (from the Kyte–Doolittle hydropathy scale) [
31]. A schematic illustrating the computational pipeline is shown in the
supplemental info (Figure S1).
2.3. VHH—Periplasmic Expression
pADL-VHH plasmids were transformed into competent E. coli BL21 (DE3) cells and grown on Luria Bertani (LB) agar plates. Single colonies were selected using pipette tips and grown in 10 mL of LB + 1% Glc + 50 µg/mL Carbenicillin starter culture overnight at 37 °C and 250 rpm to prepare the starter culture. For VHH protein expression, the starter cultures were diluted in 500 mL Terrific Broth (TB, Thermo Fisher (Waltham, MA, USA) A1374301) and grown at 37 °C, followed by induction with 1 mM isopropylthio-β-galactoside (IPTG, Thermo Fisher, 15529019) once the OD600 reached 0.6 and incubated at 30 °C overnight. After bacteria culture growth at 30 °C and 250 rpm overnight, an osmotic shock was performed to selectively release the periplasmic proteins into the supernatant. The cells were spun down at 6000 g and resuspended in 20 mL 1X HES buffer (200 mM HEPES pH 8.0, 0.65 mM EDTA, 0.5M sucrose). After 1 h of incubation on a rotating platform at 4 °C, 20 mL of 0.25X HES buffer was added and incubated for one more hour at 4 °C. The periplasmic fraction was harvested by centrifugation at 8000 rpm for 15 min. The supernatant was transferred into a new 50 mL tube, and centrifugation at 8000 rpm for 15 min was repeated. The recombinant His-tagged proteins were successively purified using HisPur™ Ni-NTA Resin (Thermo Scientific, 88223)). Four milliliters of 50% resin slurry was washed with 20 mL Ni-NTA wash buffer (50 mM HEPES, 150 mM NaCl, 10 mM imidazole) 3 times using centrifugation at 1250 rpm for 3 min after each wash. The periplasm-containing supernatant was mixed with Ni-NTA resin and incubated for 1 h at 4 °C. After that, the resin was spun down at 1250 rpm for 3 min, washed with 30 mL of Ni-NTA wash buffer 3 times, resuspended in 5–10 mL of Ni-NTA wash buffer, and transferred to a disposable column. Elution was performed with 4 × 2 mL aliquots of Ni-NTA elution buffer (50 mM HEPES pH 8.0, 150 mM NaCl, 500 mM imidazole). Eluted fractions were analyzed in NuPAGE™ Bis-Tris Mini Protein Gel (Thermo Fisher, NP0322BOX). Fractions containing the VHH protein were concentrated, and the PBS buffer was exchanged and additionally purified via small-scale size exclusion chromatography using a Superdex 200 Increase 10/300 GL column (Cytiva (Marlborough, MA, USA), 28990944) on an ÄKTA Pure instrument. The protein of interest was further eluted via isocratic elution in PBS buffer containing 25 mM trehalose, 0.5 mM MgCl2, and 0.05% Tween20, before being concentrated, aliquoted, and stored at −80 °C. The final concentration of the protein was determined using 280 nm absorbance on Nanodrop and using storage buffer as a blank. The protein concentration was then adjusted to perform characterization assays.
2.4. Enzyme Linked Immuno Sorbent Assay (ELISA)
The VHHs in their soluble form were characterized for their selective binding using ELISA performed on 96-well plates (Biolegend Nunc™ MaxiSorp™ ELISA Plates, Uncoated; Biolegend (San Diego, CA, USA) 423501). The wells were coated overnight with K-Ras G12D antigens or PBS (for negative control). The coated wells were then washed with PBS and 0.05% Tween 4 times and blocked with 2% Milk-PBS prepared in the lab using BD Difco™ Skim Milk (BD Biosciences (Franklin Lakes, NJ, USA), Cat# 232100). The VHHs were introduced in the wells and serially diluted in blocking buffer in the range from 24.4 ng/mL (~1.5 nM) to 100 ug/mL (~6.25 nM), including a blank with no protein for further background subtraction. The Anti-Ras antibody (Abcam (Cambridge, UK), ab55391) was used as the positive control in a concentration range starting from 0 to 2 μg/mL. Following incubation of the targeted antigen (just blocking buffer) with the variable VHH concentrations, the plate was washed with PBS and 0.05% Tween to remove unbound VHH. To detect the antigen-bound VHH, an anti-6x His tag primary Mab (Thermo Fisher Scientific 4E3D10H2/E3) was introduced in a 1:1000 dilution and incubated for one hour before further washes. Finally, a goat anti-mouse IgG polyclonal secondary antibody (Invitrogen (Waltham, MA, USA) A16072) was added in a 1:5000 dilution and incubated at room temperature before signal development. After the final washing steps, a horseradish peroxidase substrate was added, and the absorbance at 450 nm was read on a Tecan 20 M Spark plate reader following stoppage of the reaction with 0.16 M H2SO4. Each condition was performed in duplicate or triplicate, and absorbance signals were analyzed based on the average signal calculated in two or three wells. The standard deviation was determined based on duplicated or triplicated conditions as well.
2.5. Bio-Layer Interferometry (BLI)/Affinity Determination
Kinetic assays were carried out at 30 °C with orbital shaking at 1000 rpm on a ForteBio Octet RED384 instrument (Molecular Devices) with a running buffer of 1X Kinetics pH 7.4 buffer (Sartorius) + 0.25% BSA and using ForteBio Data Acquisition software 9.0. Random Amine chemistry with an Amine Coupling Second Generation kit (Sartorius) was used to load K-Ras G12D ligands on Amine Reactive Second Generation (AR2G) biosensors (Sartorius) according to the manufacturer’s instructions [
32]. Briefly AR2G sensors equilibrated in water for 10 min were activated with 20 mM 1-Ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride) (EDC) and 10 mM s-NHS (N-hydroxysulfosuccinimide) (NHS) for 300 s and were either mock loaded (no ligand reference sensor) or loaded with 30 μg/mL of K-Ras G12D in 10 mM sodium acetate buffer, pH 5.0, for 600 s, followed by a quench step with 1 M ethanolamine, pH 8.5, for 500 s. Each ligand-loaded sensor equilibrated in running buffer to establish a baseline was dipped into wells containing 2-fold dilutions of VHH analytes (F4.0, F4.3, F4.6, B6.0, and B6.5) in running buffer or buffer alone (reference sample). Increasing concentrations of analytes ranging from 15 µM to 0 µM were allowed to associate for 300 s, followed by a 600 s dissociation step, where the VHH-bound sensors were washed with running buffer. Binding curves were analyzed using ForteBio Data Analysis HT 10.0 evaluation software [
32]. To control for background signal and non-specific binding, raw experimental data were processed by subtracting the reference biosensor (no ligand) and reference sample (no analyte). Sensors were regenerated with 2 short 15 s pulses of 10 mM glycine, pH 1.7, followed by 180 s of washing with running buffer. For each interaction pair, processed data from 3 independent experiments of at least 5 different concentrations of analyte binding were globally fit to a 1:1 Langmuir binding model (or heterogenous ligand binding model) to calculate the association rate constant
ka and the dissociation rate constant
kd. Equilibrium dissociation constant
KD, which defines the strength of the interaction or affinity, was calculated as the kinetic dissociation rate constant divided by the kinetic association rate constant.
3. Results
Our primary goal was to develop VHH binders against the target K-Ras G12D, an oncogenic mutant GTPase. Recombinantly expressed K-Ras G12D is expected to be in its GDP-bound state. Using phage display, we performed a screen on a camelid-derived naïve VHH library and selected the clones that showed preferential binding to the target as lead hits. The sequences of the four lead clones (F4.0, F3.0, B6.0, and E4.0) and their alignments shown in
Figure 1A, confirmed the uniqueness of the clones as highlighted by substantial differences in the CDR regions. We proceeded to express these clones in bacterial expression cell cultures (see Methods
Section 2.3) and found very poor yields, as shown in
Figure 1B. Traditionally, VHHs are thought to be stable and highly soluble and hence are expected to possess high levels of expression. However, in repeated expression of our lead hits, we found very minimum yields. We hypothesized that the naïve library created by combinatorial methods potentially resulted in the low expressing clones, as the surface hydrophobicity is not controlled for in their generation.
To test the abovementioned hypothesis, we adopted a computational approach developed by Trout and co-workers for improving the stability of antibody binders. In their work, Trout and co-workers [
30] developed a molecular context-dependent measure for surface hydrophobicity of proteins, called spatial aggregation propensity (SAP), which can be characterized using MD simulation trajectories. The 3D structures of VHHs needed for this approach was obtained using a combination of MOE and I-TASSER for consensus, which was then subject to energy minimization, equilibration, and a 100 ns production run in explicit water to arrive at an ensemble of representative structures for SAP calculations. The SAP
i score for the ith structure was calculated using Equation (1), which was then used to obtain (i) the overall SAP score, a summation obtained over all the residues of the VHH (
Figure 1C) and (ii) SAP maps that identify hydrophobic hotspots, highlighting the locations that can be targeted for solubility enhancing mutations (
Figure 1D).
In
Figure 1C, the SAP score of a GFP-specific VHH, Enhancer (ENH) and a few other known VHH binders to prostate-specific antigen (PSA) were compared with the K-Ras specific lead hits from our phage display screen. Interestingly, ENH, which is commercially available and a known high expressing protein, showed a significantly lower SAP score compared to our clones. In addition to ENH, we also observed significantly lower SAP scores for the high expressing PSA-specific VHHs, indicating a correlation between SAP scores and soluble expression. This observation suggested the likelihood of hydrophobic patches on the surface of the K-Ras specific binders from our screen, which might lead to aggregation and consequently lower expression of the proteins. The SAP maps for the VHH binders shown in
Figure 1D supports this hypothesis, highlighting the hydrophobic patches on low expressing clones, especially for clones B6.0 and E4.0. These maps also provide guidance for targeted mutagenesis of the binders for soluble expression.
To improve the soluble expression of the binders, we followed two different approaches with two select clones (B6.0 and F4.0). Our first approach is purely computational and involved introducing targeted hydrophilic (charged/polar) mutations in the hydrophobic hotspots of B6.0, while minimizing the impact to the CDR regions. Specifically, we identified six positions in the B6.0 sequence, I31, I54, Y59, F69, Y106, and Y107, that form hydrophobic patches on the surface of the protein, as identified in
Figure 2C. The sequences of the mutated clones (B6.1–B6.6) are shown in
Figure 2A, containing two to six mutations, with varying levels of hydrophobicity. The soluble expression and SAP scores are shown in
Figure 2B and
Figure 2D, respectively. While two of the clones (B6.1 and B6.4) did not show any improvement in soluble expression, the other four clones (B6.2, B6.3, B6.5, and B6.6) had remarkable increases in yields (2× to 8×) and were expressed at high enough levels for further characterization. This is remarkable considering the high expression was achieved with just a few select mutations. Interestingly, we were also able to establish a strong correlation (R
2 = 0.98) between the SAP scores and the soluble expression of the VHH clones (
Figure 2E), except for clone B6.4, which implies there might be additional factors at play, such as protein folding. Nevertheless, by introducing a few hydrophilic mutations at the hydrophobic hotspots, we were able to significantly increase the expression of the VHH. Past theoretical studies focused on surface hydrophobicity at the nanoscale have predicted an asymmetric effect of large hydrophobicity changes from small hydrophilic perturbations, which are consistent with our observations here [
33].
Alternatively, following a different approach, we performed one round of random mutagenesis on F4.0 using phage display, and identified three clones (F4.1, F4.2, and F4.3) that either improved or maintained the affinity to the target (K-Ras). Phage ELISA for these clones shown in
supplemental info (Figure S2) suggest moderate increases in binding for the newly identified clones. While clones F4.1 and F4.2 had single mutations, I55N and G109D, F4.3 had two mutations, W71R and R125P (
Figure 3A,B). We determined the expression of the new clones and found a nominal increase in the expression level for the F4.2 and F4.3 clones (
Figure 3C). We modeled these clones and found that the SAP scores improved significantly for the best expressing clone (F4.3). Interestingly, this improvement is facilitated by a single amino acid hydrophilic mutation (W71R) in the middle of a hydrophobic patch, as highlighted in
Figure 3B (F4.0 vs. F4.3). We modeled three novel clones, F4.4, F4.5, and F4.6 (sequences shown in
Figure 3A), that combined the mutations from F4.1, F4.2, and F4.3. We found significant reductions in SAP scores for the novel clones, with F4.6 showing the largest decrease in surface hydrophobicity. We expressed all the novel clones and found that all of them had significant increases in expression levels, consistent with the SAP scores. Remarkably, F4.6 had a 40× increase compared to F4.3 (
Figure 3E). The surface maps of the clones colored by SAP scores clearly show elimination of the hydrophobic patches by addition of hydrophilic side chain amino acids. Due to the increased levels of expression from both these approaches, we were able to further characterize these clones on soluble ELISA and Octet.
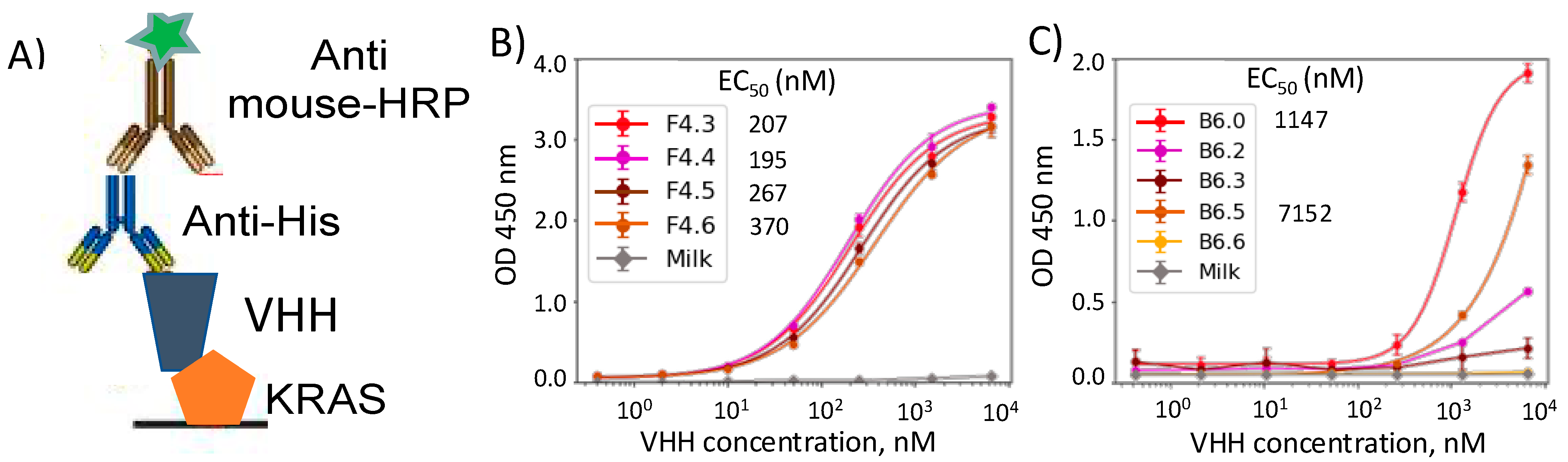
The moderate to high expressing clones (five B6 and four F4 clones) were produced in 500 mL cultures. First, we performed Enzyme Linked ImmunoSorbent Assay (ELISA) of the clones in their soluble form against the target (
Figure 4). We adsorbed K-Ras G12D onto a surface and incubated them with VHHs overnight. His tags on the VHHs which were added for purification, were also used in their recognition with anti-His antibodies. While all the F4 clones showed similar affinity towards the target in the sub μM range, most of the B6 clones (except for B6.5) showed decreased binding (
Figure 4B,C). This behavior clearly highlights the shortcomings of our first approach of performing hydrophilic mutations and indicates the need for random mutagenesis to identify potential soluble enhancing mutations. We chose B6.0, B6.5, F4.3, and F4.6 for further analysis to quantify the binding affinity.
To quantify the strength of the interactions between the VHHs and K-Ras G12D, we performed binding assays on the Octet instrument and quantified the affinity of select clones to the target. Briefly, we loaded K-Ras G12D on the sensor tip using random amine chemistry to achieve a varied number of orientations of the antigen on the surface. The VHH was then introduced at various concentrations, and binding of the VHH to the surface was then monitored over 300 s, followed by a buffer wash over 600 s with the running buffer (referenced in the methods section), where the unbinding kinetics is monitored. The curves thus obtained at various concentrations were fit to Langmuir isotherms using a 1:1 binding model, to arrive at rate constants and the equilibrium dissociation constant, K
D shown in
Table 1. Raw sensogram data are shown in the
supplemental info (Figure S3).
Consistent with the ELISA results, we saw stronger binder affinity (as indicated by K
D) for the F4 clones compared to the B6 clones. EC
50 calculated from the ELISA curves are in qualitative agreement with their corresponding K
D counterparts (
Table S1). While B6.5 showed a μM range affinity to the target, the F4.6 clone possessed an impressive sub 100 nM affinity, which is an improvement from its parent clone (F4.3). The kinetic constants revealed weaker association of both B6.5 and F4.6 compared to their respective parent clones as expected from the hydrophilic nature of the mutations, which might increase the tendency of the protein to remain in an unbound state. However, while the dissociation kinetic constant is also weaker for the B6.5 clone, the F4.6 clone gains significant interactions with the target and remains strongly bound. Therefore, the high expressing F4 clone (F4.6) holds potential to be a K-Ras binder, especially in a multimeric form. We are currently pursuing to produce affinity reagents for labelling K-Ras G12D in cellular imaging.
4. Discussion and Conclusions
In this work, we tested a novel computationally guided approach to improve soluble expression of VHH binders. Our work was motivated by poor expression of VHH binders selected from a naïve phage library screened against our target (K-Ras). Similar observations have been made by other researchers, who have proposed ways to improve the stability and expression of VHH binders [
34]. We hypothesized that the high surface hydrophobicity of the VHHs screened in our study contributes towards poor solubility, leading to poor soluble expression. To support this hypothesis, we adapted earlier work by Trout and coworkers [
30], in which they developed a computational molecular measure of surface hydrophobicity called spatial aggregation propensity (SAP). Through modeling and simulations, we were able to produce SAP maps and average SAP scores, which not only showed high SAP scores for poorly expressing clones but also established a strong correlation of surface hydrophobicity with levels of soluble expression. To our knowledge, this is the first study to establish a correlation between SAP scores and expression levels of VHH binders.
Having established this correlation, we set out to improve the soluble expression of these binders that suffer from poor expression. Based on previous studies of hydrophobicity at the molecular scale, hydrophobic hotspots can be neutralized by single amino acid mutations [
33]. Therefore, we hypothesized that introducing a few surface mutations would result in significantly lower SAP scores. We followed two different approaches to generate mutations. In the first approach, we tested a few hydrophilic mutations that are predicted to improve SAP scores. This approach is purely computational, and the mutations were arbitrarily chosen to be charged/polar. As we expected, most of these clones had higher expression, showing a strong correlation between SAP scores and expression levels. While this approach showed promise, most of the high expressing clones also lost their binding affinity to their target when they were tested on ELISA. Even though the mutation sites were carefully chosen not to be in the CDR regions, we suspect that due to the small size of the VHH, the regions proximal to the CDR regions are also involved in its binding to the target. Even for the B6.5 clone, which had comparable binding on ELISA, the equilibration dissociation constant was significantly higher than the parent clone (B6.0), indicating the loss of binding affinity.
In an alternate approach, we performed a round of random mutagenesis on phage display and selected a few clones (with one or two mutations) that showed comparable binding to the target. Some of these clones showed marginal increases in expression levels and provided us with a list of mutations that could be combined computationally and tested. In doing so, we predicted that the clone that combined all four mutations would have the highest expression level. Remarkably, the soluble expression was completely in agreement with the predictions. Importantly, these clones showed no loss in binding affinity to the target on ELISA and even showed stronger binding, indicated by the lower equilibration dissociation constant.
While we performed the two different approaches on the two different clones, we have been able to point out the possible shortcomings of the targeted hydrophilic mutation approach (the former) and the corresponding advantage of the random mutagenesis approach (the latter), which is underpinned by the significant role that a few amino acid changes can have on the affinity of the small sized VHH binder. However, a comprehensive understanding of the success rate of the two different approaches needs a high-throughput assay on multiple clones, and that will be part of our future studies. Nevertheless, the current work highlights the impact of small changes in surface chemistry on surface hydrophobicity and the consequent solubility of the binders, and most interestingly, the ability of molecular models in predicting those changes.
In conclusion, we presented a hybrid approach that combined random mutagenesis with computational predictions to improve the soluble expression of VHH binders. Even though similar studies have been conducted on antibodies (IgG) [
30], this is the first application of such an approach on VHH binders. Our study shows that VHH binders need this hybrid approach to arrive at solubility enhancing mutations due to their smaller size compared to IgGs. Even a single to a few amino acid mutations around the CDR regions, while improving expression levels, might lead to loss of binding and hence need to be chosen carefully through random mutagenesis.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}